Camel Hemorphins Exhibit a More Potent Angiotensin-I Converting Enzyme Inhibitory Activity than Other Mammalian Hemorphins: An In Silico and In Vitro Study


Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Structure Pre-Processing
2.2. Active Site Identification and Grid Generation
2.3. Peptide Docking
2.4. Analysis of Docking Results and Binding Free Energy Calculation
2.5. Molecular Dynamics (MD) Simulations
2.6. In Vitro ACE Inhibition Assay
2.7. Statistical Analysis
3. Results
3.1. Molecular Docking
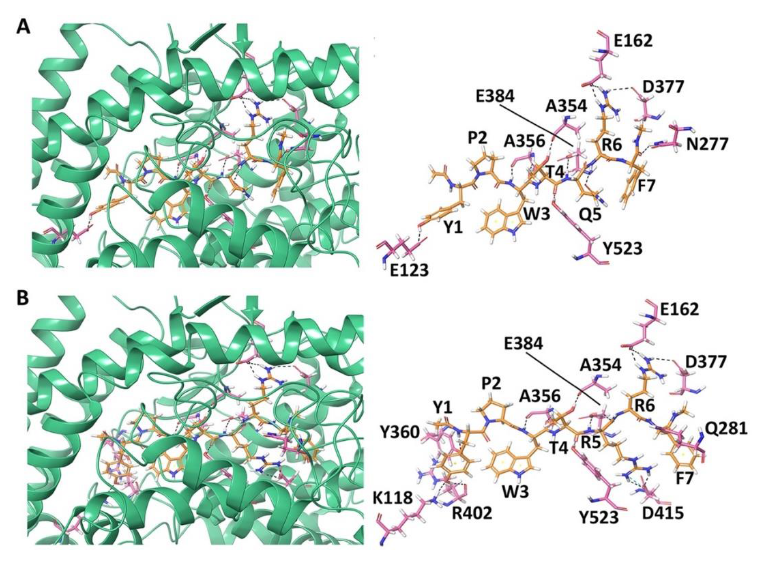
3.1.1. Docking of Non-Camel and Camel LVV-Hemorphin-7 to ACE
3.1.2. Docking of Non-Camel and Camel Hemorphin-7 to ACE
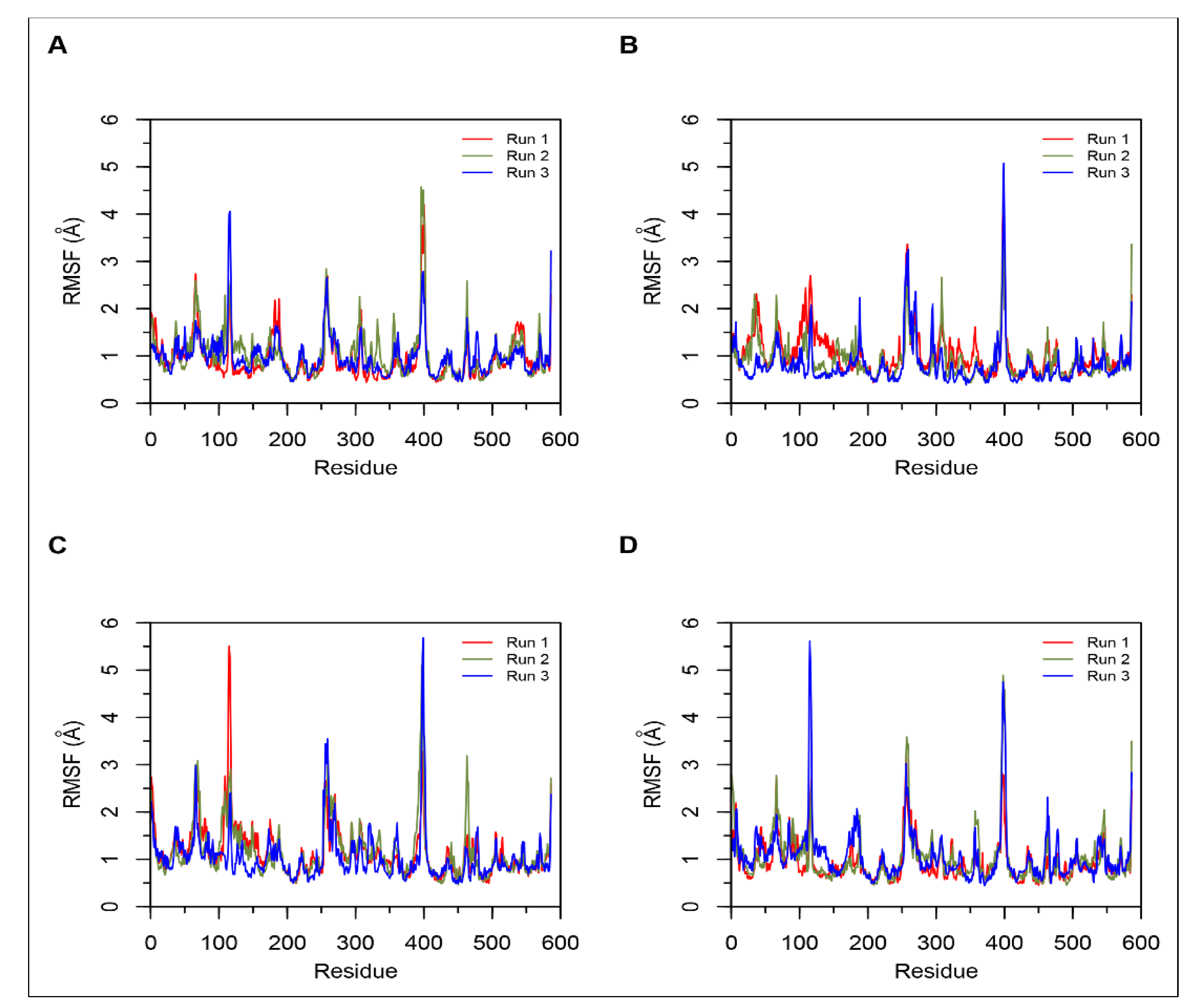
3.2. Molecular Dynamics Simulations
Simulations of Non-Camel and Camel Hemorphins Bound to ACE
3.3. In Vitro ACE Inhibition Assay
3.3.1. ACE Inhibitory Activity of Non-Camel and Camel Hemorphins
3.3.2. Comparison of the ACE Inhibition Potential of Camel and Non-Camel Hemorphins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lawes, C.M.; Vander Hoorn, S.; Rodgers, A. International Society of, H. Global burden of blood-pressure-related disease, 2001. Lancet 2008, 371, 1513–1518. [Google Scholar] [CrossRef]
- Kearney, P.M.; Whelton, M.; Reynolds, K.; Muntner, P.; Whelton, P.K.; He, J. Global burden of hypertension: Analysis of worldwide data. Lancet 2005, 365, 217–223. [Google Scholar] [CrossRef]
- Sawicka, K.; Szczyrek, M.; Jastrzebska, I.; Prasal, M.; Zwolak, A.; Daniluk, J. Hypertension–The silent killer. J. Pre-Clin. Clin. Res. 2011, 5, 43–46. [Google Scholar]
- Schütten, M.T.; Houben, A.J.; de Leeuw, P.W.; Stehouwer, C.D. The link between adipose tissue renin-angiotensin-aldosterone system signaling and obesity-associated hypertension. Physiology 2017, 32, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ondetti, M.A.; Rubin, B.; Cushman, D.W. Design of specific inhibitors of angiotensin-converting enzyme: New class of orally active antihypertensive agents. Science 1977, 196, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Priyanto, A.D.; Doerksen, R.J.; Chang, C.I.; Sung, W.C.; Widjanarko, S.B.; Kusnadi, J.; Lin, Y.C.; Wang, T.C.; Hsu, J.L. Screening, discovery, and characterization of angiotensin-I converting enzyme inhibitory peptides derived from proteolytic hydrolysate of bitter melon seed proteins. J. Proteom. 2015, 128, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Chen, Y.; Zhao, W.; Li, J.; Liu, J.; Chen, F. Identification and molecular docking study of novel angiotensin--converting enzyme inhibitory peptides from Salmo salar using in silico methods. J. Sci. Food Agric. 2018, 98, 3907–3914. [Google Scholar] [CrossRef]
- Iwaniak, A.; Minkiewicz, P.; Darewicz, M. Food-originating ACE inhibitors, including antihypertensive peptides, as preventive food components in blood pressure reduction. Compr. Rev. Food Sci. Food Saf. 2014, 13, 114–134. [Google Scholar] [CrossRef]
- Wang, X.; Chen, H.; Fu, X.; Li, S.; Wei, J. A novel antioxidant and ACE inhibitory peptide from rice bran protein: Biochemical characterization and molecular docking study. LWT 2017, 75, 93–99. [Google Scholar] [CrossRef]
- Lee, J.; Albiston, A.L.; Allen, A.M.; Mendelsohn, F.A.; Ping, S.E.; Barrett, G.L.; Murphy, M.; Morris, M.J.; McDowall, S.G.; Chai, S.Y. Effect of I.C.V. injection of AT4 receptor ligands, NLE1-angiotensin IV and LVV-hemorphin 7, on spatial learning in rats. Neuroscience 2004, 124, 341–349. [Google Scholar] [CrossRef]
- Brantl, V.; Gramsch, C.; Lottspeich, F.; Mertz, R.; Jaeger, K.H.; Herz, A. Novel opioid peptides derived from hemoglobin: Hemorphins. Eur. J. Pharmacol. 1986, 125, 309–310. [Google Scholar] [CrossRef]
- Piot, J.M.; Zhao, Q.; Guillochon, D.; Ricart, G.; Thomas, D. Isolation and characterization of two opioid peptides from a bovine hemoglobin peptic hydrolysate. Biochem. Biophys. Res. Commun. 1992, 189, 101–110. [Google Scholar] [CrossRef]
- Moisan, S.; Harvey, N.; Beaudry, G.; Forzani, P.; Burhop, K.E.; Drapeau, G.; Rioux, F. Structural requirements and mechanism of the pressor activity of Leu-Val-Val-hemorphin-7, a fragment of hemoglobin beta-chain in rats. Peptides 1998, 19, 119–131. [Google Scholar] [CrossRef]
- Glamsta, E.L.; Meyerson, B.; Silberring, J.; Terenius, L.; Nyberg, F. Isolation of a hemoglobin-derived opioid peptide from cerebrospinal fluid of patients with cerebrovascular bleedings. Biochem. Biophys. Res. Commun. 1992, 184, 1060–1066. [Google Scholar] [CrossRef]
- Karelin, A.A.; Philippova, M.M.; Karelina, E.V.; Ivanov, V.T. Isolation of endogenous hemorphin-related hemoglobin fragments from bovine brain. Biochem. Biophys. Res. Commun. 1994, 202, 410–415. [Google Scholar] [CrossRef]
- Moeller, I.; Lew, R.A.; Mendelsohn, F.A.; Smith, A.I.; Brennan, M.E.; Tetaz, T.J.; Chai, S.Y. The globin fragment LVV--hemorphin--7 is an endogenous ligand for the AT4 receptor in the brain. J. Neurochem. 1997, 68, 2530–2537. [Google Scholar] [CrossRef]
- Hughes, J.; Smith, T.W.; Kosterlitz, H.W.; Fothergill, L.A.; Morgan, B.A.; Morris, H.R. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature 1975, 258, 577–580. [Google Scholar] [CrossRef]
- Albiston, A.L.; Pederson, E.S.; Burns, P.; Purcell, B.; Wright, J.W.; Harding, J.W.; Mendelsohn, F.A.; Weisinger, R.S.; Chai, S.Y. Attenuation of scopolamine-induced learning deficits by LVV-hemorphin-7 in rats in the passive avoidance and water maze paradigms. Behav. Brain. Res. 2004, 154, 239–243. [Google Scholar] [CrossRef]
- Cheng, B.C.; Tao, P.L.; Cheng, Y.Y.; Huang, E.Y. LVV-hemorphin 7 and angiotensin IV in correlation with antinociception and anti-thermal hyperalgesia in rats. Peptides 2012, 36, 9–16. [Google Scholar] [CrossRef]
- Ali, A.; Palakkott, A.; Ashraf, A.; Al Zamel, I.; Baby, B.; Vijayan, R.; Ayoub, M.A. Positive modulation of angiotensin II type 1 receptor–mediated signaling by LVV–hemorphin-7. Front. Pharmacol. 2019, 10, 1258. [Google Scholar] [CrossRef]
- Cejka, J.; Zelezna, B.; Velek, J.; Zicha, J.; Kunes, J. LVV-hemorphin-7 lowers blood pressure in spontaneously hypertensive rats: Radiotelemetry study. Physiol. Res. 2004, 53, 603–607. [Google Scholar] [PubMed]
- Lantz, I.; Glämsta, E.-L.; Talbäck, L.; Nyberg, F. Hemorphins derived from hemoglobin have an inhibitory action on angiotensin converting enzyme activity. FEBS Lett. 1991, 287, 39–41. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Piot, J.M. Investigation of inhibition angiotensin-converting enzyme (ACE) activity and opioid activity of two hemorphins, LVV-hemorphin-5 and VV-hemorphin-5, isolated from a defined peptic hydrolysate of bovine hemoglobin. Neuropeptides 1997, 31, 147–153. [Google Scholar] [CrossRef]
- Fruitier-Arnaudin, I.; Cohen, M.; Bordenave, S.; Sannier, F.; Piot, J.M. Comparative effects of angiotensin IV and two hemorphins on angiotensin-converting enzyme activity. Peptides 2002, 23, 1465–1470. [Google Scholar] [CrossRef]
- Ali, A.; Baby, B.; Soman, S.S.; Vijayan, R. Molecular insights into the interaction of hemorphin and its targets. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, A.; Baby, B.; Vijayan, R. From desert to medicine: A review of camel genomics and therapeutic products. Front. Genet. 2019, 10, 17. [Google Scholar] [CrossRef]
- Jirimutu, T.B.C.G.; Wang, Z.; Ding, G.; Chen, G.; Sun, Y.; Sun, Z.; Zhang, H.; Wang, L.; Hasi, S.; Zhang, Y.; et al. Genome sequences of wild and domestic bactrian camels. Nat. Commun. 2012, 3, 1202. [Google Scholar] [CrossRef]
- Agrawal, R.P.; Jain, S.; Shah, S.; Chopra, A.; Agarwal, V. Effect of camel milk on glycemic control and insulin requirement in patients with type 1 diabetes: 2-years randomized controlled trial. Eur. J. Clin. Nutr. 2011, 65, 1048–1052. [Google Scholar] [CrossRef] [Green Version]
- Muyldermans, S.; Baral, T.N.; Retamozzo, V.C.; De Baetselier, P.; De Genst, E.; Kinne, J.; Leonhardt, H.; Magez, S.; Nguyen, V.K.; Revets, H.; et al. Camelid immunoglobulins and nanobody technology. Vet. Immunol. Immunopathol. 2009, 128, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Tagliazucchi, D.; Shamsia, S.; Conte, A. Release of angiotensin converting enzyme-inhibitory peptides during in vitro gastro-intestinal digestion of camel milk. Int. Dairyj. 2016, 56, 119–128. [Google Scholar] [CrossRef]
- Akif, M.; Schwager, S.L.; Anthony, C.S.; Czarny, B.; Beau, F.; Dive, V.; Sturrock, E.D.; Acharya, K.R. Novel mechanism of inhibition of human angiotensin-I-converting enzyme (ACE) by a highly specific phosphinic tripeptide. Biochem. J. 2011, 436, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger Release 2016-4: Schrödinger Suite 2016-4, P.P.W.E., S., LLC: New York, NY, USA, 2016. Impact, Schrödinger, LLC: New York, NY, USA, 2016. Prime, Schrödinger, LLC: New York, NY, USA, 2016.
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Watts, K.S.; Dalal, P.; Murphy, R.B.; Sherman, W.; Friesner, R.A.; Shelley, J.C. ConfGen: A conformational search method for efficient generation of bioactive conformers. J. Chem. Inf. Model. 2010, 50, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger Release 2016-4: Maestro; Schrödinger LLC: New York, NY, USA, 2016.
- Schrödinger Release 2016-4: Prime; Schrödinger LLC: New York, NY, USA, 2016.
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [Green Version]
- Desmond Molecular Dynamics System 2015; D.E.S.R., New York, NY, 2015. Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2015.
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Zhang, J. LBFGS Quasi-Newtonian Methods for Molecular Modeling Prion AGAAAAGA Amyloid Fibrils. In Molecular Structures and Structural Dynamics of Prion Proteins and Prions; Springer: Dordrecht, The Netherlands, 2015; pp. 291–307. [Google Scholar]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Humphreys, D.D.; Friesner, R.A.; Berne, B.J. A multiple-time-step molecular dynamics algorithm for macromolecules. J. Chem. Phys. 1994, 98, 6885–6892. [Google Scholar] [CrossRef]
- Cohen, M.; Fruitier-Arnaudin, I.; Piot, J. Hemorphins: Substrates and/or inhibitors of dipeptidyl peptidase IV: Hemorphins N-terminus sequence influence on the interaction between hemorphins and DPPIV. Biochimie 2004, 86, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Garreau, I.; Zhao, Q.; Pejoan, C.; Cupo, A.; Piot, J. VV-hemorphin-7 and LVV-hemorphin-7 released during in vitro peptic hemoglobin hydrolysis are morphinomimetic peptides. Neuropeptides 1995, 28, 243–250. [Google Scholar] [CrossRef]
- Fleming, I. Signaling by the angiotensin-converting enzyme. Circ. Res. 2006, 98, 887–896. [Google Scholar] [CrossRef] [Green Version]
- Abdelhedi, O.; Nasri, R.; Mora, L.; Jridi, M.; Toldra, F.; Nasri, M. In silico analysis and molecular docking study of angiotensin I-converting enzyme inhibitory peptides from smooth-hound viscera protein hydrolysates fractionated by ultrafiltration. Food. Chem. 2018, 239, 453–463. [Google Scholar] [CrossRef]
- Ma, F.F.; Wang, H.; Wei, C.K.; Thakur, K.; Wei, Z.J.; Jiang, L. Three Novel ACE Inhibitory Peptides Isolated From Ginkgo biloba Seeds: Purification, Inhibitory Kinetic and Mechanism. Front. Pharmacol. 2018, 9, 1579. [Google Scholar] [CrossRef]
- Natesh, R.; Schwager, S.L.; Evans, H.R.; Sturrock, E.D.; Acharya, K.R. Structural details on the binding of antihypertensive drugs captopril and enalaprilat to human testicular angiotensin I-converting enzyme. Biochemistry 2004, 43, 8718–8724. [Google Scholar] [CrossRef]
- Wang, X.; Wu, S.; Xu, D.; Xie, D.; Guo, H. Inhibitor and substrate binding by angiotensin-converting enzyme: Quantum mechanical/molecular mechanical molecular dynamics studies. J. Chem. Inf. Model. 2011, 51, 1074–1082. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Sannier, F.; Garreau, I.; Guillochon, D.; Piot, J.M. Inhibition and inhibition kinetics of angiotensin converting enzyme activity by hemorphins, isolated from a peptic bovine hemoglobin hydrolysate. Biochem. Biophys. Res. Commun. 1994, 204, 216–223. [Google Scholar] [CrossRef]
- Silvestre, M.P.C.; Silva, M.R.; Silva, V.D.M.; Souza, M.W.S.d.; Junior, L.; de Oliveira, C.; Afonso, W.d.O. Analysis of whey protein hydrolysates: Peptide profile and ACE inhibitory activity. Braz. J. Pharm. Sci. 2012, 48, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wu, S.; Zhou, L.; Wang, F.; Lan, X.; Sun, J.; Tong, Z.; Liao, D. Separation and Characterization of Angiotensin I Converting Enzyme (ACE) Inhibitory Peptides from Saurida elongata Proteins Hydrolysate by IMAC-Ni(2). Mar. Drugs 2017, 15, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Zhu, Y.; Chen, J.; Wu, H.; Shi, L.; Wang, X.; Wang, L. Characterization of ACE inhibitory peptides from Mactra veneriformis hydrolysate by nano-liquid chromatography electrospray ionization mass spectrometry (Nano-LC-ESI-MS) and molecular docking. Mar. Drugs 2014, 12, 3917–3928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohmura, M.; Nio, N.; Kubo, K.; Minoshima, Y.; Munekata, E.; Ariyoshi, Y. Inhibition of angiotensin-converting enzyme by synthetic peptides of human β-casein. Agric. Biol. Chem. 1989, 53, 2107–2114. [Google Scholar]
- Moayedi, A.; Mora, L.; Aristoy, M.-C.; Hashemi, M.; Safari, M.; Toldrá, F. ACE-inhibitory and antioxidant activities of peptide fragments obtained from tomato processing by-products fermented using Bacillus subtilis: Effect of amino acid composition and peptides molecular mass distribution. Appl. Biochem. Biotechnol. 2017, 181, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Li, L.; Liu, G.; Hu, S.Q. Inhibition mechanism and model of an angiotensin I-converting enzyme (ACE)-inhibitory hexapeptide from yeast (Saccharomyces cerevisiae). PLoS ONE 2012, 7, e37077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyagi, T.; Koshimizu, T.-a.; Tanoue, A. Vasopressin regulation of blood pressure and volume: Findings from V1a receptor–deficient mice. Kidney Int. 2009, 76, 1035–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arendse, L.B.; Danser, A.H.J.; Poglitsch, M.; Touyz, R.M.; Burnett, J.C., Jr.; Llorens-Cortes, C.; Ehlers, M.R.; Sturrock, E.D. Novel Therapeutic Approaches Targeting the Renin-Angiotensin System and Associated Peptides in Hypertension and Heart Failure. Pharmacol. Rev. 2019, 71, 539–570. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Glide Docking Score-GScore (kcal/mol) | MM-GBSA (kcal/mol) | Residues Forming Hydrogen Bonds | Residues Forming Hydrophobic Interactions | Residues Forming π-π Stacking or Cation-π Interactions |
|---|---|---|---|---|---|
| LVVYPWTQRF | −14.045 | −134.860 | Asn70, Glu123, Arg124, Glu162, Thr282, Ala354, Ala356, Glu384, Tyr523 | Tyr51, Trp59, Tyr62, Ala63, Ile88, Ala89, Val119, Leu122, Ala125, Leu139, Leu140, Cys352, Trp357, Tyr360, Cys370, Val379, Val380, Phe391, Tyr394, Ala418, Phe457, Phe512, Val518, Tyr520, Phe527 | His410 |
| LVVYPWTRRF | −18.824 | −147.566 | Trp59, Glu123, Thr166, Glu162, Trp220, Ala354, Ala356, Glu384, Asp415, Tyr523 | Tyr62, Ala63, Ile88, Leu139, Leu161, Ala204, Ala207, Ala216, Met223, Trp279, Val379, Val380, Phe391, Tyr394, Pro407, Phe457, Phe512, Val518, Pro519, Phe527 | Tyr360, His410 |
| YPWTQRF | −15.955 | −112.525 | Glu123, Glu162, Asn277, Ala354, Ala356, Asn377, Glu384 | Tyr62, Ala63, Trp279, Cys352, Trp357, Phe359, Tyr360, Cys370, Val379, Val380, Phe391, Tyr394, Pro407, Phe457, Phe512, Tyr520, Phe527 | Trp59, His410 |
| YPWTRRF | −17.202 | −130.392 | Lys118, Glu162, Gln281, Ala354, Ala356, Tyr360, Asn377, Glu384, Arg402, Asp415, Tyr523 | Tyr62, Ala63, Ile88, Trp279, Trp357, Cys370, Val379, Val380, Phe391, Tyr394, Pro407, Phe457, Phe512, Val518, Phe527 | Trp59, His410 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.; Alzeyoudi, S.A.R.; Almutawa, S.A.; Alnajjar, A.N.; Al Dhaheri, Y.; Vijayan, R. Camel Hemorphins Exhibit a More Potent Angiotensin-I Converting Enzyme Inhibitory Activity than Other Mammalian Hemorphins: An In Silico and In Vitro Study. Biomolecules 2020, 10, 486. https://doi.org/10.3390/biom10030486
Ali A, Alzeyoudi SAR, Almutawa SA, Alnajjar AN, Al Dhaheri Y, Vijayan R. Camel Hemorphins Exhibit a More Potent Angiotensin-I Converting Enzyme Inhibitory Activity than Other Mammalian Hemorphins: An In Silico and In Vitro Study. Biomolecules. 2020; 10(3):486. https://doi.org/10.3390/biom10030486
Chicago/Turabian StyleAli, Amanat, Seham Abdullah Rashed Alzeyoudi, Shamma Abdulla Almutawa, Alya Nasir Alnajjar, Yusra Al Dhaheri, and Ranjit Vijayan. 2020. "Camel Hemorphins Exhibit a More Potent Angiotensin-I Converting Enzyme Inhibitory Activity than Other Mammalian Hemorphins: An In Silico and In Vitro Study" Biomolecules 10, no. 3: 486. https://doi.org/10.3390/biom10030486