
Natural Product Isoliquiritigenin Activates GABAB Receptors to Decrease Voltage-Gate Ca2+ Channels and Glutamate Release in Rat Cerebrocortical Nerve Terminals

 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drugs
2.2. Animals
2.3. Preparation of Synaptosomes
2.4. Glutamate Release
2.5. Cytosolic Free Ca2+ Concentration ([Ca2+]C)
2.6. Immunohistochemistry
2.7. Molecular Docking Study
2.8. Western Blotting
2.9. Statistical Analysis
3. Results
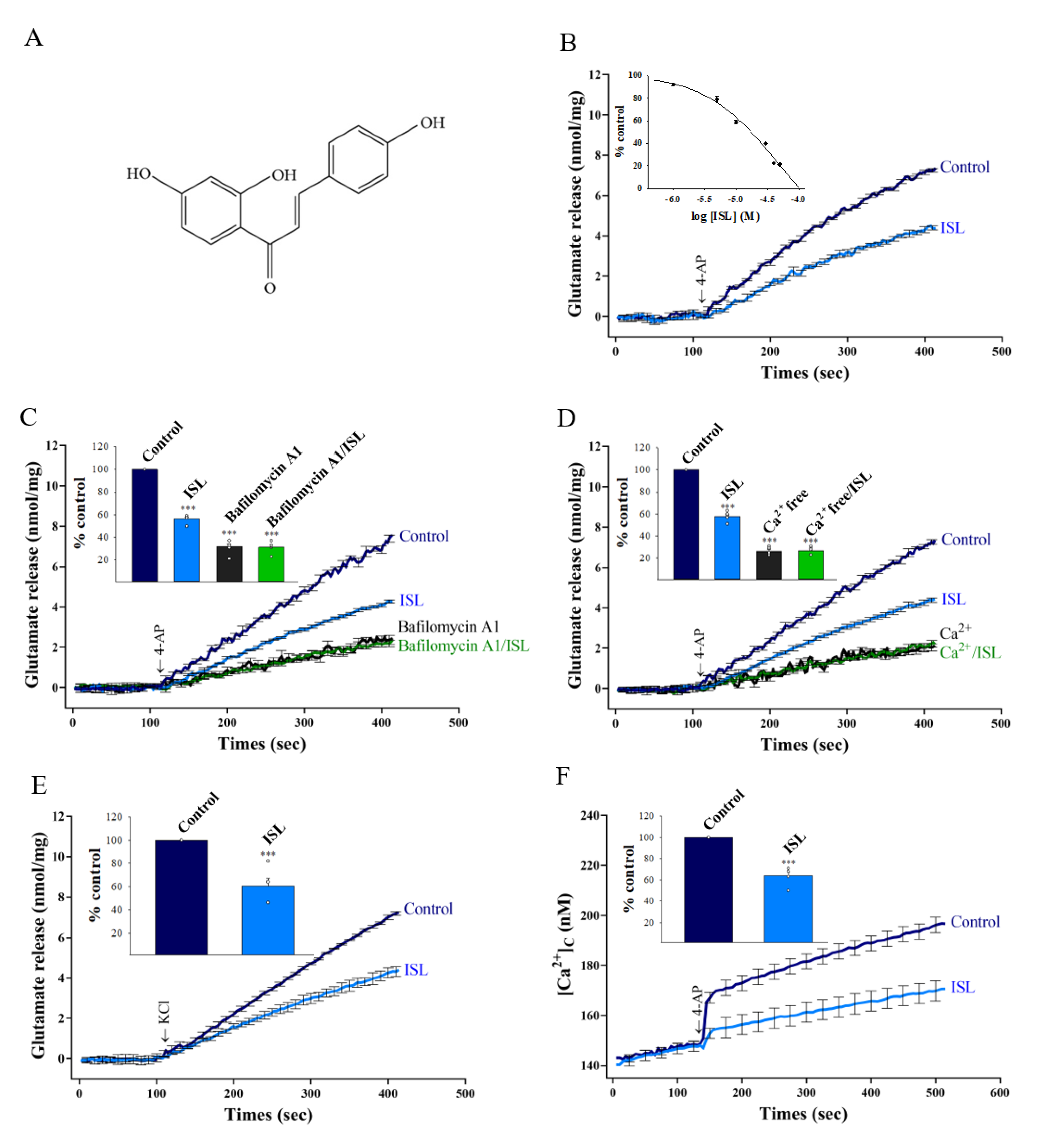
3.1. ISL Inhibits 4-AP-evoked Glutamate Release from Nerve Terminals in the Cerebral Cortex through Ca2+ Influx Reduction
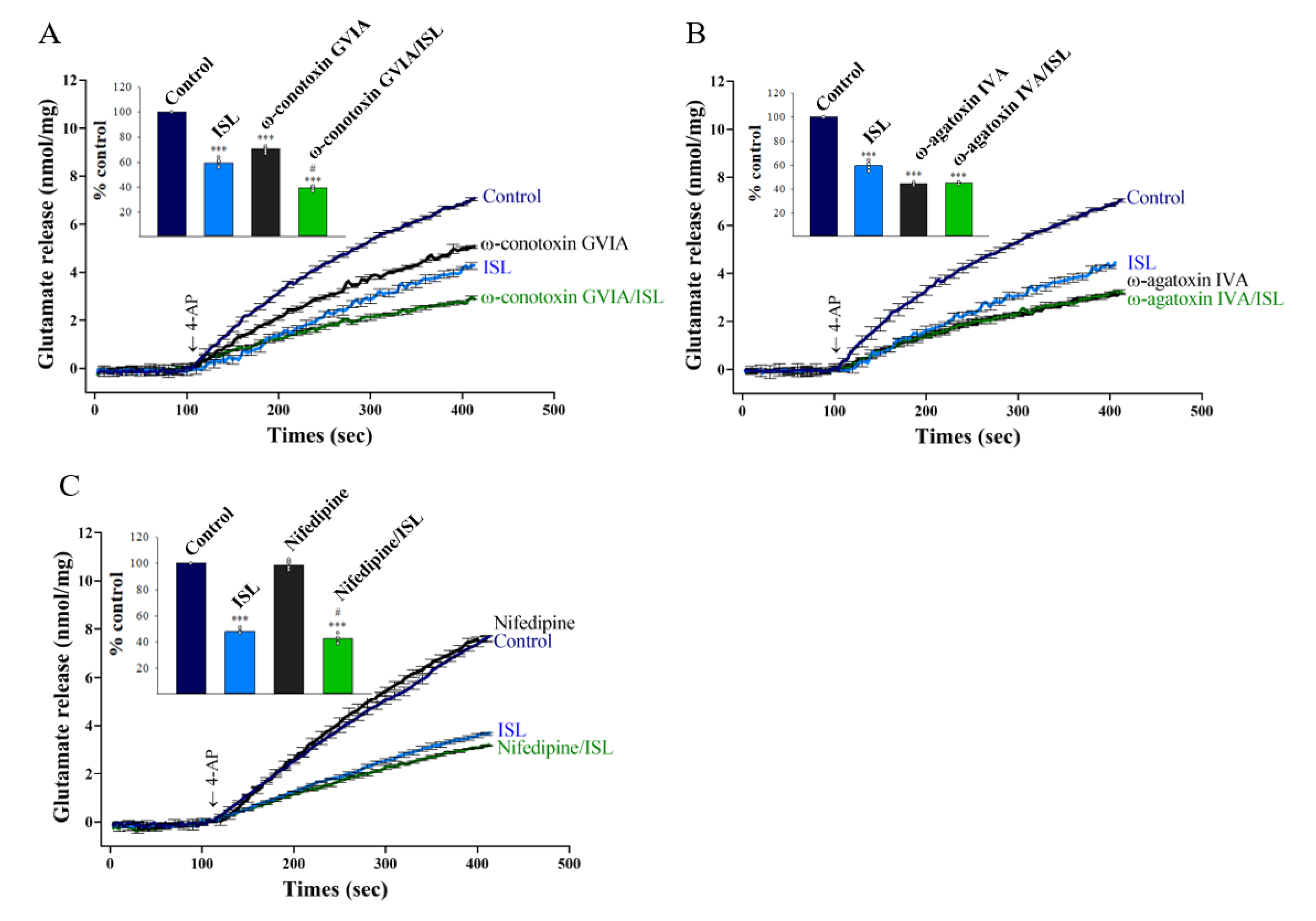
3.2. Specifics of VGCCs Involved in ISL Inhibition of Glutamate Release
3.3. Protein Kinase C Suppression Is Involved in the ISL-mediated Inhibition of Glutamate Release
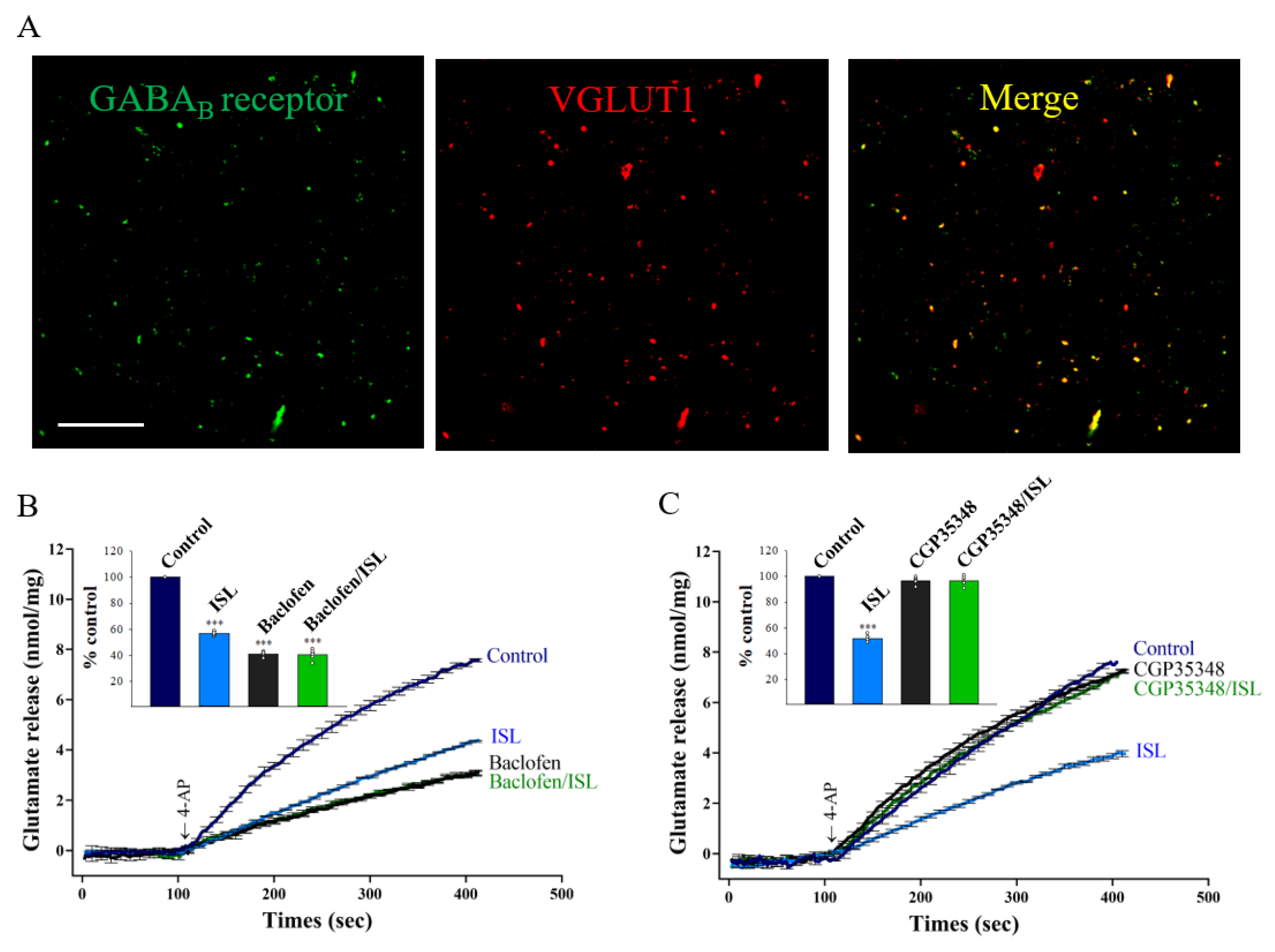
3.4. Presynaptic GABAB Receptors Mediate ISL’s Effect on Glutamate Release
3.5. ISL Interacts with the GABAB Receptors
3.6. Gβγ-coupling Mechanism Is Involved in the ISL-mediated Inhibition of Glutamate Release
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peng, F.; Du, Q.; Peng, C.; Wang, N.; Tang, H.; Xie, X.; Shen, J.; Chen, J. A Review: The Pharmacology of Isoliquiritigenin. Phytother. Res. 2015, 29, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, M.; Kim, H.; Lee, Y.; Lee, Y.I. Phytochemical and Pharmacological Role of Liquiritigenin and Isoliquiritigenin From Radix Glycyrrhizae in Human Health and Disease Models. Front. Aging Neurosci. 2018, 10, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamura, Y.; Hitomi, S.; Omiya, Y.; Ujihara, I.; Kokabu, S.; Morimoto, Y.; Ono, K. Isoliquiritigenin, an active ingredient of Glycyrrhiza, elicits antinociceptive effects via inhibition of Nav channels. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ye, M.; Zhang, Y.; Huang, M.; Xu, W.; Chu, K.; Chen, L.; Que, J. Blood-brain barrier permeability of Gualou Guizhi granules and neuroprotective effects in ischemia/reperfusion injury. Mol. Med. Rep. 2015, 12, 1272–1278. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Xiong, X.; Sui, Y. Isoliquiritigenin Attenuates Neuroinflammation in Traumatic Brain Injury in Young Rats. Neuroimmunomodulation 2019, 26, 102–110. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, J.; Chen, S.; Xue, J.; Huang, S.; Wang, Y.; Chen, O. Isoliquiritigenin attenuates lipopolysaccharide-induced cognitive impairment through antioxidant and anti-inflammatory activity. BMC Neurosci. 2019, 20, 41. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Liu, J.; Huang, S.; Zhu, W.; Wang, Y.; Chen, O.; Xue, J. Neuroprotective effects of isoliquiritigenin against cognitive impairment via suppression of synaptic dysfunction, neuronal injury, and neuroinflammation in rats with kainic acid-induced seizures. Int. Immunopharmacol. 2019, 72, 358–366. [Google Scholar] [CrossRef]
- Shi, D.; Yang, J.; Jiang, Y.; Wen, L.; Wang, Z.; Yang, B. The antioxidant activity and neuroprotective mechanism of isoliquiritigenin. Free Radic. Biol. Med. 2020, 152, 207–215. [Google Scholar] [CrossRef]
- Yang, E.J.; Min, J.S.; Ku, H.Y.; Choi, H.S.; Park, M.K.; Kim, M.K.; Song, K.S.; Lee, D.S. Isoliquiritigenin isolated from Glycyrrhiza uralensis protects neuronal cells against glutamate-induced mitochondrial dysfunction. Biochem. Biophys. Res. Commun. 2012, 421, 658–664. [Google Scholar] [CrossRef]
- Selvaraj, B.; Kim, D.W.; Huh, G.; Lee, H.; Kang, K.; Lee, J.W. Synthesis and biological evaluation of isoliquiritigenin derivatives as a neuroprotective agent against glutamate mediated neurotoxicity in HT22 cells. Bioorg. Med. Chem. Lett. 2020, 30, 127058. [Google Scholar] [CrossRef]
- Zeng, J.; Chen, Y.; Ding, R.; Feng, L.; Fu, Z.; Yang, S.; Deng, X.; Xie, Z.; Zheng, S. Isoliquiritigenin alleviates early brain injury after experimental intracerebral hemorrhage via suppressing ROS- and/or NF-κB-mediated NLRP3 inflammasome activation by promoting Nrf2 antioxidant pathway. J. Neuroinflamm. 2017, 14, 119. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Jia, J. Isoliquiritigenin Confers Neuroprotection and Alleviates Amyloid-β42-Induced Neuroinflammation in Microglia by Regulating the Nrf2/NF-κB Signaling. Front. Neurosci. 2021, 15, 638772. [Google Scholar] [CrossRef] [PubMed]
- Headley, P.M.; Grillner, S. Excitatory amino acids and synaptic transmission: The evidence for a physiological function. Trends Pharmacol. Sci. 1990, 11, 205–211. [Google Scholar] [CrossRef]
- Javitt, D.C. Glutamate as a therapeutic target in psychiatric disorders. Mol. Psychiatry 2004, 9, 984–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meldrum, B.; Garthwaite, J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol. Sci. 1990, 11, 379–387. [Google Scholar] [CrossRef]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, J.C.; Egea, J.; Del Carmen Godino, M.; Fernandez-Gomez, F.J.; Sánchez-Prieto, J.; Gandía, L.; García, A.G.; Jordán, J.; Hernández-Guijo, J.M. Neuroprotectant minocycline depresses glutamatergic neurotransmission and Ca2+ signalling in hippocampal neurons. Eur. J. Neurosci. 2007, 26, 2481–2495. [Google Scholar] [CrossRef] [PubMed]
- Lazarevic, V.; Yang, Y.; Ivanova, D.; Fejtova, A.; Svenningsson, P. Riluzole attenuates the efficacy of glutamatergic transmission by interfering with the size of the readily releasable neurotransmitter pool. Neuropharmacology 2018, 143, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhao, B.; She, Y.; Song, X. Dexmedetomidine ameliorates lidocaine-induced spinal neurotoxicity via inhibiting glutamate release and the PKC pathway. Neurotoxicology 2018, 69, 77–83. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, S.; Kan, J.; Zhang, J.; Zhou, L.; Huang, Y.; Zhang, Y. Chinese Herbal Medicine Interventions in Neurological Disorder Therapeutics by Regulating Glutamate Signaling. Curr. Neuropharmacol. 2020, 18, 260–276. [Google Scholar] [CrossRef]
- Hsu, S.K.; Hung, C.F.; Yang, H.C.; Weng, J.R.; Wang, S.J. TCD, a triterpenoid isolated from wild bitter gourd, reduces synaptosomal release of glutamate and protects against kainic acid-induced neuronal death. Food Founction 2020, 11, 9858–9867. [Google Scholar] [CrossRef]
- Lu, C.W.; Lin, T.Y.; Chiu, K.M.; Lee, M.Y.; Huang, J.H.; Wang, S.J. Silymarin Inhibits Glutamate Release and Prevents against Kainic Acid-Induced Excitotoxic Injury in Rats. Biomedicines 2020, 8, 486. [Google Scholar] [CrossRef]
- Dar, N.J.; Satti, N.K.; Dutt, P.; Hamid, A.; Ahmad, M. Attenuation of Glutamate-Induced Excitotoxicity by Withanolide-A in Neuron-Like Cells: Role for PI3K/Akt/MAPK Signaling Pathway. Mol. Neurobiol. 2018, 55, 2725–2739. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Sihra, T.S. Synaptosomes possess an exocytotic pool of glutamate. Nature 1986, 321, 772–773. [Google Scholar] [CrossRef]
- Lu, C.W.; Lin, T.Y.; Huang, S.K.; Wang, S.J. Echinacoside Inhibits Glutamate Release by Suppressing Voltage-Dependent Ca2+ Entry and Protein Kinase C in Rat Cerebrocortical Nerve Terminals. Int. J. Mol. Sci. 2016, 17, 1006. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.C.; Hsieh, P.W.; Kuo, J.R.; Wang, S.J. Rosmarinic Acid, a Bioactive Phenolic Compound, Inhibits Glutamate Release from Rat Cerebrocortical Synaptosomes through GABA(A) Receptor Activation. Biomolecules 2021, 11, 1029. [Google Scholar] [CrossRef]
- Sihra, T.S.; Bogonez, E.; Nicholls, D.G. Localized Ca2+ entry preferentially effects protein dephosphorylation, phosphorylation, and glutamate release. J. Biol. Chem. 1992, 267, 1983–1989. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]
- Wang, C.C.; Kuo, J.R.; Wang, S.J. Fingolimod inhibits glutamate release through activation of S1P1 receptors and the G protein βγ subunit-dependent pathway in rat cerebrocortical nerve terminals. Neuropharmacology 2021, 185, 108451. [Google Scholar] [CrossRef]
- Wang, C.C.; Kuo, J.R.; Huang, S.K.; Wang, S.J. Metabotropic glutamate 7 receptor agonist AMN082 inhibits glutamate release in rat cerebral cortex nerve terminal. Eur. J. Pharmacol. 2018, 823, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G. Presynaptic modulation of glutamate release. Prog. Brain Res. 1998, 116, 15–22. [Google Scholar] [CrossRef]
- Barrie, A.P.; Nicholls, D.G.; Sanchez-Prieto, J.; Sihra, T.S. An ion channel locus for the protein kinase C potentiation of transmitter glutamate release from guinea pig cerebrocortical synaptosomes. J. Neurochem. 1991, 57, 1398–1404. [Google Scholar] [CrossRef]
- Coffey, E.T.; Sihra, T.S.; Nicholls, D.G.; Pocock, J.M. Phosphorylation of synapsin I and MARCKS in nerve terminals is mediated by Ca2+ entry via an Aga-GI sensitive Ca2+ channel which is coupled to glutamate exocytosis. FEBS Lett. 1994, 353, 264–268. [Google Scholar] [CrossRef] [Green Version]
- Jang, E.Y.; Choe, E.S.; Hwang, M.; Kim, S.C.; Lee, J.R.; Kim, S.G.; Jeon, J.P.; Buono, R.J.; Yang, C.H. Isoliquiritigenin suppresses cocaine-induced extracellular dopamine release in rat brain through GABAB receptor. Eur. J. Pharmacol. 2008, 587, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Perkinton, M.S.; Sihra, T.S. Presynaptic GABAB receptor modulation of glutamate exocytosis from rat cerebrocortical nerve terminals: Receptor decoupling by protein kinase C. J. Neurochem. 1998, 70, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Cortés, H.; Paz, F.; Erlij, D.; Aceves, J.; Florán, B. GABAB receptors modulate depolarization-stimulated [³H] glutamate release in slices of the pars reticulata of the rat substantia nigra. Eur. J. Pharmacol. 2010, 649, 161–167. [Google Scholar] [CrossRef]
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural Products for Drug Discovery in the 21st Century: Innovations for Novel Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.M.; Roy, K.K.; Hockerman, G.H.; Doerksen, R.J.; Colby, D.A. Activation of the γ-Aminobutyric Acid Type B (GABAB) Receptor by Agonists and Positive Allosteric Modulators. J. Med. Chem. 2015, 58, 6336–6347. [Google Scholar] [CrossRef]
- Geng, Y.; Bush, M.; Mosyak, L.; Wang, F.; Fan, Q.R. Structural mechanism of ligand activation in human GABAB receptor. Nature 2013, 504, 254–259. [Google Scholar] [CrossRef] [Green Version]
- Nair, P.C.; McKinnon, R.A.; Miners, J.O.; Bastiampillai, T. Binding of clozapine to the GABAB receptor: Clinical and structural insights. Mol. Psychiatry 2020, 25, 1910–1919. [Google Scholar] [CrossRef]
- Padgett, C.L.; Slesinger, P.A. GABAB receptor coupling to G-proteins and ion channels. Adv. Pharmacol. 2010, 58, 123–147. [Google Scholar] [CrossRef]
- Yamane, H.K.; Fung, B.K. Covalent modifications of G-proteins. Annu. Rev. Pharmacol. Toxicol. 1993, 32, 201–241. [Google Scholar] [CrossRef]
- Turner, T.J.; Dunlap, K. Pharmacological characterization of presynaptic calcium channels using subsecond biochemical measurements of synaptosomal neurosecretion. Neuropharmacology 1995, 34, 1469–1478. [Google Scholar] [CrossRef]
- Vázquez, E.; Sánchez-Prieto, J. Presynaptic modulation of glutamate release targets different calcium channels in rat cerebrocortical nerve terminals. Eur. J. Neurosci. 1997, 9, 2009–2018. [Google Scholar] [CrossRef] [PubMed]
- Millán, C.; Sánchez-Prieto, J. Differential coupling of N- and P/Q-type calcium channels to glutamate exocytosis in the rat cerebral cortex. Neurosci. Lett. 2002, 330, 29–32. [Google Scholar] [CrossRef]
- Wu, L.G.; Saggau, P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997, 20, 204–212. [Google Scholar] [CrossRef]
- Zhu, G.; Ma, S.; Li, X.; Zhang, P.; Tang, L.; Cao, L.; Liu, A.; Sugita, T.; Tomoda, T. The effect of ethanol extract of Glycyrrhiza uralensis on the voltage-gated sodium channel subtype 1.4. J. Pharmacol. Sci. 2018, 136, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, C.; Yang, J.; Sakamoto, K.; Maeda, R.; Takahashi, K.; Takasugi, H.; Ono, T.; Murakawa, M.; Kimura, J. Inhibitory effects of isoliquiritigenin and licorice extract on voltage-dependent K+ currents in H9c2 cells. J. Pharmacol. Sci. 2008, 108, 439–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, D.G.; Sihra, T.S.; Sanchez-Prieto, J. Calcium-dependent and -independent release of glutamate from synaptosomes monitored by continuous fluorometry. J. Neurochem. 1987, 49, 50–57. [Google Scholar] [CrossRef]
- Attwell, D.; Barbour, B.; Szatkowski, M. Nonvesicular release of neurotransmitter. Neuron 1993, 11, 401–407. [Google Scholar] [CrossRef]
- Bettler, B.; Kaupmann, K.; Mosbacher, J.; Gassmann, M. Molecular structure and physiological functions of GABAB receptors. Physiol. Rev. 2004, 84, 835–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.G.; Saggau, P. GABAB receptor-mediated presynaptic inhibition in guinea-pig hippocampus is caused by reduction of presynaptic Ca2+ influx. J. Physiol. 1995, 485, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Dittman, J.S.; Regehr, W.G. Contributions of calcium-dependent and calcium-independent mechanisms to presynaptic inhibition at a cerebellar synapse. J. Neurosci. 1996, 16, 1623–1633. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Kajikawa, Y.; Tsujimoto, T. G-Protein-coupled modulation of presynaptic calcium currents and transmitter release by a GABAB receptor. J. Neurosci. 1998, 18, 3138–3146. [Google Scholar] [CrossRef] [Green Version]
- Kajikawa, Y.; Saitoh, N.; Takahashi, T. GTP-binding protein beta gamma subunits mediate presynaptic calcium current inhibition by GABAB receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 8054–8058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zurawski, Z.; Yim, Y.Y.; Alford, S.; Hamm, H.E. The expanding roles and mechanisms of G protein-mediated presynaptic inhibition. J. Biol. Chem. 2019, 294, 1661–1670. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, J.H.; Thelen, M.; Rosen, A.; Janmey, P.A.; Nairn, A.C.; Aderem, A. MARCKS is an actin filament crosslinking protein regulated by protein kinase C and calcium-calmodulin. Nature 1992, 356, 618–622. [Google Scholar] [CrossRef]
- Barclay, J.W.; Craig, T.J.; Fisher, R.J.; Ciufo, L.F.; Evans, G.J.; Morgan, A.; Burgoyne, R.D. Phosphorylation of Munc18 by protein kinase C regulates the kinetics of exocytosis. J. Biol. Chem. 2003, 278, 10538–10545. [Google Scholar] [CrossRef] [Green Version]
- Morgan, A.; Burgoyne, R.D.; Barclay, J.W.; Craig, T.J.; Prescott, G.R.; Ciufo, L.F.; Evans, G.J.; Graham, M.E. Regulation of exocytosis by protein kinase C. Biochem. Soc. Trans. 2005, 33, 1341–1344. [Google Scholar] [CrossRef]
- Kawakami, Z.; Ikarashi, Y.; Kase, Y. Isoliquiritigenin is a novel NMDA receptor antagonist in kampo medicine yokukansan. Cell. Mol. Neurobiol. 2011, 31, 1203–1212. [Google Scholar] [CrossRef]
- Meldrum, B.S. Glutamate as a neurotransmitter in the brain: Review of physiology and pathology. J. Nutr. 2000, 130, 1007–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.K.; Yang, E.J.; Kim, J.Y.; Song, K.S.; Seong, Y.H. Inhibitory effects of Glycyrrhizae radix and its active component, isoliquiritigenin, on Aβ (25–35)-induced neurotoxicity in cultured rat cortical neurons. Arch. Pharmacal Res. 2012, 35, 897–904. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, T.-Y.; Lu, C.-W.; Hsieh, P.-W.; Chiu, K.-M.; Lee, M.-Y.; Wang, S.-J. Natural Product Isoliquiritigenin Activates GABAB Receptors to Decrease Voltage-Gate Ca2+ Channels and Glutamate Release in Rat Cerebrocortical Nerve Terminals. Biomolecules 2021, 11, 1537. https://doi.org/10.3390/biom11101537
Lin T-Y, Lu C-W, Hsieh P-W, Chiu K-M, Lee M-Y, Wang S-J. Natural Product Isoliquiritigenin Activates GABAB Receptors to Decrease Voltage-Gate Ca2+ Channels and Glutamate Release in Rat Cerebrocortical Nerve Terminals. Biomolecules. 2021; 11(10):1537. https://doi.org/10.3390/biom11101537
Chicago/Turabian StyleLin, Tzu-Yu, Cheng-Wei Lu, Pei-Wen Hsieh, Kuan-Ming Chiu, Ming-Yi Lee, and Su-Jane Wang. 2021. "Natural Product Isoliquiritigenin Activates GABAB Receptors to Decrease Voltage-Gate Ca2+ Channels and Glutamate Release in Rat Cerebrocortical Nerve Terminals" Biomolecules 11, no. 10: 1537. https://doi.org/10.3390/biom11101537