
Alcohol Metabolism Enriches Squamous Cell Carcinoma Cancer Stem Cells That Survive Oxidative Stress via Autophagy

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and 3D Organoid Culture
2.2. EtOH Treatment, Pharmacological and Genetic Modifications of Cells
2.3. Flow Cytometry to Evaluate Cell Surface CD44 Expression, Proliferation, Apoptosis, Mitochondrial Mass, and Mitochondrial Membrane Potential
2.4. Mitochondrial Superoxide Assays
2.5. Quantitative Reverse-Transcription Polymerase Chain Reaction (qRT-PCR)
2.6. Xenograft Transplantation Experiment
2.7. Statistical Analyses
3. Results
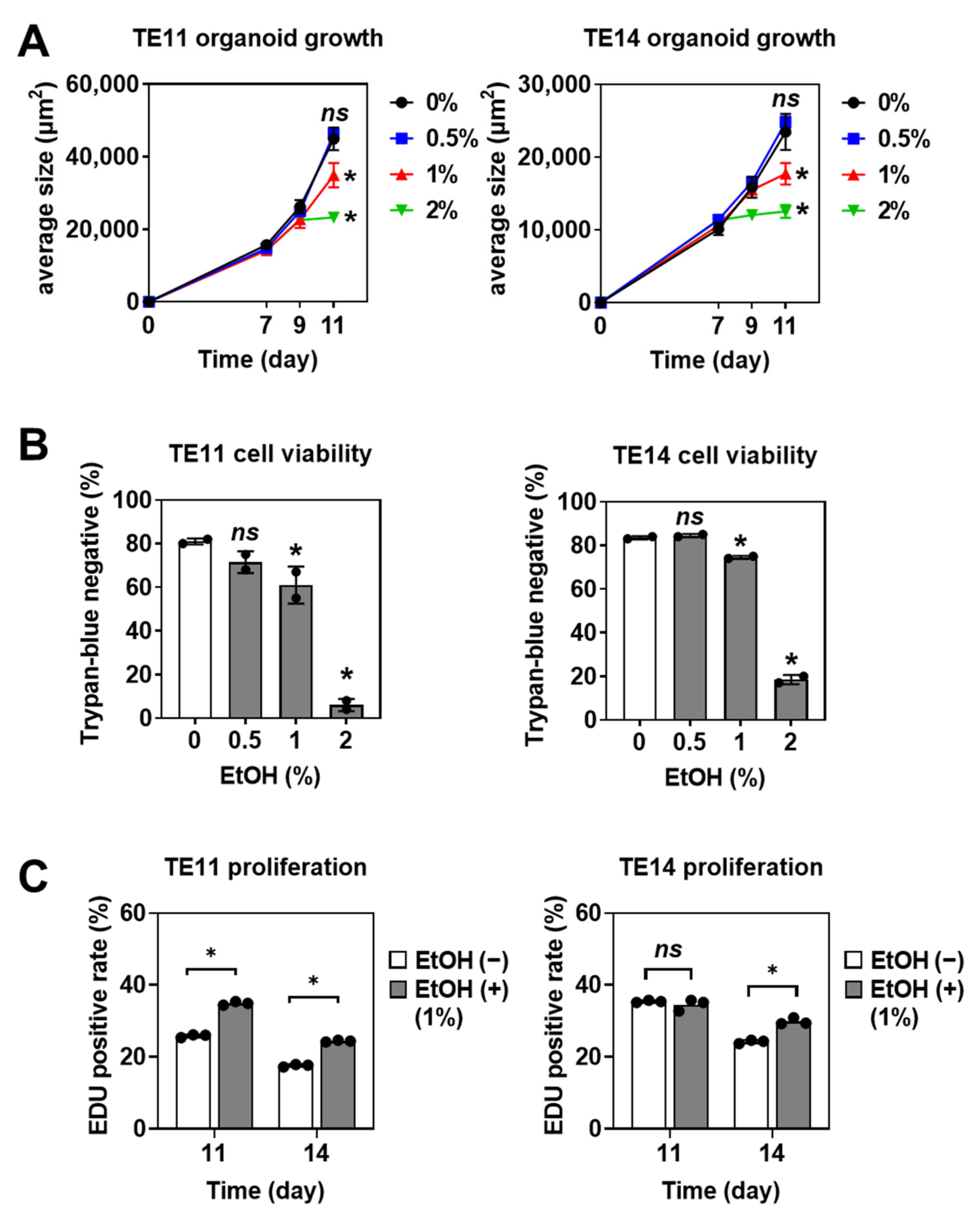
3.1. EtOH Increases the Organoid Formation Capability of SCC Cells
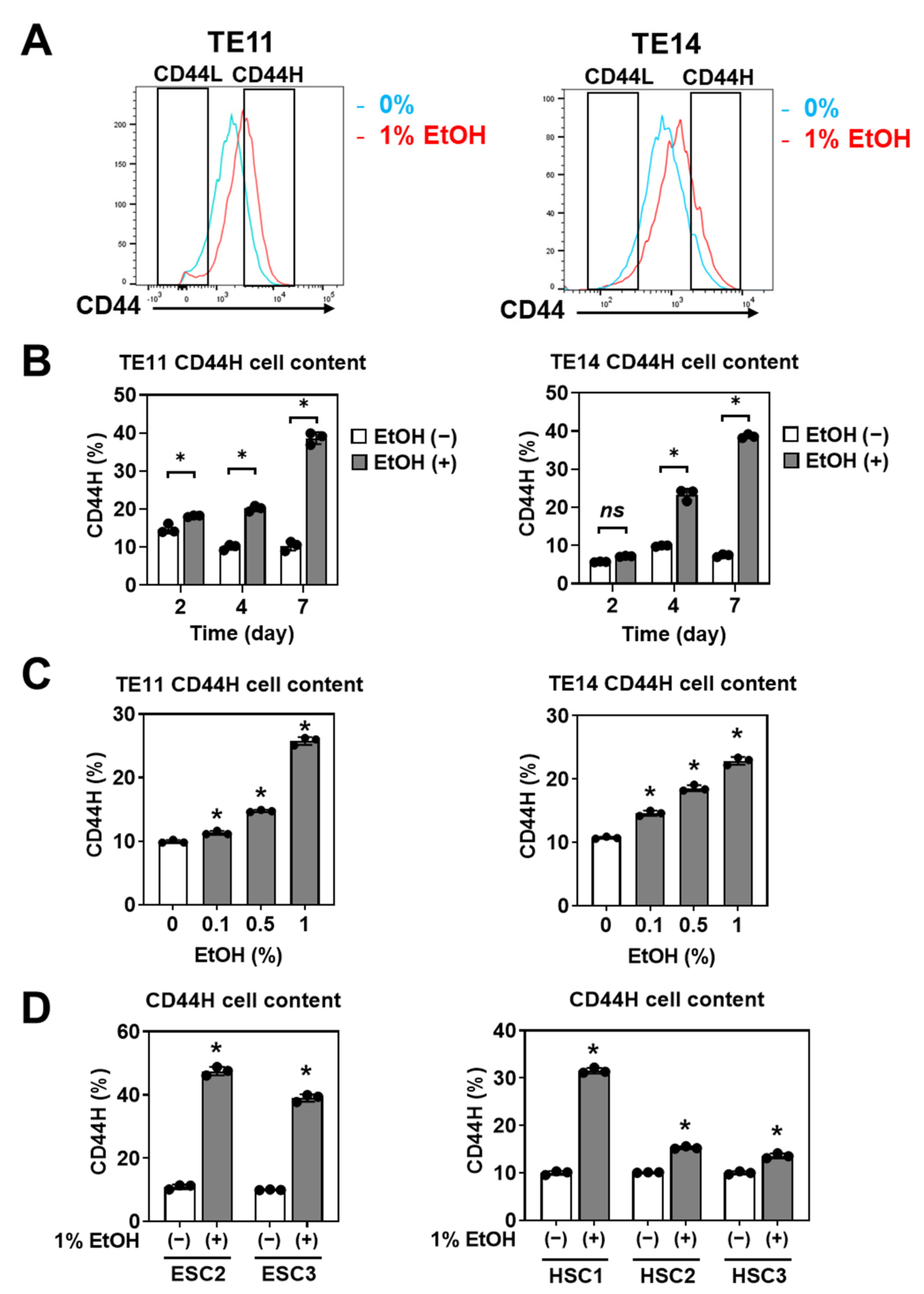
3.2. EtOH Enriches CD44H Cells within Primary SCC Organoids
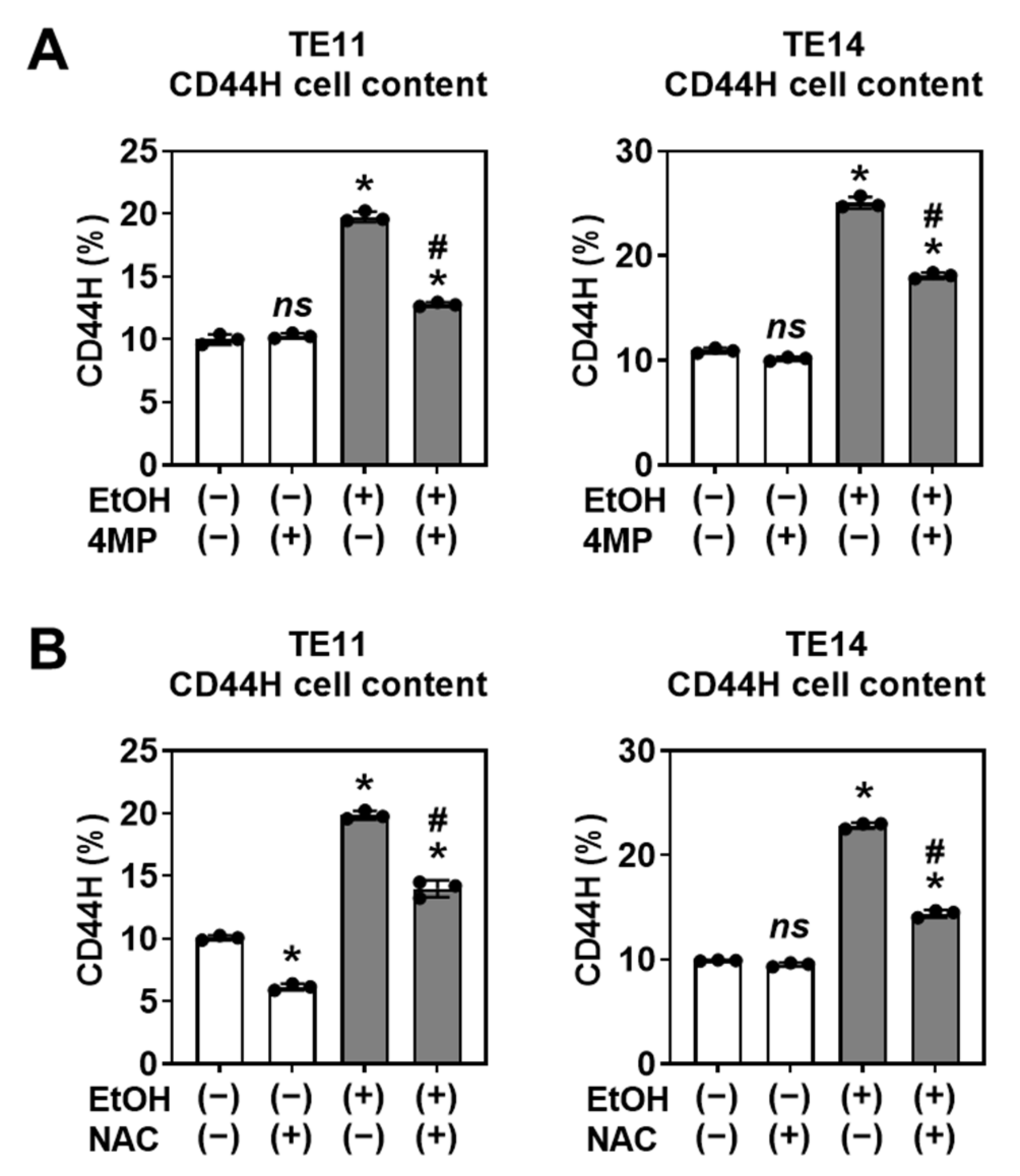
3.3. CD44H Cell Enrichment Involves EtOH Oxidation and Oxidative Stress
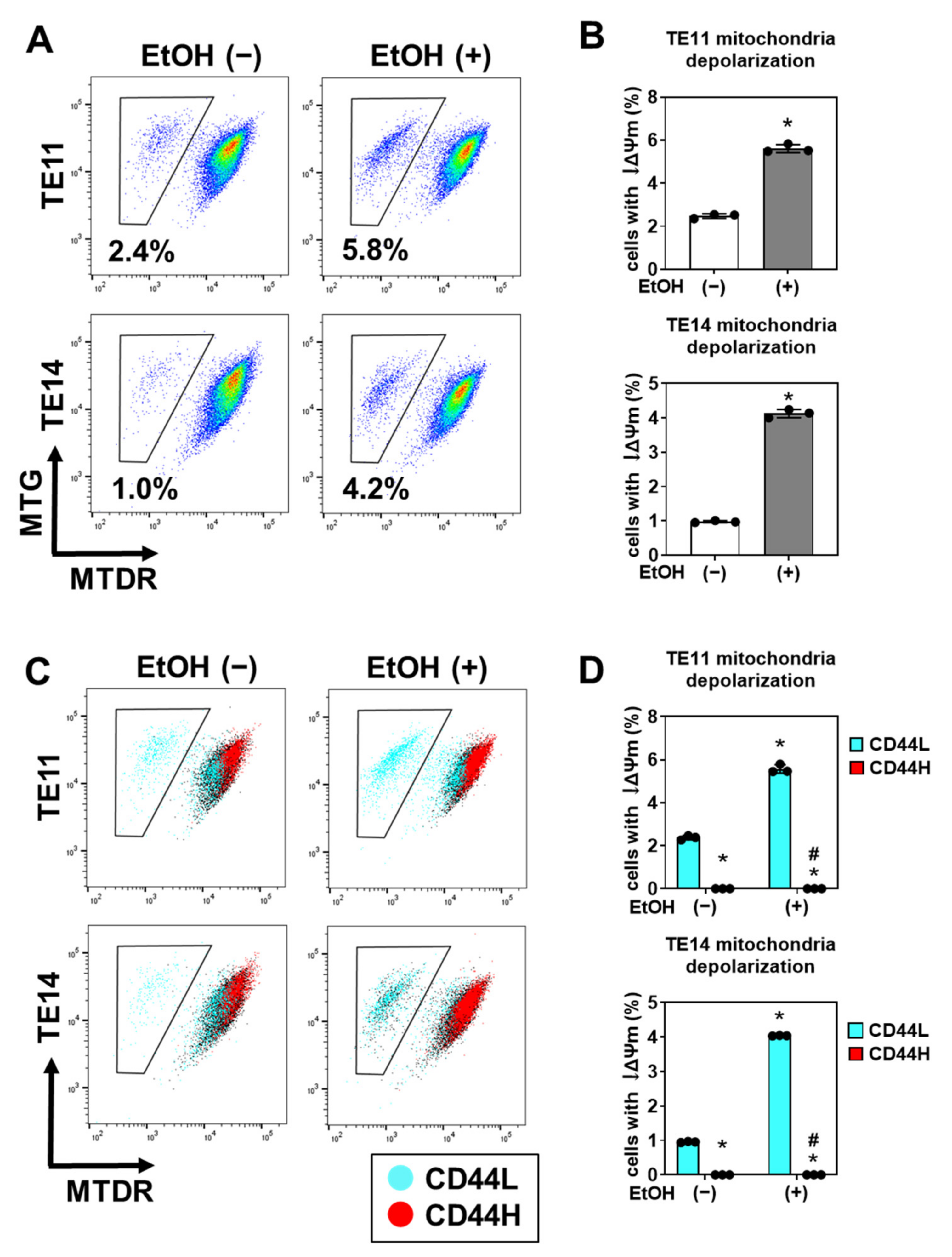
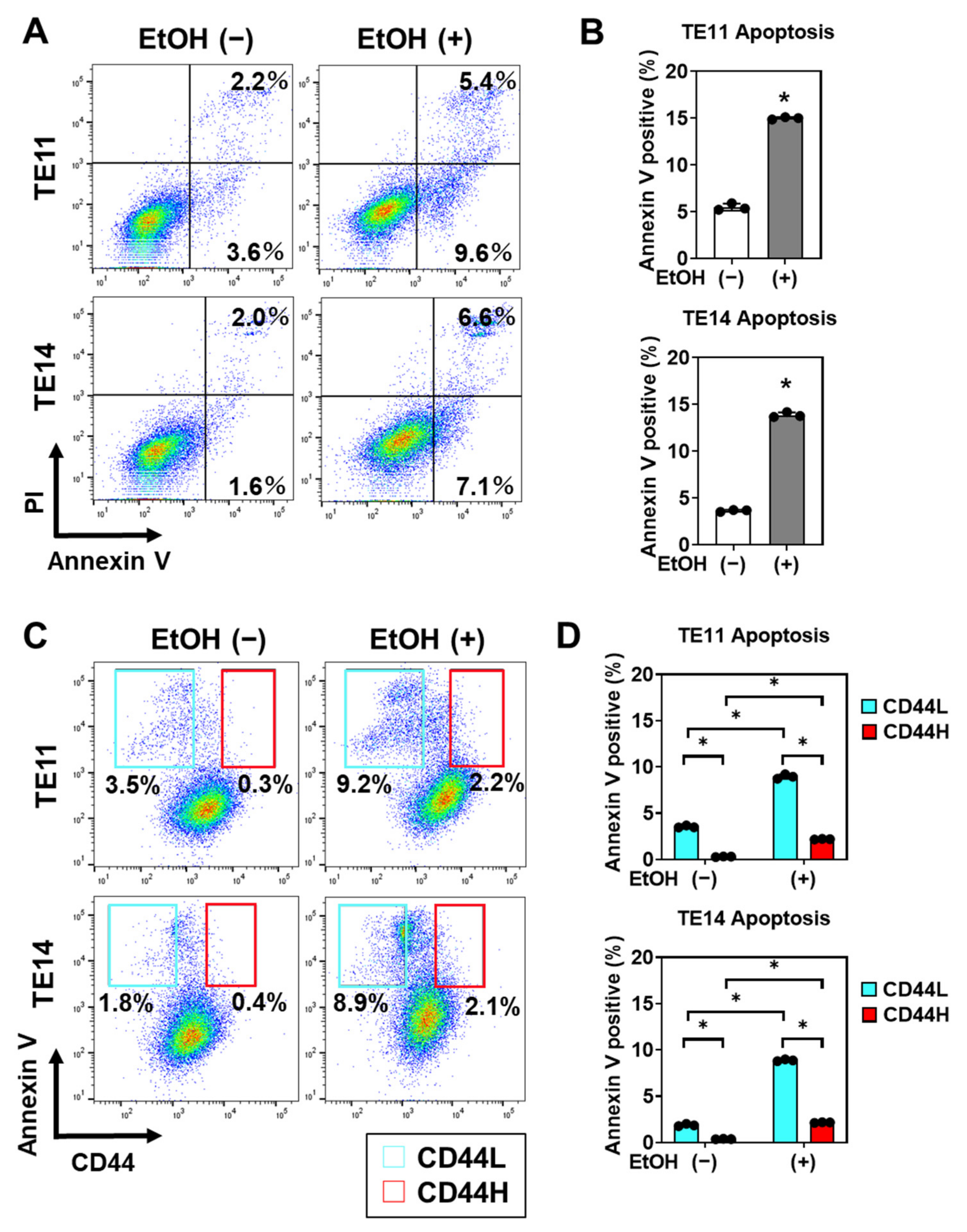
3.4. EtOH-Induced Mitochondrial Dysfunction and Apoptosis Are Limited in CD44H Cells
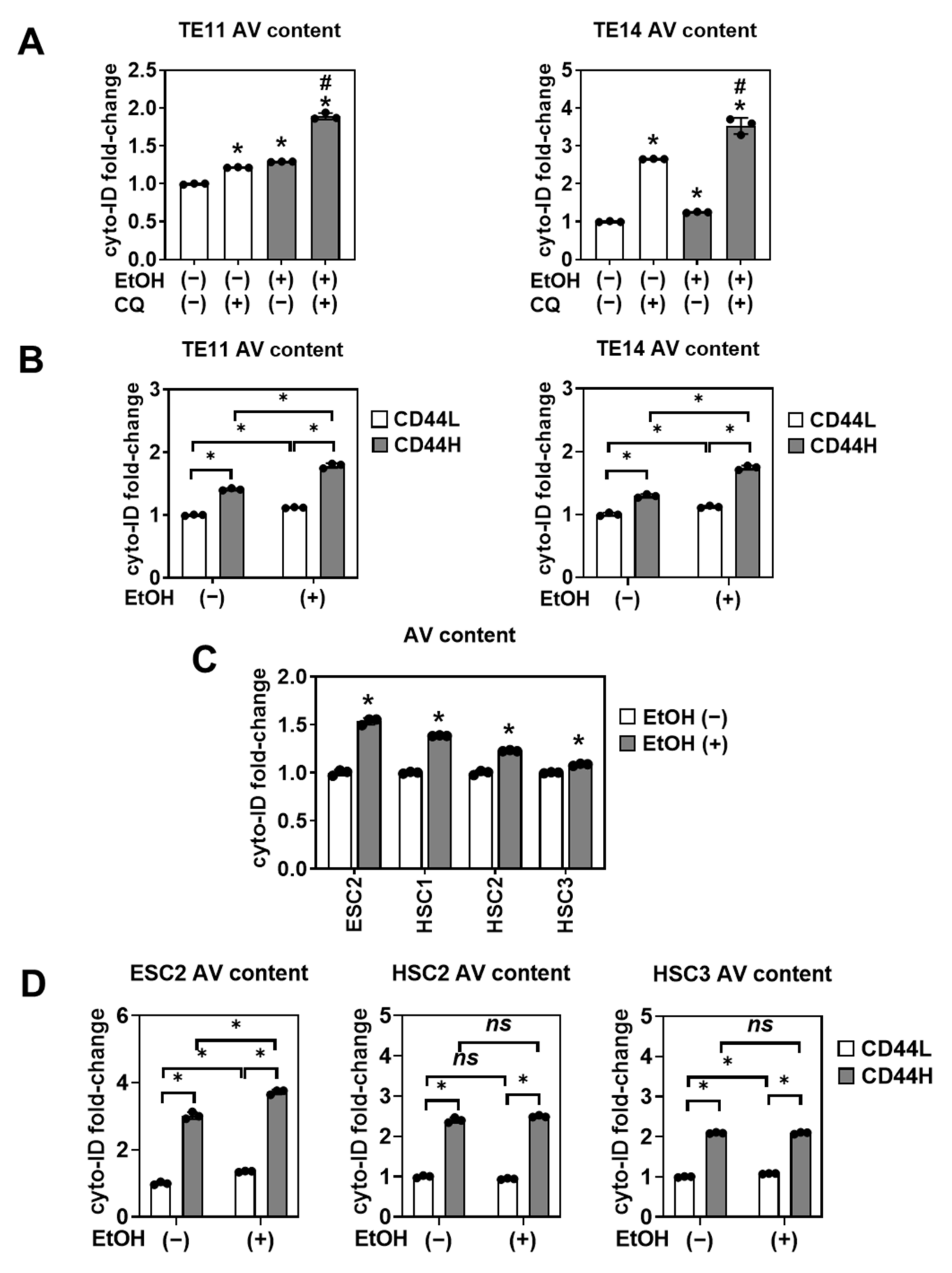
3.5. CD44H Cells Survive EtOH-Induced Oxidative Stress by Autophagy
3.6. Alcohol Drinking Enriches Intratumoral CD44H Cells via Autophagy to Promote Tumor Growth
4. Discussion
4.1. The 3D Organoid and Xenograft Models Shed Light upon the Role of EtOH in Tumor Biology
4.2. 3D Organoids Reveal HNSCC and ESCC CSCs Homeostasis under EtOH Exposure
4.3. Limitations of the 3D Organoid Model to Study Cancer Cell Response to EtOH
4.4. Alcohol Metabolism, Mitochondrial Oxidative Stress, and Autophagy in SCC Cells
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baan, R.; Straif, K.; Grosse, Y.; Lauby-Secretan, B.; El Ghissassi, F.; Bouvard, V.; Altieri, A.; Cogliano, V. Carcinogenicity of alcoholic beverages. Lancet Oncol. 2007, 8, 292–293. [Google Scholar] [CrossRef]
- Bagnardi, V.; Rota, M.; Botteri, E.; Tramacere, I.; Islami, F.; Fedirko, V.; Scotti, L.; Jenab, M.; Turati, F.; Pasquali, E.; et al. Alcohol consumption and site-specific cancer risk: A comprehensive dose–response meta-analysis. Br. J. Cancer 2015, 112, 580–593. [Google Scholar] [CrossRef]
- Matejcic, M.; Gunter, M.J.; Ferrari, P. Alcohol metabolism and oesophageal cancer: A systematic review of the evidence. Carcinogenesis 2017, 38, 859–872. [Google Scholar] [CrossRef]
- Pennathur, A.; Gibson, M.K.; Jobe, B.A.; Luketich, J.D. Oesophageal carcinoma. Lancet 2013, 381, 400–412. [Google Scholar] [CrossRef] [Green Version]
- Rustgi, A.K.; El-Serag, H.B. Esophageal Carcinoma. N. Engl. J. Med. 2014, 371, 2499–2509. [Google Scholar] [CrossRef]
- Fisher, R.S.; Malmud, L.S.; Applegate, G.; Rock, E.; Lorber, S.H. Effect of bolus composition on esophageal transit: Concise communication. J. Nucl. Med. 1982, 23, 878–882. [Google Scholar]
- Jones, A.W.; Sternebring, B. Kinetics of ethanol and methanol in alcoholics during detoxification. Alcohol Alcohol. 1992, 27, 641–647. [Google Scholar] [CrossRef]
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef]
- Amanuma, Y.; Ohashi, S.; Itatani, Y.; Tsurumaki, M.; Matsuda, S.; Kikuchi, O.; Nakai, Y.; Miyamoto, S.; Oyama, T.; Kawamoto, T.; et al. Protective role of ALDH2 against acetaldehyde-derived DNA damage in oesophageal squamous epithelium. Sci. Rep. 2015, 5, 14142. [Google Scholar] [CrossRef] [Green Version]
- Chandramouleeswaran, P.M.; Guha, M.; Shimonosono, M.; Whelan, K.A.; Maekawa, H.; Sachdeva, U.M.; Ruthel, G.; Mukherjee, S.; Engel, N.; Gonzalez, M.V.; et al. Autophagy mitigates ethanol-induced mitochondrial dysfunction and oxidative stress in esophageal keratinocytes. PLoS ONE 2020, 15, e0239625. [Google Scholar] [CrossRef]
- Hao, J.-J.; Lin, D.-C.; Dinh, H.; Mayakonda, A.; Jiang, Y.-Y.; Chang, C.; Jiang, Y.; Lu, C.-C.; Shi, Z.-Z.; Xu, X.; et al. Spatial intratumoral heterogeneity and temporal clonal evolution in esophageal squamous cell carcinoma. Nat. Genet. 2016, 48, 1500–1507. [Google Scholar] [CrossRef]
- Qi, Z.; Barrett, T.; Parikh, A.S.; Tirosh, I.; Puram, S.V. Single-cell sequencing and its applications in head and neck cancer. Oral Oncol. 2019, 99, 104441. [Google Scholar] [CrossRef]
- Natsuizaka, M.; Whelan, K.A.; Kagawa, S.; Tanaka, K.; Giroux, V.; Chandramouleeswaran, P.M.; Long, A.; Sahu, V.; Darling, D.S.; Veronique, G.; et al. Interplay between Notch1 and Notch3 promotes EMT and tumor initiation in squamous cell carcinoma. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.-S.; Li, W.-J.; Ge, D.; Zhang, P.; Li, J.-J.; Lu, C.-L.; Ji, X.-D.; Guan, D.-X.; Gao, H.; Xu, L.-Y.; et al. Tumor Initiating Cells in Esophageal Squamous Cell Carcinomas Express High Levels of CD44. PLoS ONE 2011, 6, e21419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelan, K.A.; Chandramouleeswaran, P.M.; Tanaka, K.; Natsuizaka, M.; Guha, M.; Srinivasan, S.; Darling, D.; Kita, Y.; Natsugoe, S.; Winkler, J.D.; et al. Autophagy supports generation of cells with high CD44 expression via modulation of oxidative stress and Parkin-mediated mitochondrial clearance. Oncogene 2017, 36, 4843–4858. [Google Scholar] [CrossRef] [Green Version]
- Kinugasa, H.; Whelan, K.A.; Tanaka, K.; Natsuizaka, M.; Long, A.; Guo, A.; Chang, S.; Kagawa, S.; Srinivasan, S.; Guha, M.; et al. Mitochondrial SOD2 regulates epithelial–mesenchymal transition and cell populations defined by differential CD44 expression. Oncogene 2015, 34, 5229–5239. [Google Scholar] [CrossRef] [Green Version]
- Maehara, O.; Suda, G.; Natsuizaka, M.; Ohnishi, S.; Komatsu, Y.; Sato, F.; Nakai, M.; Sho, T.; Morikawa, K.; Ogawa, K.; et al. Fibroblast growth factor-2–mediated FGFR/Erk signaling supports maintenance of cancer stem-like cells in esophageal squamous cell carcinoma. Carcinogenesis 2017, 38, 1073–1083. [Google Scholar] [CrossRef] [Green Version]
- Sato, F.; Kubota, Y.; Natsuizaka, M.; Maehara, O.; Hatanaka, Y.; Marukawa, K.; Terashita, K.; Suda, G.; Ohnishi, S.; Shimizu, Y.; et al. EGFR inhibitors prevent induction of cancer stem-like cells in esophageal squamous cell carcinoma by suppressing epithelial-mesenchymal transition. Cancer Biol. Ther. 2015, 16, 933–940. [Google Scholar] [CrossRef]
- Natsuizaka, M.; Kinugasa, H.; Kagawa, S.; Whelan, K.A.; Naganuma, S.; Subramanian, H.; Chang, S.; Nakagawa, K.J.; Rustgi, N.L.; Kita, Y.; et al. IGFBP3 promotes esophageal cancer growth by suppressing oxidative stress in hypoxic tumor microenvironment. Am. J. Cancer Res. 2014, 4, 29–41. [Google Scholar]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, M.; Ke, Z.-J.; Luo, J. Cellular and molecular mechanisms underlying alcohol-induced aggressiveness of breast cancer. Pharmacol. Res. 2016, 115, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-S.; Lee, Y.-R.; Chen, C.-S.; Tu, S.-H.; Wang, Y.-J.; Lee, C.-H.; Chen, L.-C.; Chang, H.-W.; Chang, C.-H.; Chih-Ming, S.; et al. Long-term ethanol exposure causes human liver cancer cells to become resistant to mitomycin C treatment through the inactivation of bad-mediated apoptosis. Mol. Carcinog. 2010, 49, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Kijima, T.; Nakagawa, H.; Shimonosono, M.; Chandramouleeswaran, P.M.; Hara, T.; Sahu, V.; Kasagi, Y.; Kikuchi, O.; Tanaka, K.; Giroux, V.; et al. Three-Dimensional Organoids Reveal Therapy Resistance of Esophageal and Oropharyngeal Squamous Cell Carcinoma Cells. Cell. Mol. Gastroenterol. Hepatol. 2018, 7, 73–91. [Google Scholar] [CrossRef] [Green Version]
- Karakasheva, T.A.; Kijima, T.; Shimonosono, M.; Maekawa, H.; Sahu, V.; Gabre, J.T.; Cruz-Acuña, R.; Giroux, V.; Sangwan, V.; Whelan, K.A.; et al. Generation and Characterization of Patient-Derived Head and Neck, Oral, and Esophageal Cancer Organoids. Curr. Protoc. Stem Cell Biol. 2020, 53, e109. [Google Scholar] [CrossRef] [PubMed]
- Nishihira, T.; Hashimoto, Y.; Katayama, M.; Mori, S.; Kuroki, T. Molecular and cellular features of esophageal cancer cells. J. Cancer Res. Clin. Oncol. 1993, 119, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Facompre, N.D.; Rajagopalan, P.; Sahu, V.; Pearson, A.T.; Montone, K.T.; James, C.D.; Gleber-Netto, F.O.; Weinstein, G.S.; Jalaly, J.; Lin, A.; et al. Identifying predictors of HPV -related head and neck squamous cell carcinoma progression and survival through patient-derived models. Int. J. Cancer 2020, 147, 3236–3249. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, S.; Natsuizaka, M.; Whelan, K.A.; Facompre, N.; Naganuma, S.; Ohashi, S.; Kinugasa, H.; Egloff, A.M.; Basu, D.; Gimotty, P.A.; et al. Cellular senescence checkpoint function determines differential Notch1-dependent oncogenic and tumor-suppressor activities. Oncogene 2015, 34, 2347–2359. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Whelan, K.A.; Chandramouleeswaran, P.M.; Kagawa, S.; Rustgi, S.L.; Noguchi, C.; Guha, M.; Srinivasan, S.; Amanuma, Y.; Ohashi, S.; et al. ALDH2 modulates autophagy flux to regulate acetaldehyde-mediated toxicity thresholds. Am. J. Cancer Res. 2016, 6, 781–796. [Google Scholar]
- Whelan, K.A.; Merves, J.F.; Giroux, V.; Tanaka, K.; Guo, A.; Chandramouleeswaran, P.M.; Benitez, A.J.; Dods, K.; Que, J.; Masterson, J.C.; et al. Autophagy mediates epithelial cytoprotection in eosinophilic oesophagitis. Gut 2017, 66, 1197–1207. [Google Scholar] [CrossRef]
- Machida, K.; Tsukamoto, H.; Mkrtchyan, H.; Duan, L.; Dynnyk, A.; Liu, H.M.; Asahina, K.; Govindarajan, S.; Ray, R.; Ou, J.-H.J.; et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc. Natl. Acad. Sci. USA 2009, 106, 1548–1553. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Ren, Z.; Wang, X.; Comer, A.; Frank, J.A.; Ke, Z.-J.; Huang, Y.; Zhang, Z.; Shi, X.; Wang, S.; et al. ErbB2 and p38γ MAPK mediate alcohol-induced increase in breast cancer stem cells and metastasis. Mol. Cancer 2016, 15, 52. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Ma, Y.; Shankar, S.; Srivastava, R.K. Chronic ethanol exposure of human pancreatic normal ductal epithelial cells induces cancer stem cell phenotype through SATB2. J. Cell. Mol. Med. 2018, 22, 3920–3928. [Google Scholar] [CrossRef]
- Nakagawa, H.; Kasagi, Y.; Karakasheva, T.A.; Hara, T.; Aaron, B.; Shimonosono, M.; Kijima, T.; Giroux, V.; Bailey, D.; Wilkins, B.; et al. Modeling Epithelial Homeostasis and Reactive Epithelial Changes in Human and Murine Three-Dimensional Esophageal Organoids. Curr. Protoc. Stem Cell Biol. 2020, 52, e106. [Google Scholar] [CrossRef] [PubMed]
- Whelan, K.A.; Muir, A.B.; Nakagawa, H. Esophageal 3D Culture Systems as Modeling Tools in Esophageal Epithelial Pathobiology and Personalized Medicine. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 461–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadland, S.E.; Levy, S. Objective Testing. Child Adolesc. Psychiatr. Clin. N. Am. 2016, 25, 549–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holloway, R.H.; Lange, R.C.; Plankey, M.W.; McCallum, R.W. Detection of esophageal motor disorders by radionuclide transit studies. Dig. Dis. Sci. 1989, 34, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, F.; Hesse, B.; Tromholt, N.; Højgaard, L.; Stubgaard, M. Esophageal scintigraphy: Reproducibility and normal ranges. J. Nucl. Med. 1992, 33, 2106–2109. [Google Scholar]
- Maurer, A.H. Gastrointestinal Motility, Part 1: Esophageal Transit and Gastric Emptying. J. Nucl. Med. Technol. 2016, 44, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Ni, F.; Xu, M.; Yang, J.; Chen, J.; Chen, Z.; Wang, X.; Luo, J.; Wang, S. Alcohol promotes mammary tumor growth through activation of VEGF-dependent tumor angiogenesis. Oncol. Lett. 2014, 8, 673–678. [Google Scholar] [CrossRef] [Green Version]
- Krishnamachary, B.; Penet, M.-F.; Nimmagadda, S.; Mironchik, Y.; Raman, V.; Solaiyappan, M.; Semenza, G.L.; Pomper, M.G.; Bhujwalla, Z.M. Hypoxia Regulates CD44 and Its Variant Isoforms through HIF-1α in Triple Negative Breast Cancer. PLoS ONE 2012, 7, e44078. [Google Scholar] [CrossRef]
- Liang, G.; Li, S.; Du, W.; Ke, Q.; Cai, J.; Yang, J. Hypoxia regulates CD44 expression via hypoxia-inducible factor-1α in human gastric cancer cells. Oncol. Lett. 2017, 13, 967–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, D.; Ma, Y.; Liu, J.; Trope, C.G.; Holm, R.; Nesland, J.M.; Suo, Z. The hypoxic microenvironment upgrades stem-like properties of ovarian cancer cells. BMC Cancer 2012, 12, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natsuizaka, M.; Naganuma, S.; Kagawa, S.; Ohashi, S.; Ahmadi, A.; Subramanian, H.; Chang, S.; Nakagawa, K.J.; Ji, X.; Liebhaber, S.A.; et al. Hypoxia induces IGFBP3 in esophageal squamous cancer cells through HIF-1α-mediated mRNA transcription and continuous protein synthesis. FASEB J. 2012, 26, 2620–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.J.; Natsuizaka, M.; Ohashi, S.; Wong, G.S.; Takaoka, M.; Michaylira, C.Z.; Budo, D.; Tobias, J.W.; Kanai, M.; Shirakawa, Y.; et al. Hypoxia activates the cyclooxygenase-2–prostaglandin E synthase axis. Carcinogenesis 2009, 31, 427–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meadows, G.G.; Zhang, H. Effects of Alcohol on Tumor Growth, Metastasis, Immune Response, and Host Survival. Alcohol Res. Curr. Rev. 2015, 37, 311–322. [Google Scholar]
- Ganne-Carrié, N.; Chaffaut, C.; Bourcier, V.; Archambeaud, I.; Perarnau, J.-M.; Oberti, F.; Roulot, D.; Moreno, C.; Louvet, A.; Dao, T.; et al. Estimate of hepatocellular carcinoma incidence in patients with alcoholic cirrhosis. J. Hepatol. 2018, 69, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-I.; Jang, S.I.; Hong, J.; Kim, C.H.; Kwon, S.S.; Park, J.S.; Lim, J.-B. Cancer-initiating cells in human pancreatic cancer organoids are maintained by interactions with endothelial cells. Cancer Lett. 2021, 498, 42–53. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.A.; Goyal, A.; Terry, M.B. Alcohol Intake and Breast Cancer Risk: Weighing the Overall Evidence. Curr. Breast Cancer Rep. 2013, 5, 208–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, P.J.; Enoch, M.-A.; Goldman, D.; Li, T.-K.; Yokoyama, A. The Alcohol Flushing Response: An Unrecognized Risk Factor for Esophageal Cancer from Alcohol Consumption. PLoS Med. 2009, 6, e1000050. [Google Scholar] [CrossRef]
- Yoshida, A.; Huang, I.Y.; Ikawa, M. Molecular abnormality of an inactive aldehyde dehydrogenase variant commonly found in Orientals. Proc. Natl. Acad. Sci. USA 1984, 81, 258–261. [Google Scholar] [CrossRef] [Green Version]
- Crabb, D.W.; Matsumoto, M.; Chang, D.; You, M. Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc. Nutr. Soc. 2004, 63, 49–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamada, M.; Nagano, O.; Tateyama, S.; Ohmura, M.; Yae, T.; Ishimoto, T.; Sugihara, E.; Onishi, N.; Yamamoto, T.; Yanagawa, H.; et al. Modulation of Glucose Metabolism by CD44 Contributes to Antioxidant Status and Drug Resistance in Cancer Cells. Cancer Res. 2012, 72, 1438–1448. [Google Scholar] [CrossRef] [Green Version]
- Ishimoto, T.; Nagano, O.; Yae, T.; Tamada, M.; Motohara, T.; Oshima, H.; Oshima, M.; Ikeda, T.; Asaba, R.; Yagi, H.; et al. CD44 Variant Regulates Redox Status in Cancer Cells by Stabilizing the xCT Subunit of System xc− and Thereby Promotes Tumor Growth. Cancer Cell 2011, 19, 387–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryoo, I.-G.; Choi, B.-H.; Ku, S.-K.; Kwak, M.-K. High CD44 expression mediates p62-associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: Implications for cancer stem cell resistance. Redox Biol. 2018, 17, 246–258. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shimonosono, M.; Tanaka, K.; Flashner, S.; Takada, S.; Matsuura, N.; Tomita, Y.; Sachdeva, U.M.; Noguchi, E.; Sangwan, V.; Ferri, L.; et al. Alcohol Metabolism Enriches Squamous Cell Carcinoma Cancer Stem Cells That Survive Oxidative Stress via Autophagy. Biomolecules 2021, 11, 1479. https://doi.org/10.3390/biom11101479
Shimonosono M, Tanaka K, Flashner S, Takada S, Matsuura N, Tomita Y, Sachdeva UM, Noguchi E, Sangwan V, Ferri L, et al. Alcohol Metabolism Enriches Squamous Cell Carcinoma Cancer Stem Cells That Survive Oxidative Stress via Autophagy. Biomolecules. 2021; 11(10):1479. https://doi.org/10.3390/biom11101479
Chicago/Turabian StyleShimonosono, Masataka, Koji Tanaka, Samuel Flashner, Satoshi Takada, Norihiro Matsuura, Yasuto Tomita, Uma M. Sachdeva, Eishi Noguchi, Veena Sangwan, Lorenzo Ferri, and et al. 2021. "Alcohol Metabolism Enriches Squamous Cell Carcinoma Cancer Stem Cells That Survive Oxidative Stress via Autophagy" Biomolecules 11, no. 10: 1479. https://doi.org/10.3390/biom11101479