Antiproliferative Effect of Acridine Chalcone Is Mediated by Induction of Oxidative Stress

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
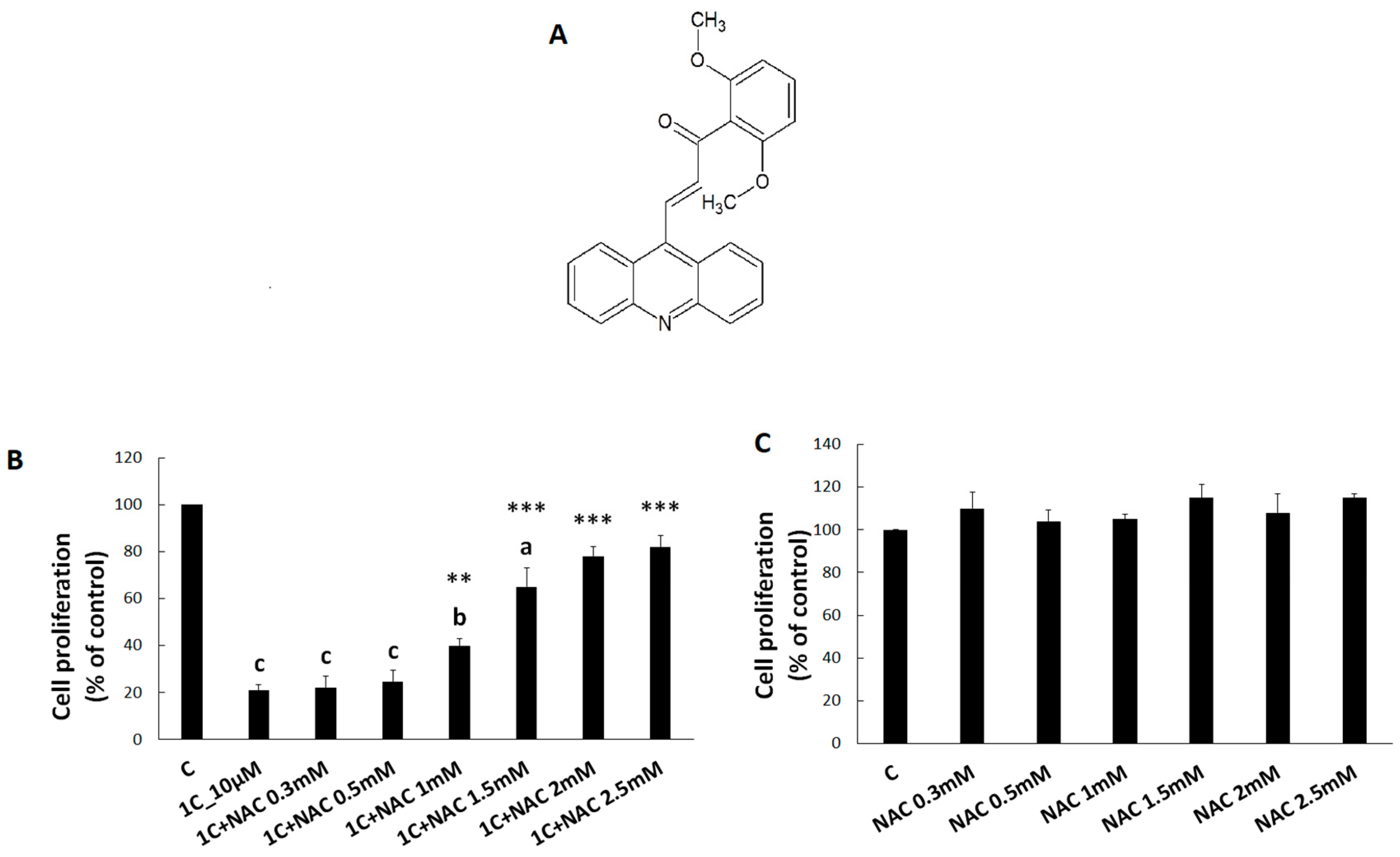
2.1. Tested Compound
2.2. Cell Culture
2.3. Viability Test
2.4. Flow Cytometry Analyses
2.4.1. Analysis of Cell Cycle
2.4.2. Flow Cytometry Analysis of Free Radicals, Apoptosis, Signalling Pathways, and DNA Damage
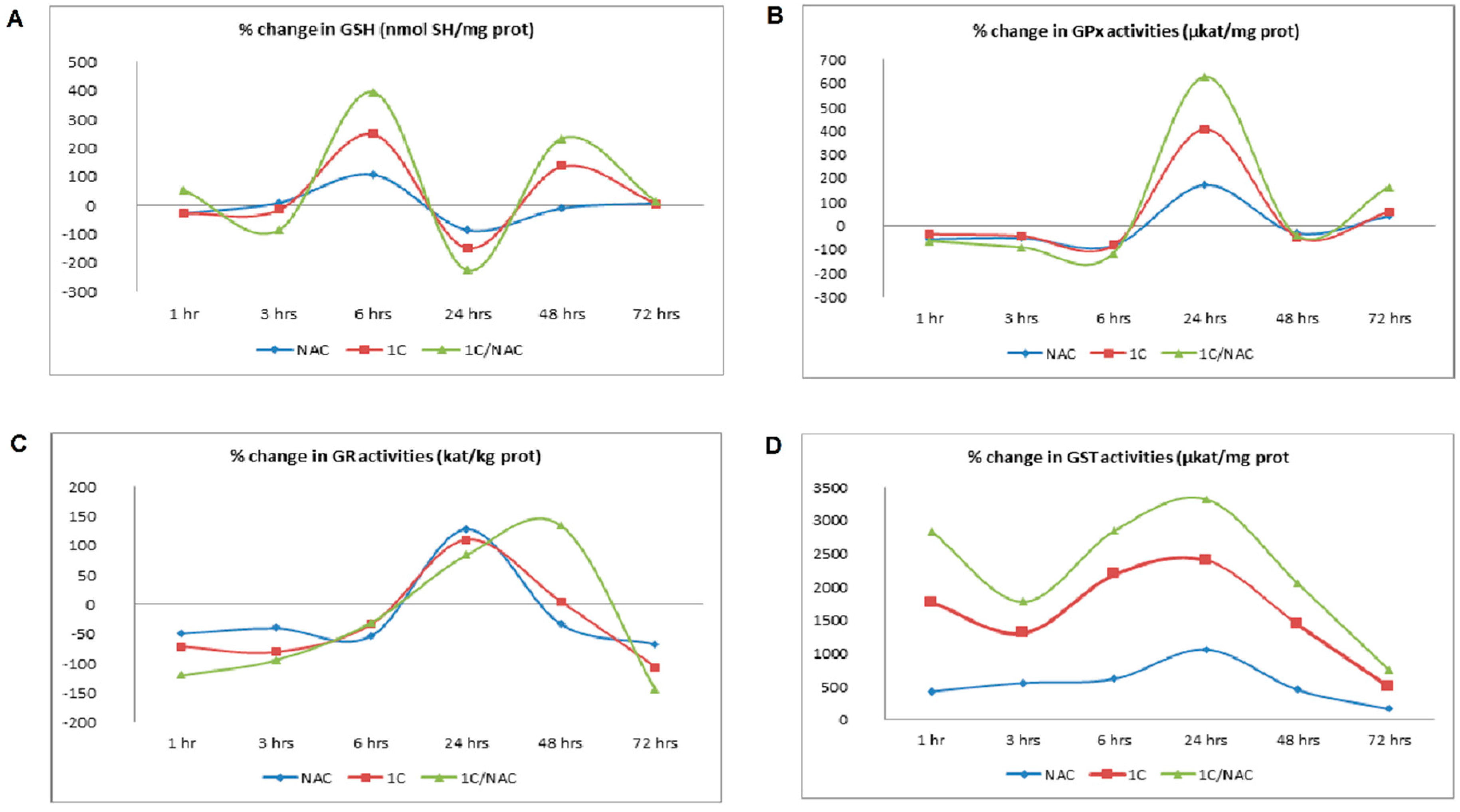
2.5. Antioxidant Enzyme Activities and Glutathione Content Measurement
2.6. Western Blot Analysis
2.7. Statistical Analysis
3. Results
3.1. Viability of HCT116 Cells after 1C and NAC Treatment
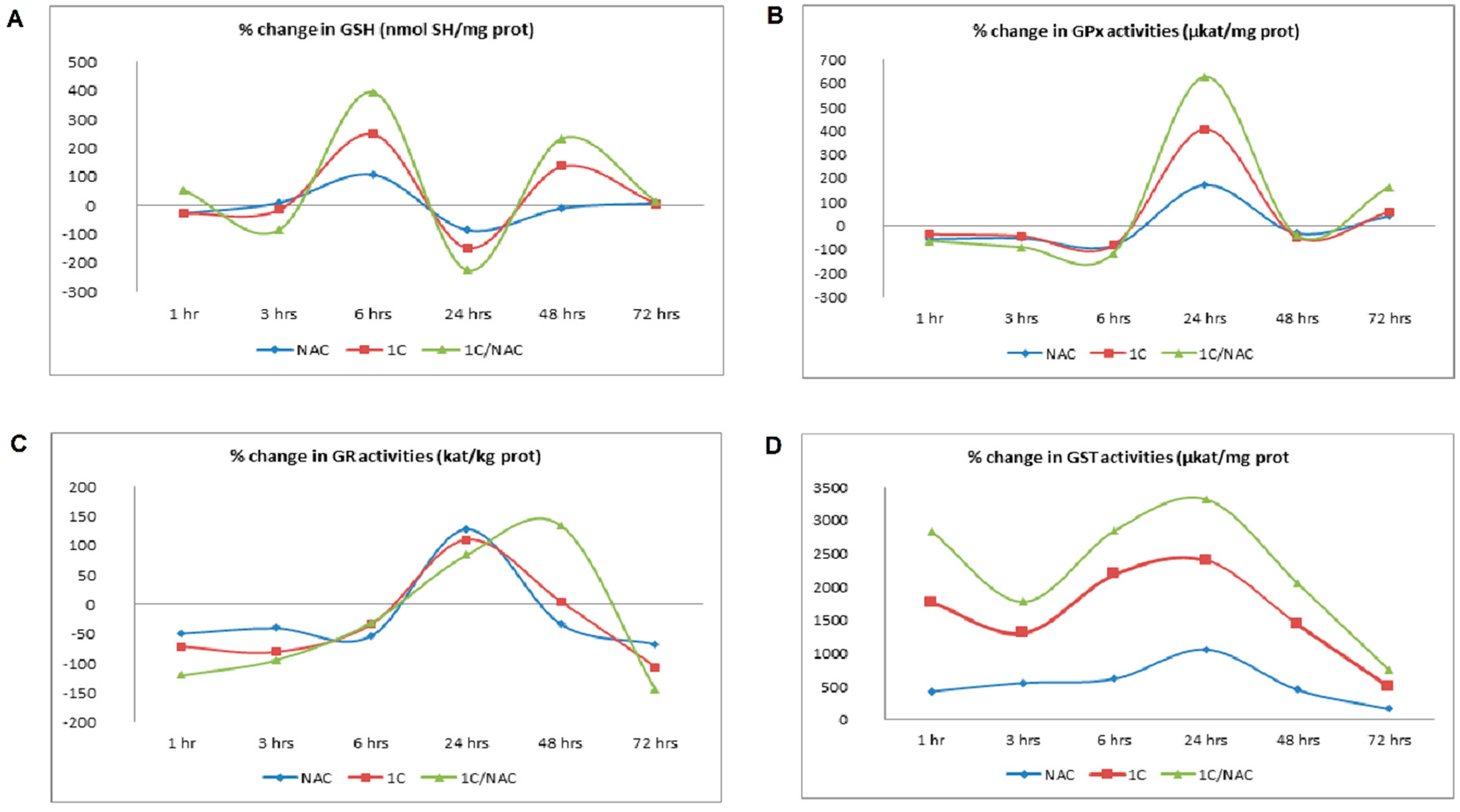
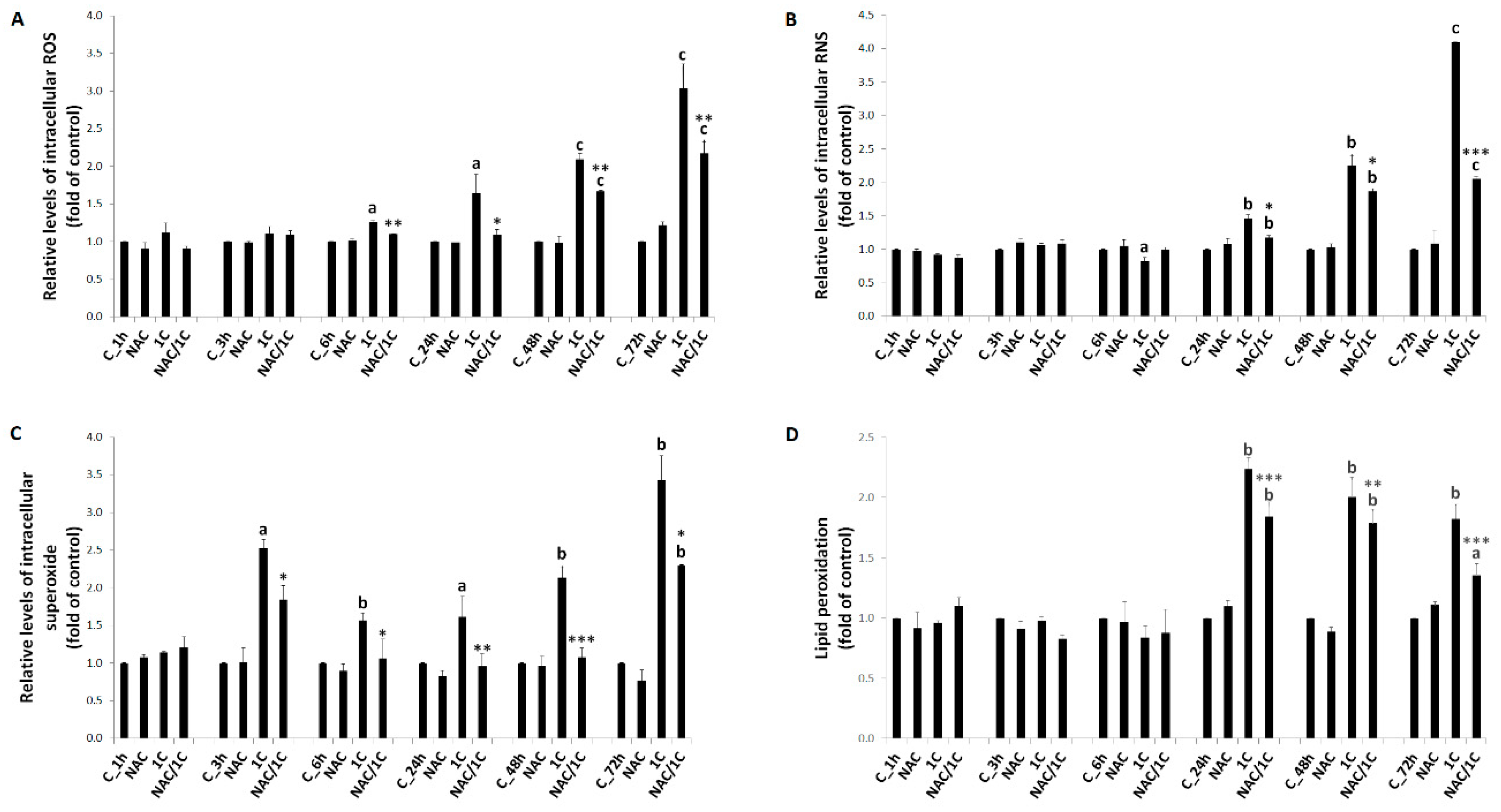
3.2. NAC and 1C-Induced Oxidative Stress
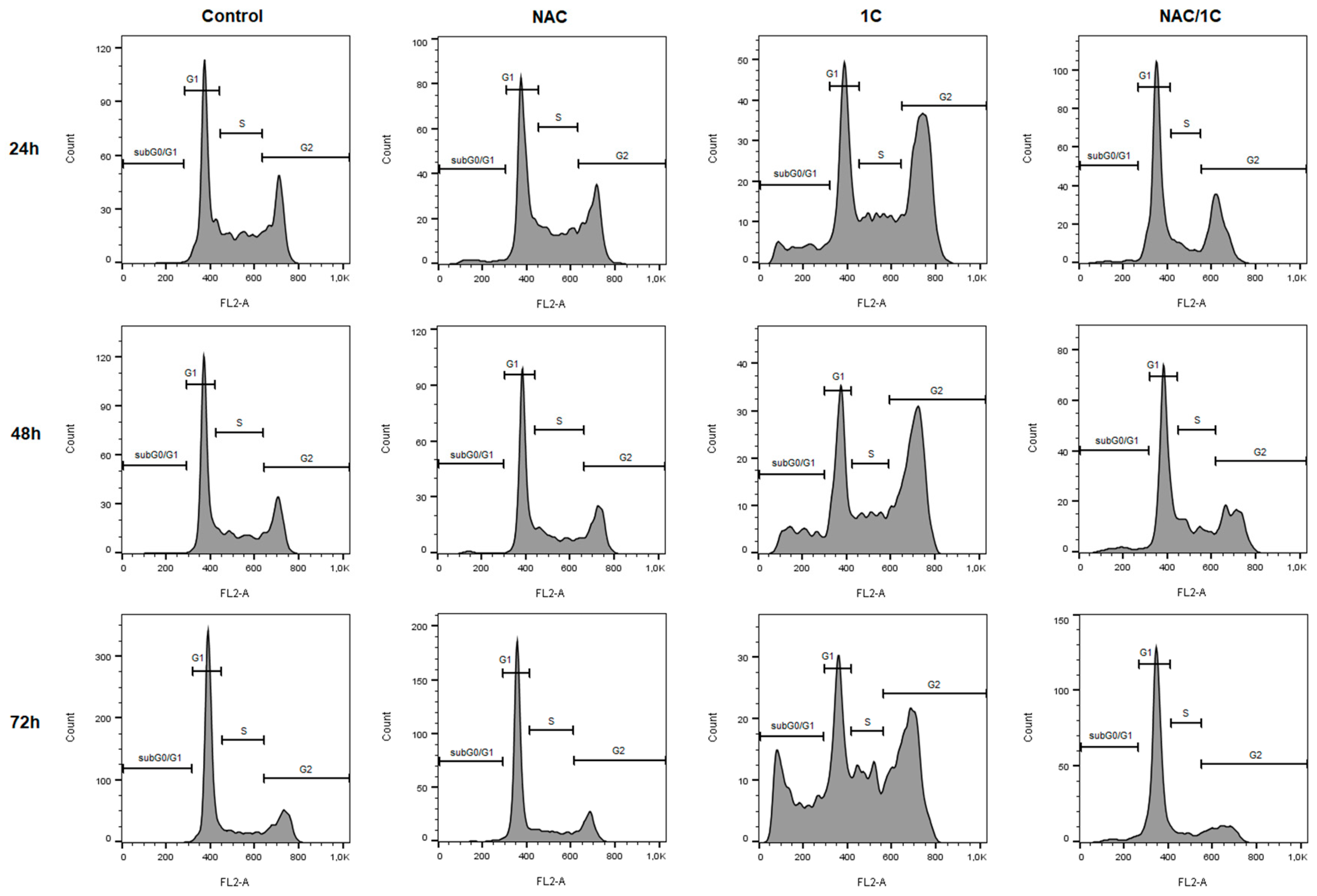
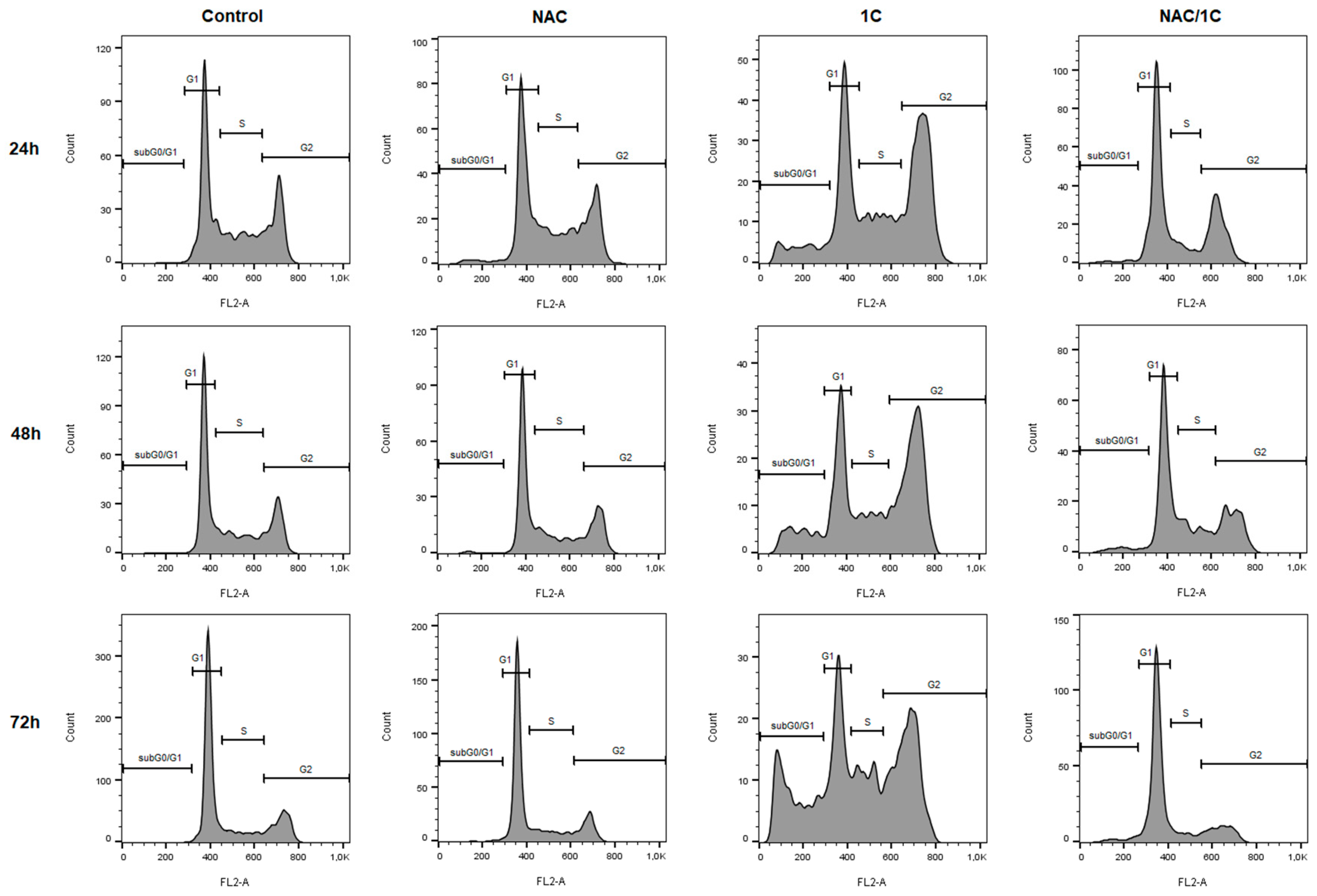
3.3. Effect of NAC on Cell Cycle
3.4. Effect of NAC on the Presence of Apoptotic Markers
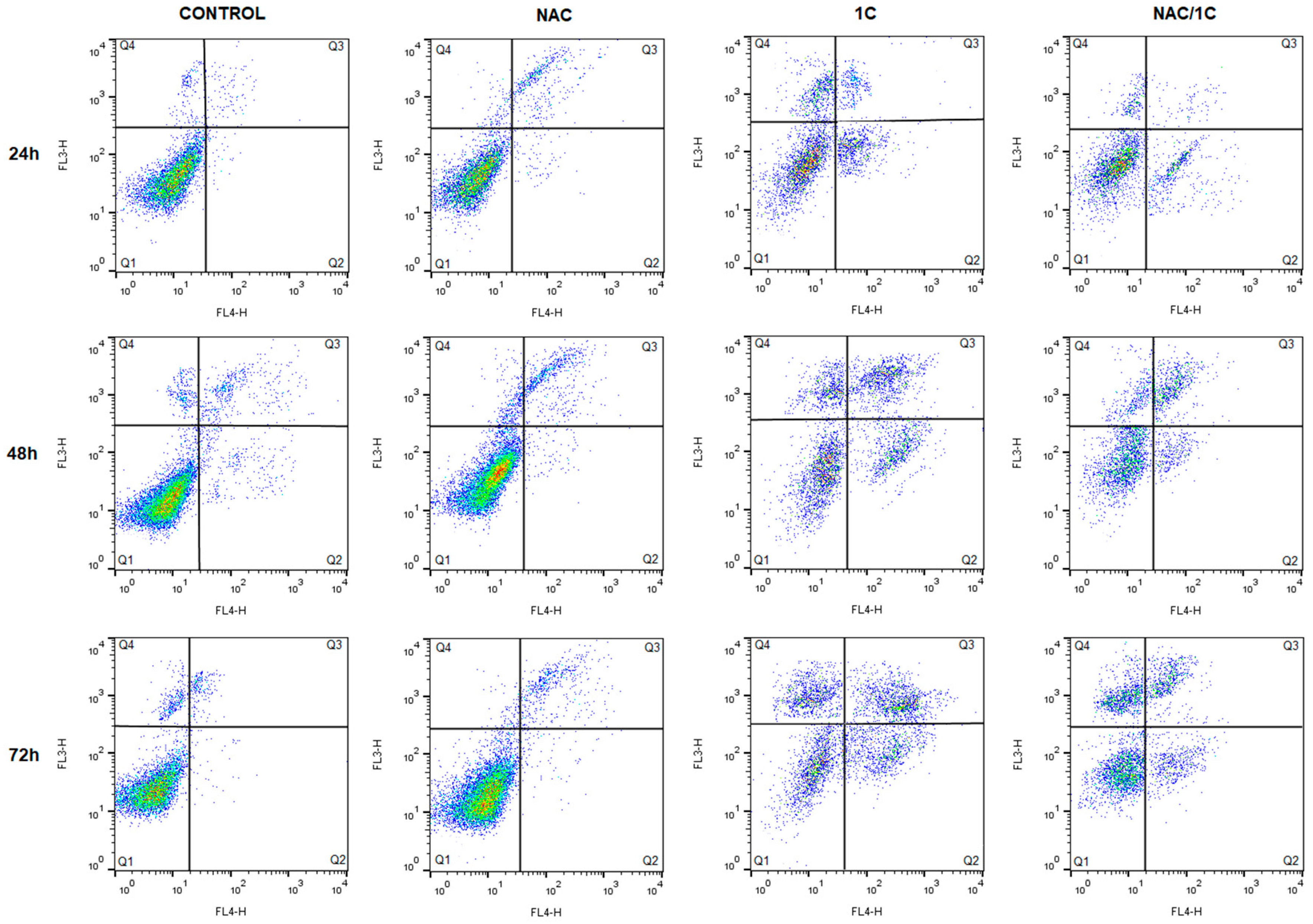
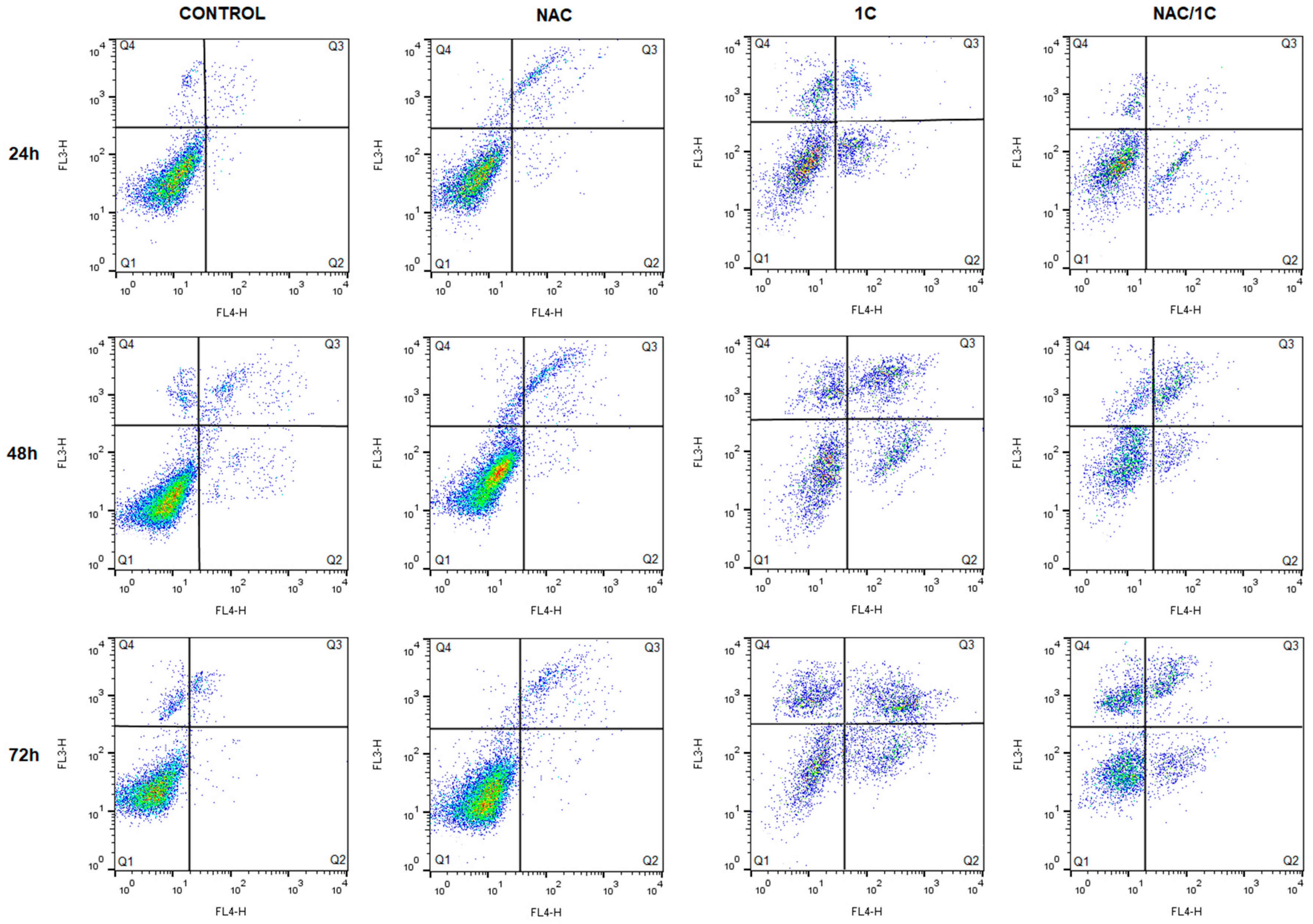
3.4.1. Phosphatidylserine Externalization
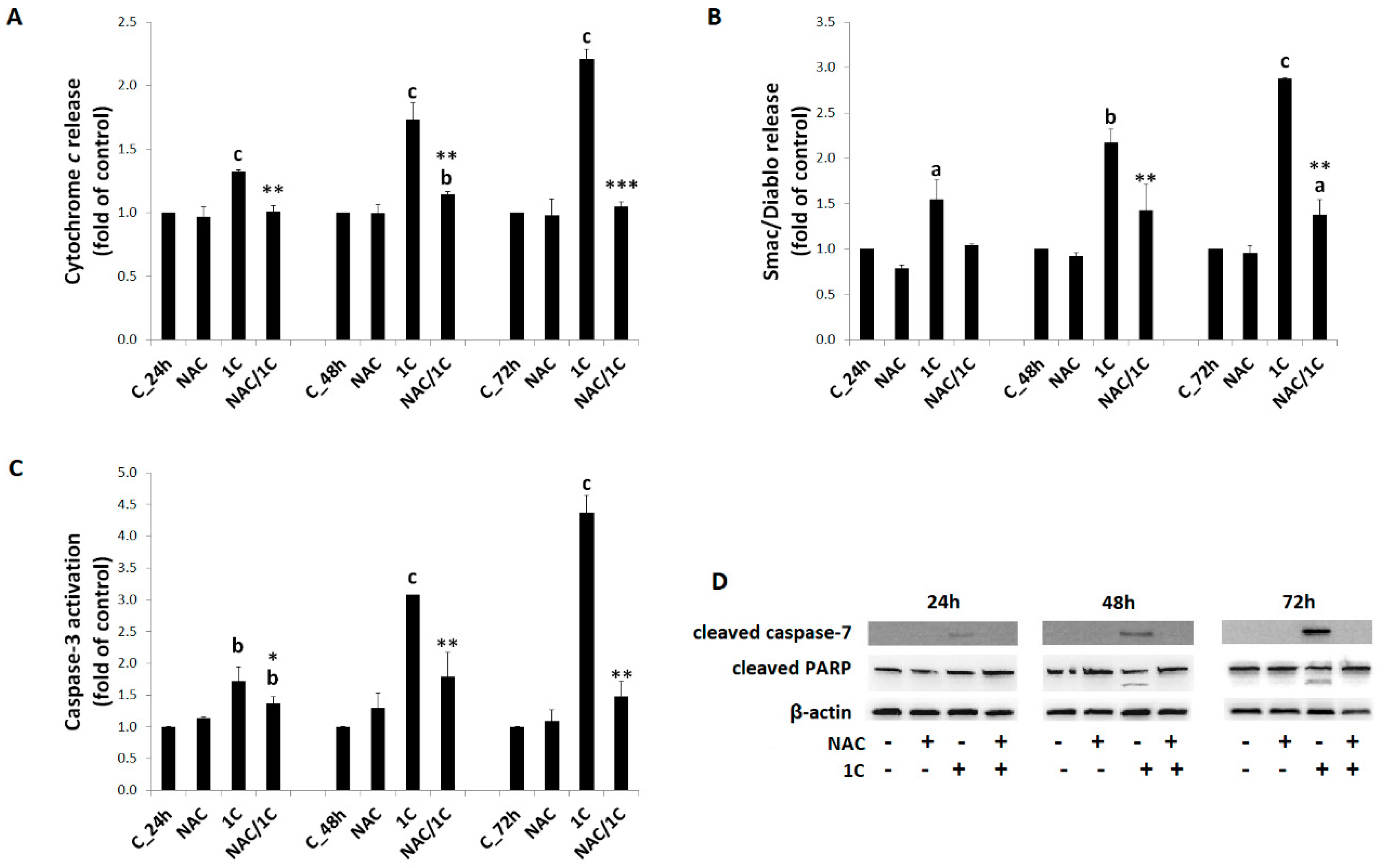
3.4.2. Cytochrome c and Smac/DIABLO Release
3.4.3. Caspase-3 and -7 Activation
3.4.4. PARP Cleavage
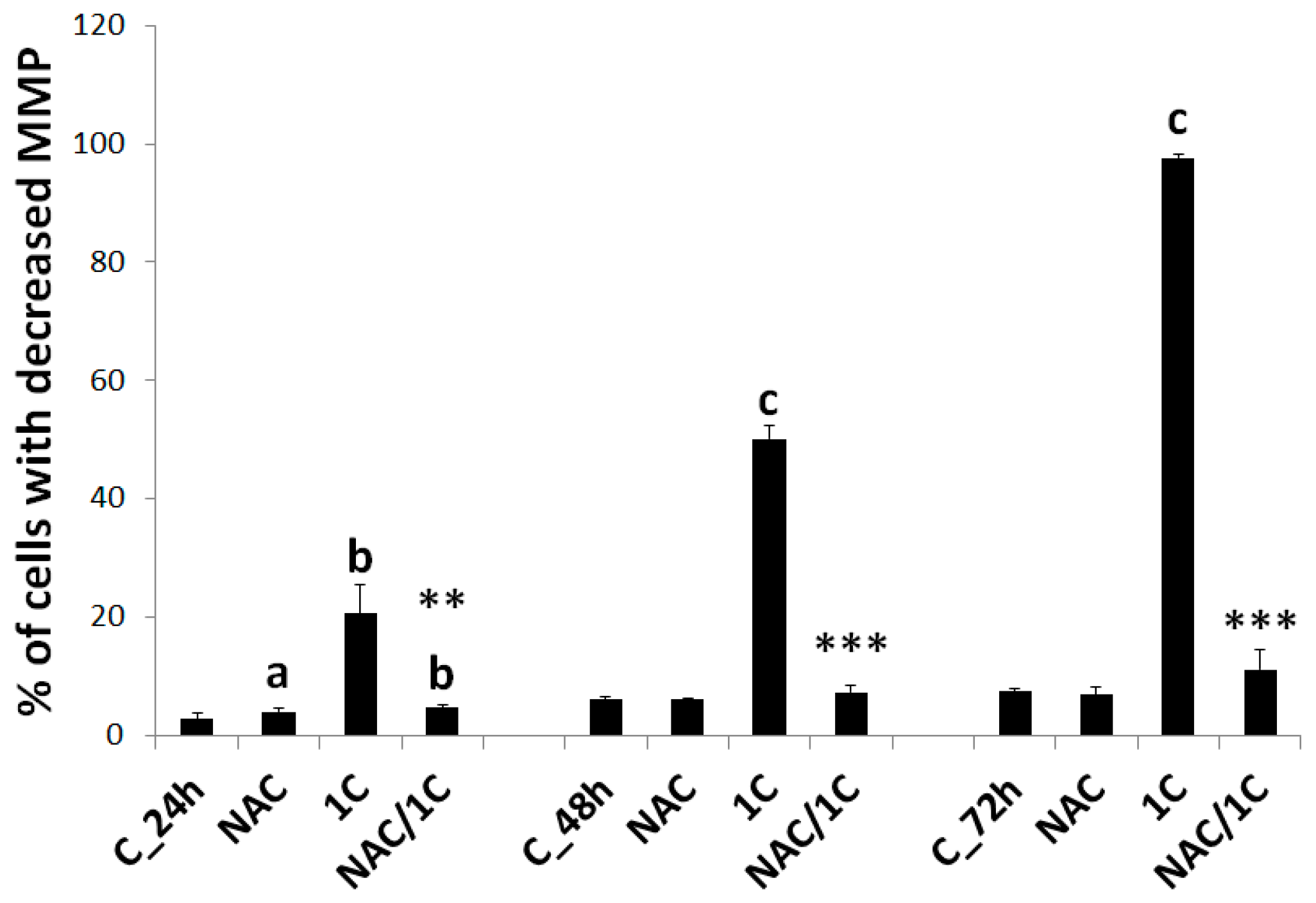
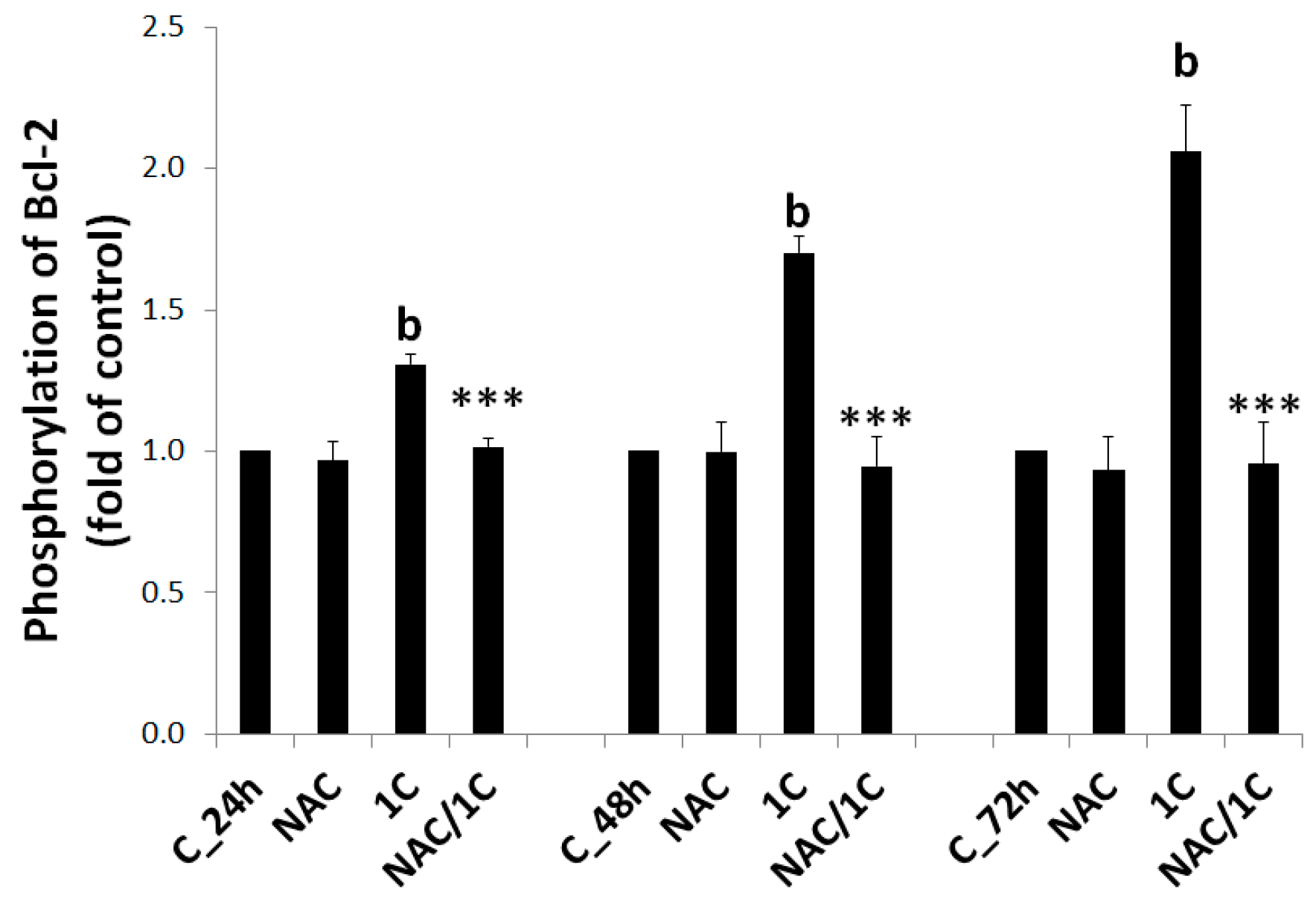
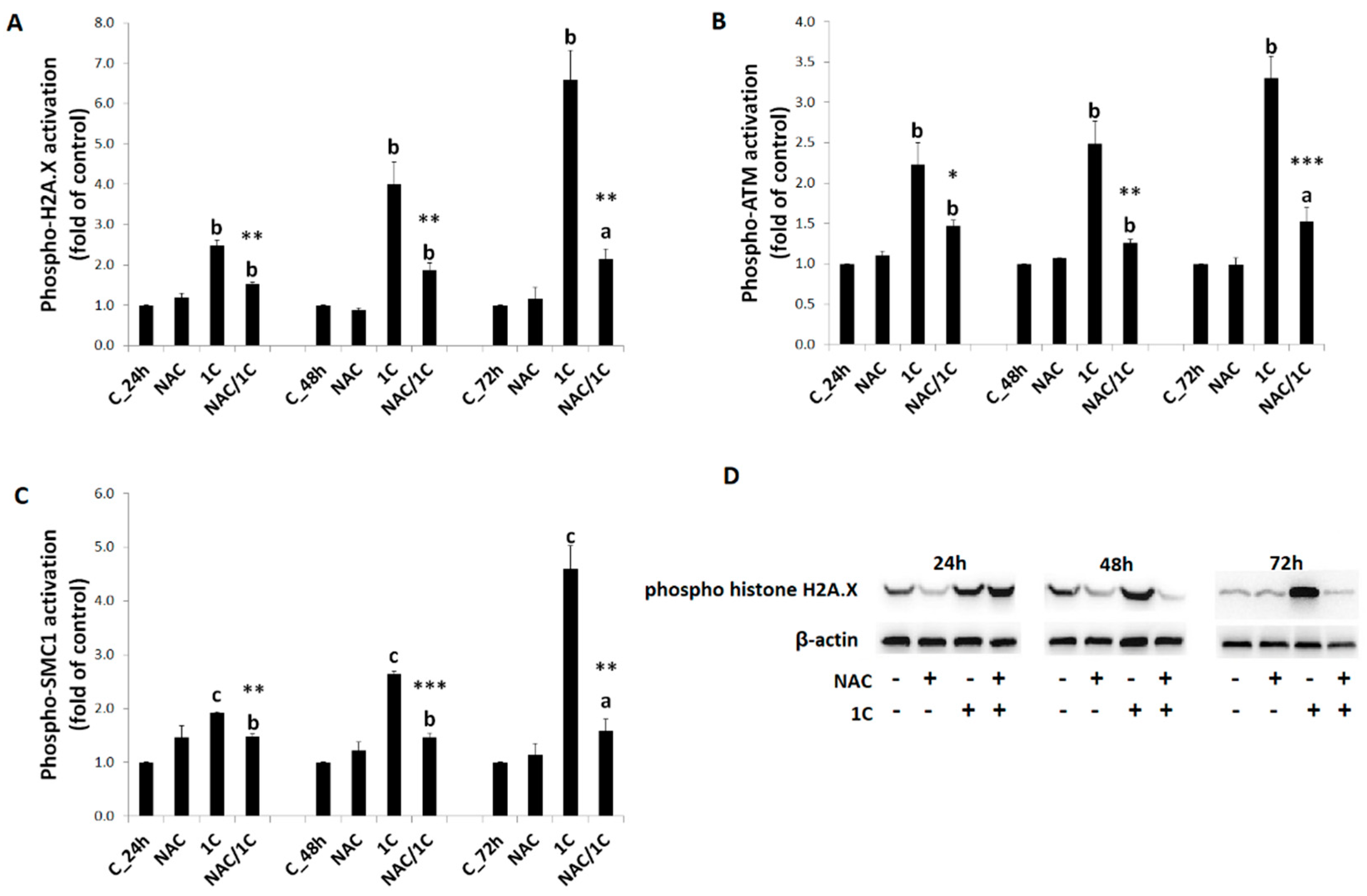
3.5. Effect of NAC on 1C-Induced Mitochondrial Dysfunction, Bcl-2 Phosphorylation, and DNA Damage
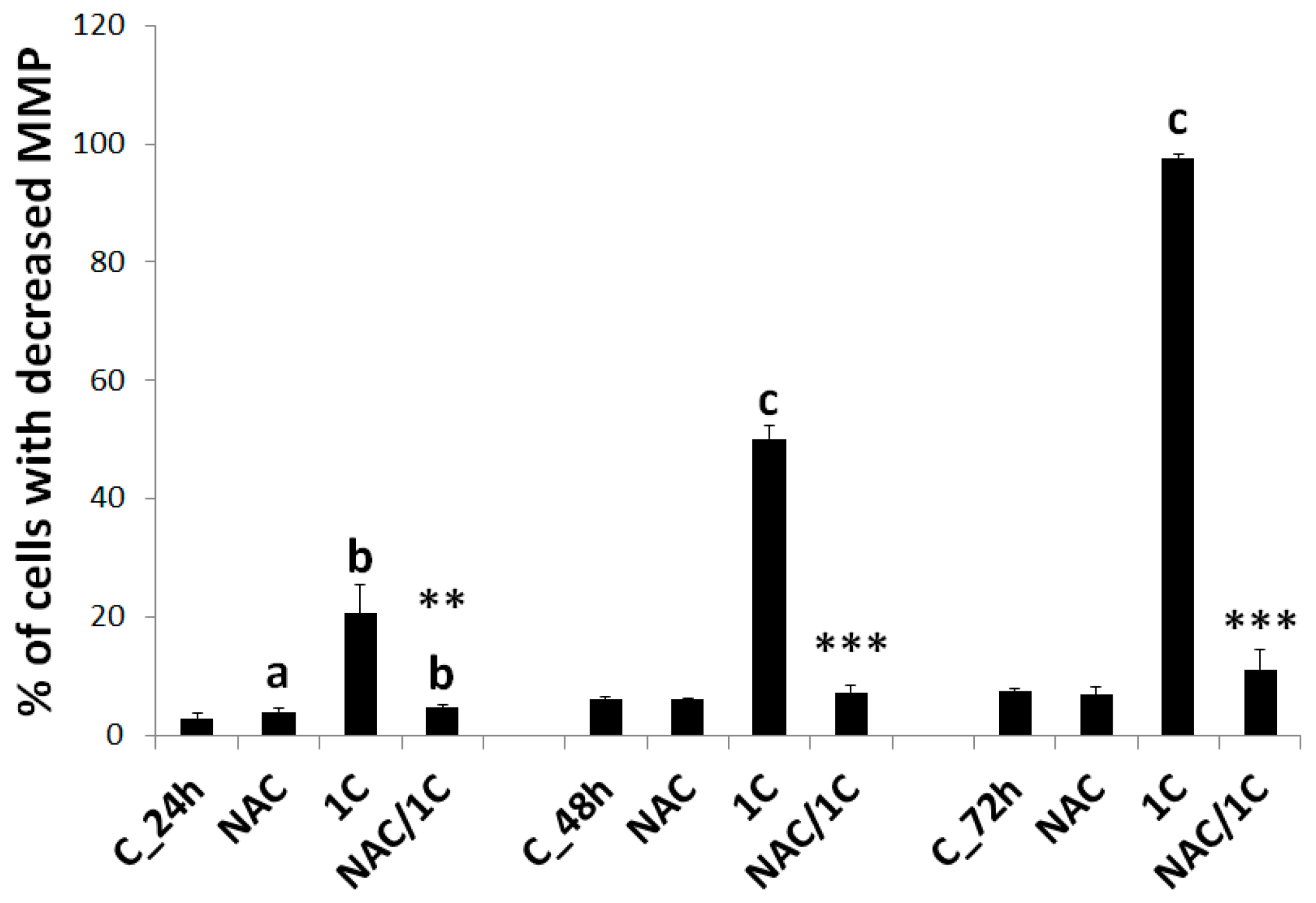
3.5.1. Mitochondrial Membrane Potential
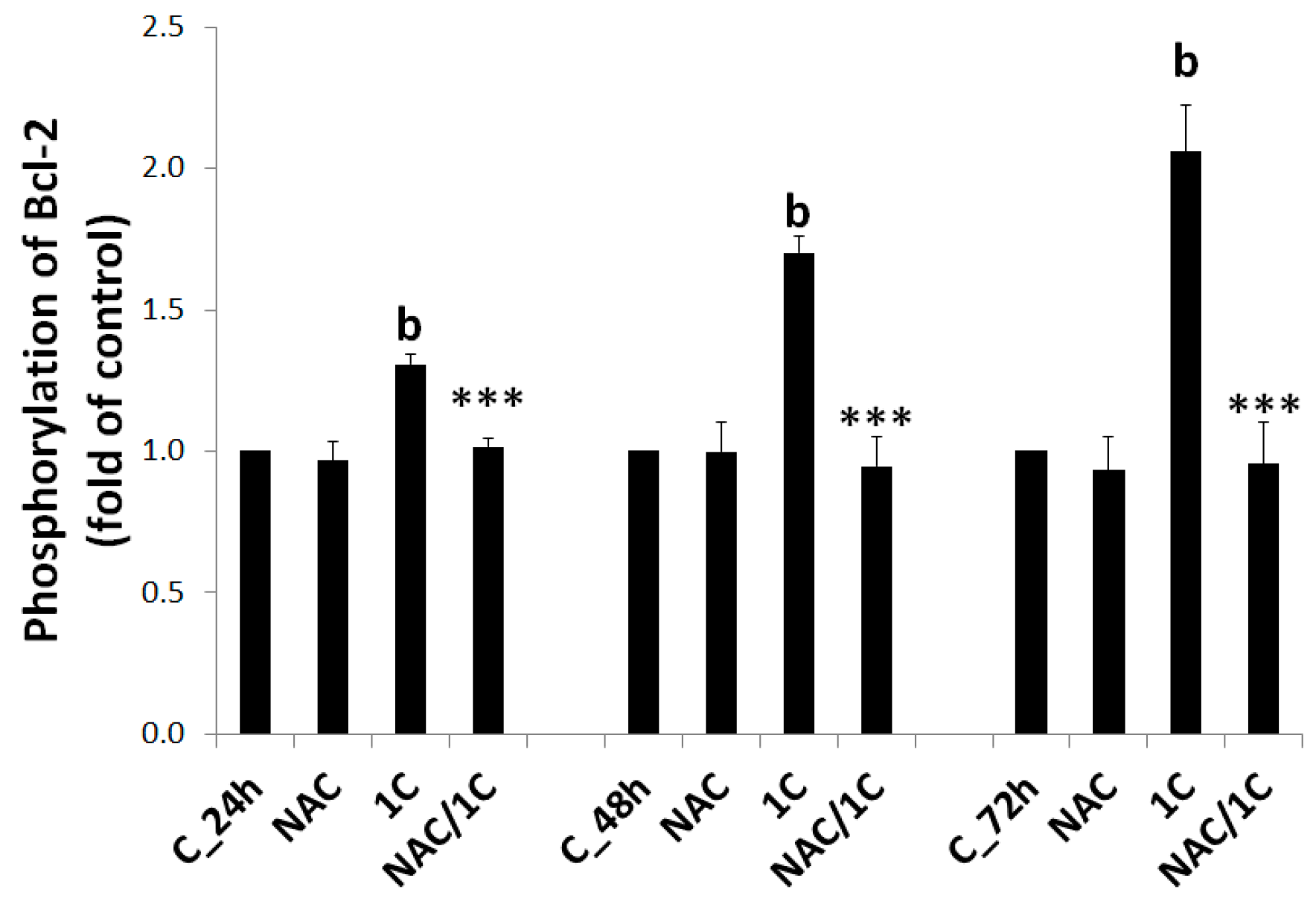
3.5.2. Bcl-2 Phosphorylation
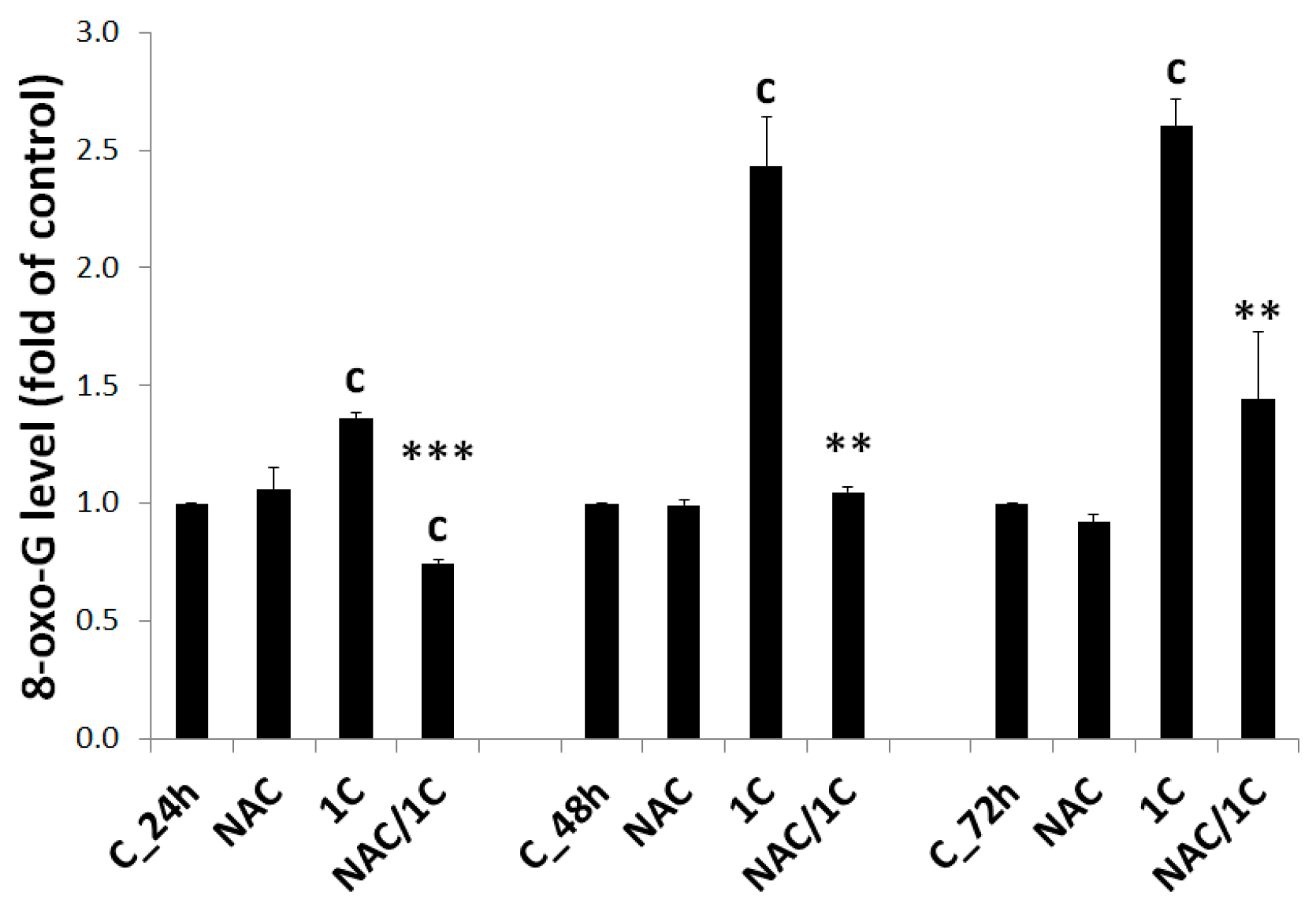
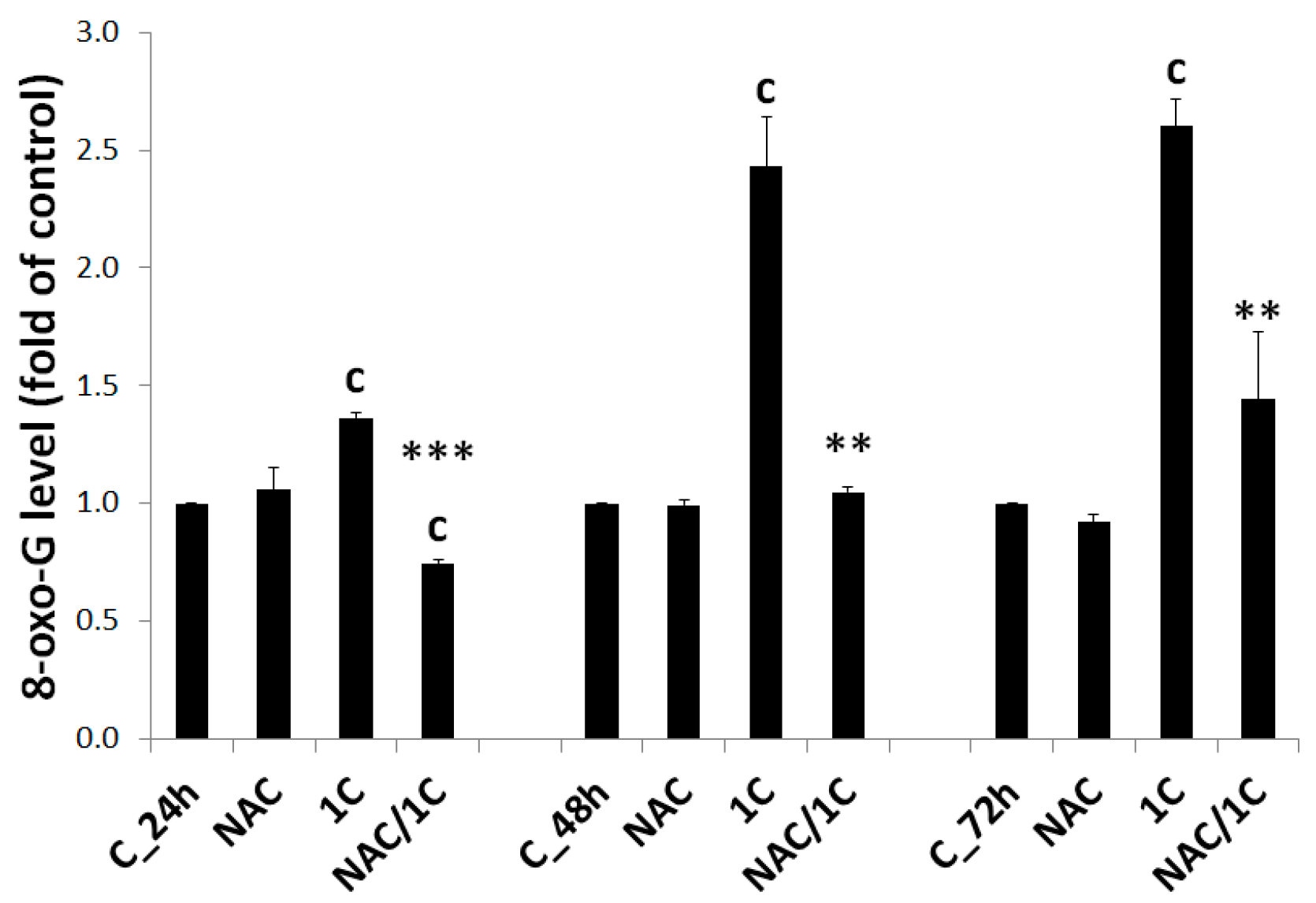
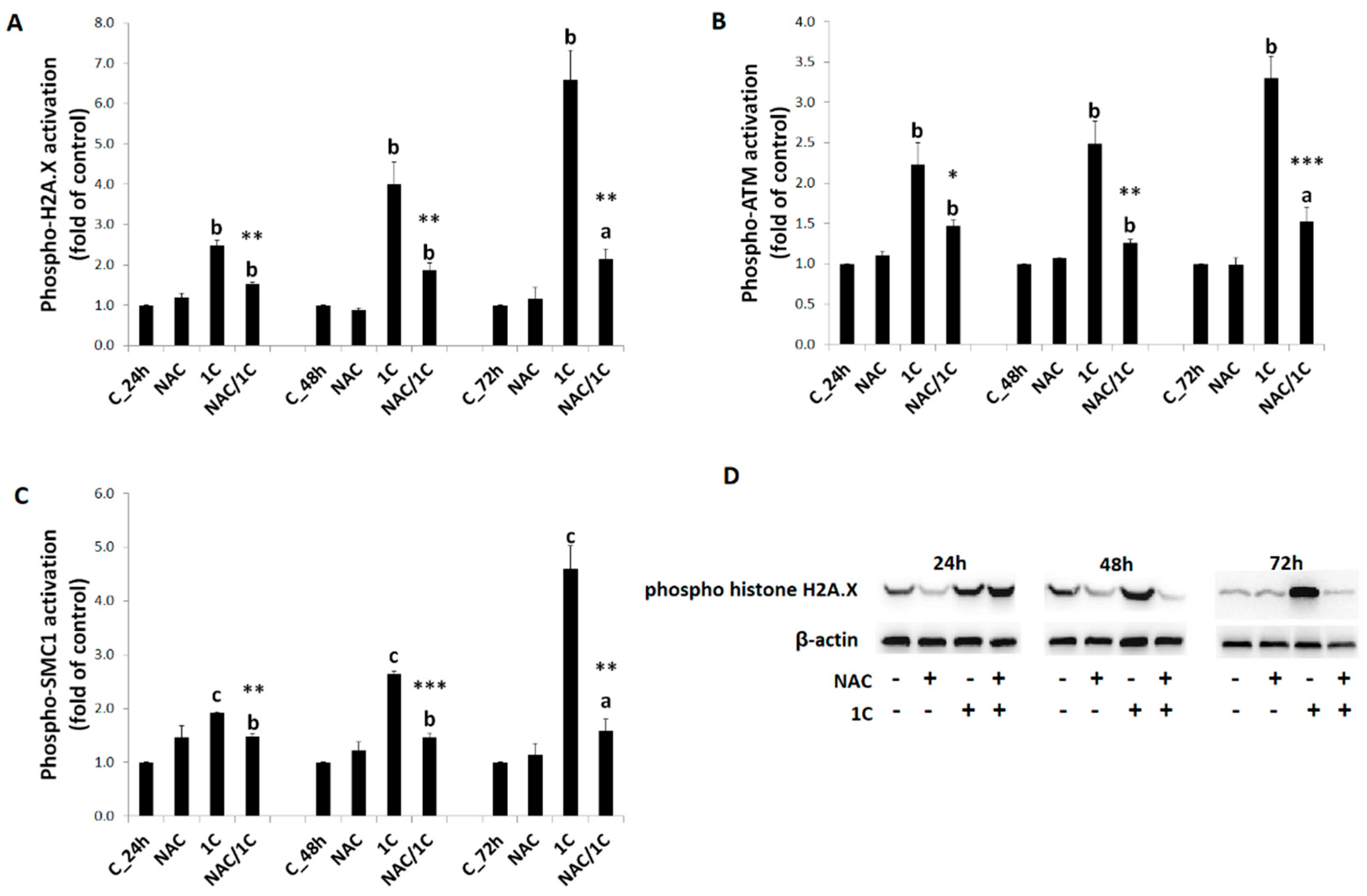
3.5.3. DNA Damage
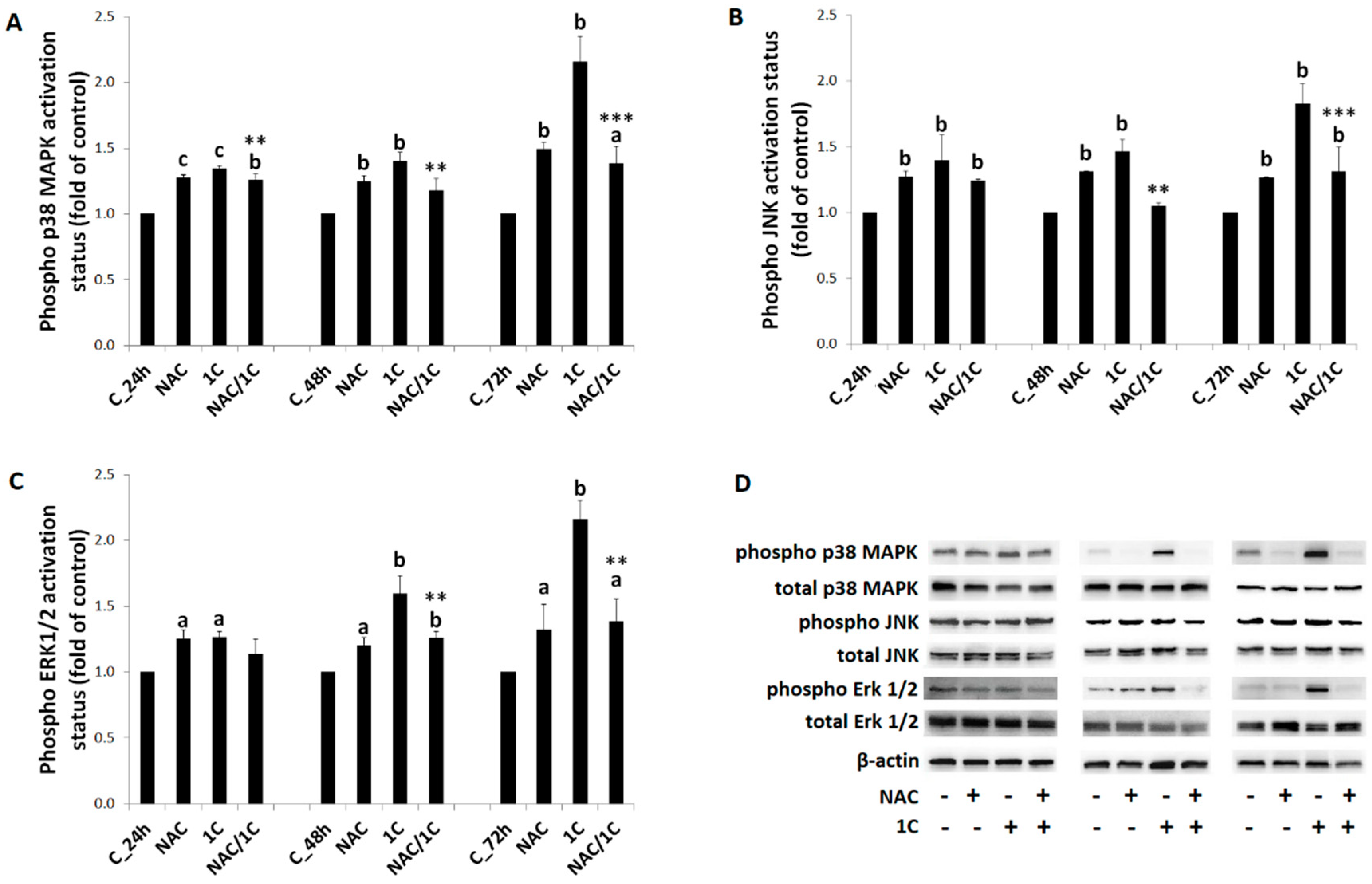
3.6. Phosphorylation of p38 MAPK, JNK, and ERK1/2 Was Reduced by NAC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ferreyra, M.L.F.; Rius, S.; Fernie, A.R. Flavonoids: Biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 2012, 3, 222. [Google Scholar]
- Sökmen, M.; Khan, M.A. The antioxidant activity of some curcuminoids and chalcones. Inflammopharmacology 2016, 24, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Chalcone Derivatives: Anti-inflammatory Potential and Molecular Targets Perspectives. Curr. Top. Med. Chem. 2017, 17, 3146–3169. [Google Scholar] [CrossRef]
- Tratrat, C.; Haroun, M.; Xenikakis, I.; Liaras, K.; Tsolaki, E.; Eleftheriou, P.; Petrou, A.; Aldhubiab, B.; Attimarad, M.; Venugopala, K.N.; et al. Design, Synthesis, Evaluation of Antimicrobial Activity and Docking Studies of New Thiazole-based Chalcones. Curr. Top. Med. Chem. 2019, 19, 356–375. [Google Scholar] [CrossRef]
- Trein, M.R.; Oliveira, L.R.E.; Rigo, G.V.; Garcia, M.A.R.; Petro-Silveira, B.; Trentin, D.D.S.; Macedo, A.J.; Regasini, L.O.; Tasca, T. Anti-Trichomonas vaginalis activity of chalcone and amino-analogues. Parasitol. Res. 2018, 118, 607–615. [Google Scholar] [CrossRef]
- Fontes, L.B.A.; Dias, D.D.S.; De Carvalho, L.S.A.; Mesquita, H.L.; Reis, L.D.S.; Dias, A.T.; Filho, A.A.D.S.; Corrêa, J.O.D.A. Immunomodulatory effects of licochalcone A on experimental autoimmune encephalomyelitis. J. Pharm. Pharmacol. 2014, 66, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Mirossay, L.; Varinska, L.; Mojzis, J. Antiangiogenic Effect of Flavonoids and Chalcones: An Update. Int. J. Mol. Sci. 2017, 19, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varinska, L.; Van Wijhe, M.; Belleri, M.; Mitola, S.M.F.; Perjesi, P.; Presta, M.; Koolwijk, P.; Ivanová, L.; Mojzis, J. Anti-angiogenic activity of the flavonoid precursor 4-hydroxychalcone. Eur. J. Pharmacol. 2012, 691, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Champelovier, P.; Chauchet, X.; Puch, F.; Vergnaud, S.; Garrel, C.; Laporte, F.; Boutonnat, J.; Boumendjel, A. Cellular and molecular mechanisms activating the cell death processes by chalcones: Critical structural effects. Toxicol. In Vitro 2013, 27, 2305–2315. [Google Scholar] [CrossRef] [PubMed]
- Rozmer, Z.; Berki, T.; Maász, G.; Perjesi, P. Different effects of two cyclic chalcone analogues on redox status of Jurkat T cells. Toxicol. In Vitro 2014, 28, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Takac, P.; Kello, M.; Pilatova, M.B.; Kudlickova, Z.; Vilkova, M.; Slepcikova, P.; Petik, P.; Mojzis, J. New chalcone derivative exhibits antiproliferative potential by inducing G2/M cell cycle arrest, mitochondrial-mediated apoptosis and modulation of MAPK signalling pathway. Chem.-Biol. Interact. 2018, 292, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Varghese, E.; Samuel, S.M.; Abotaleb, M.; Cheema, S.; Mamtani, R.; Büsselberg, D. The “Yin and Yang” of Natural Compounds in Anticancer Therapy of Triple-Negative Breast Cancers. Cancers 2018, 10, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, E.; Chiaradia-Delatorre, L.D.; Silva, E.; Nunes, R.J.; Yunes, R.A.; Creczynski-Pasa, T.B. Involvement of extrinsic and intrinsic apoptotic pathways together with endoplasmic reticulum stress in cell death induced by naphthylchalcones in a leukemic cell line: Advantages of multi-target action. Toxicol. In Vitro 2014, 28, 769–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Xiangyu, C.; Ya, L.; Yu, W.; Feng, X. An orally antitumor chalcone hybrid inhibited HepG2 cells growth and migration as the tubulin binding agent. Investig. New Drugs 2019, 37, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-C.; Wang, Z.-X.; Jin, S.-J.; Zhang, Y.-X.; Hu, G.-Q.; Cui, D.-T.; Wang, J.-S.; Wang, M.; Wang, F.-Q.; Zhao, Z.-J. Dual Inhibition of Topoisomerase II and Tyrosine Kinases by the Novel Bis-Fluoroquinolone Chalcone-Like Derivative HMNE3 in Human Pancreatic Cancer Cells. PLoS ONE 2016, 11, e0162821. [Google Scholar] [CrossRef] [Green Version]
- Cong, H.; Zhao, X.; Castle, B.T.; Pomeroy, E.J.; Zhou, B.; Lee, J.; Wang, Y.; Bian, T.; Miao, Z.; Zhang, W.; et al. An Indole–Chalcone Inhibits Multidrug-Resistant Cancer Cell Growth by Targeting Microtubules. Mol. Pharm. 2018, 15, 3892–3900. [Google Scholar] [CrossRef]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Anti-cancer chalcones: Structural and molecular target perspectives. Eur. J. Med. Chem. 2015, 98, 69–114. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, X.; Wang, Y.; Sun, Q.; Chen, M.; Liu, S.; Zou, X. Licochalcone A suppresses hexokinase 2-mediated tumor glycolysis in gastric cancer via downregulation of the Akt signaling pathway. Oncol. Rep. 2017, 39, 1181–1190. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.-H.; Chiu, Y.-F.; Wu, W.-J.; Wu, P.-L.; Lin, C.-Y.; Lin, C.-L.; Hsieh, Y.-H.; Liu, C.-J. Synergistic antimetastatic effect of cotreatment with licochalcone A and sorafenib on human hepatocellular carcinoma cells through the inactivation of MKK4/JNK and uPA expression. Environ. Toxicol. 2018, 33, 1237–1244. [Google Scholar] [CrossRef]
- Drutovic, D.; Chripkova, M.; Pilátová, M.; Kruzliak, P.; Perjesi, P.; Šarišský, M.; Lupi, M.; Damia, G.; Broggini, M.; Mojzis, J. Benzylidenetetralones, cyclic chalcone analogues, induce cell cycle arrest and apoptosis in HCT116 colorectal cancer cells. Tumor Biol. 2014, 35, 9967–9975. [Google Scholar] [CrossRef]
- Kello, M.; Drutovic, D.; Pilatova, M.B.; Tischlerova, V.; Perjesi, P.; Mojzis, J. Chalcone derivatives cause accumulation of colon cancer cells in the G2/M phase and induce apoptosis. Life Sci. 2016, 150, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Riaz, S.; Iqbal, M.; Ullah, R.; Zahra, R.; Chotana, G.A.; Faisal, A.; Saleem, R. Synthesis and evaluation of novel α-substituted chalcones with potent anti-cancer activities and ability to overcome multidrug resistance. Bioorganic Chem. 2019, 87, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Dong, A.; Wang, R.; Cao, H. Ursolic acid benzaldehyde chalcone, leads to inhibition of cell proliferation and arrests cycle in G1/G0 phase in colon cancer. Saudi J. Biol. Sci. 2018, 25, 1762–1766. [Google Scholar] [CrossRef] [PubMed]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Kalani, A.; Rai, S.; Swarnkar, S.; Tota, S.; Nath, C.; Tyagi, N. Mechanism of Oxidative Stress and Synapse Dysfunction in the Pathogenesis of Alzheimer’s Disease: Understanding the Therapeutics Strategies. Mol. Neurobiol. 2014, 53, 648–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rani, V.; Deep, G.; Singh, R.; Palle, K.; Yadav, U.C. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Bin Lee, S.; Won, J.; Choi, H.Y.; Kim, K.; Yang, G.-M.; Dayem, A.A.; Cho, S.-G. Correlation between Oxidative Stress, Nutrition, and Cancer Initiation. Int. J. Mol. Sci. 2017, 18, 1544. [Google Scholar] [CrossRef] [Green Version]
- Kwak, J.; Kim, M.-J.; Choi, K.-C.; Choi, H.-K.; Jun, W.; Park, H.-J.; Lee, Y.-H.; Yoon, H.-G. The chalcone derivative Chana 1 protects against amyloid beta peptide-induced oxidative stress and cognitive impairment. Int. J. Mol. Med. 2012, 30, 193–198. [Google Scholar]
- Mahapatra, D.K.; Asati, V.; Bharti, S.K. Chalcones and their therapeutic targets for the management of diabetes: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2015, 92, 839–865. [Google Scholar] [CrossRef]
- Mahapatra, D.K.; Bharti, S.K. Therapeutic potential of chalcones as cardiovascular agents. Life Sci. 2016, 148, 154–172. [Google Scholar] [CrossRef]
- Lau, J.T.F.; Jiang, X.-J.; Zhou, Y.; Lo, P.-C. A disulfide-linked conjugate of ferrocenyl chalcone and silicon(iv) phthalocyanine as an activatable photosensitiser. Chem. Commun. 2013, 49, 4274–4276. [Google Scholar] [CrossRef] [PubMed]
- Hseu, Y.-C.; Huang, Y.-C.; Thiyagarajan, V.; Mathew, D.; Lin, K.-Y.; Chen, S.-C.; Liu, J.-Y.; Hsu, L.-S.; Li, M.-L.; Yang, H.-L. Anticancer activities of chalcone flavokawain B from Alpinia pricei Hayata in human lung adenocarcinoma (A549) cells via induction of reactive oxygen species-mediated apoptotic and autophagic cell death. J. Cell. Physiol. 2019, 234, 17514–17526. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, T.; Zhang, L.; Wang, X.; Dong, H.; Li, L.; Fu, D.; Li, Y.; Zi, X.; Liu, H.-M.; et al. A novel chalcone derivative S17 induces apoptosis through ROS dependent DR5 up-regulation in gastric cancer cells. Sci. Rep. 2017, 7, 9873. [Google Scholar] [CrossRef] [PubMed]
- Strathmann, J.; Klimo, K.; Sauer, S.W.; Okun, J.G.; Prehn, J.H.; Gerhauser, C. Xanthohumol-induced transient superoxide anion radical formation triggers cancer cells into apoptosis via a mitochondria-mediated mechanism. FASEB J. 2010, 24, 2938–2950. [Google Scholar] [CrossRef] [Green Version]
- Guzy, J.; Kubálková, J.; Rozmer, Z.; Fodor, K.; Mareková, M.; Poškrobová, M.; Perjesi, P. Activation of oxidative stress response by hydroxyl substituted chalcones and cyclic chalcone analogues in mitochondria. FEBS Lett. 2009, 584, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.S.; Galati, G.; Pannala, A.S.; Rice-Evans, C.; O’Brien, P.J. Simultaneous detection of the antioxidant and pro-oxidant activity of dietary polyphenolics in a peroxidase system. Free. Radic. Res. 2003, 37, 787–794. [Google Scholar] [CrossRef]
- Carlberg, I.; Mannervik, B. Glutathione reductase. Methods Enzymol. 1985, 113, 484–490. [Google Scholar]
- Zagrodzki, P.; Nicol, F.; McCoy, M.A.; Smyth, J.; Kennedy, D.; Beckett, G.; Arthur, J. Iodine deficiency in cattle: Compensatory changes in thyroidal selenoenzymes. Res. Vet. Sci. 1998, 64, 209–211. [Google Scholar] [CrossRef]
- Floreani, M.; Petrone, M.; Debetto, P.; Palatini, P. A comparison between different methods for the determination of reduced and oxidized glutathione in mammalian tissues. Free. Radic. Res. 1997, 26, 449–455. [Google Scholar] [CrossRef]
- Niki, E. Oxidative stress and antioxidants: Distress or eustress? Arch. Biochem. Biophys. 2016, 595, 19–24. [Google Scholar] [CrossRef]
- Kehrer, J.P.; Klotz, L.-O. Free radicals and related reactive species as mediators of tissue injury and disease: Implications for Health. Crit. Rev. Toxicol. 2015, 45, 765–798. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chikara, S.; Nagaprashantha, L.D.; Singhal, J.; Horne, D.; Awasthi, S.; Singhal, S.S. Oxidative stress and dietary phytochemicals: Role in cancer chemoprevention and treatment. Cancer Lett. 2018, 413, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Bubols, G.B.; Vianna, D.d.R.; Medina-Remon, A.; von Poser, G.; Lamuela-Raventos, R.M.; Eifler-Lima, V.L.; Garcia, S.C. The antioxidant activity of coumarins and flavonoids. Mini Rev. Med. Chem. 2013, 13, 318–334. [Google Scholar] [PubMed]
- Leskovec, J.; Rezar, V.; Svete, A.N.; Salobir, J.; Levart, A. Antioxidative Effects of Olive Polyphenols Compared to Vitamin E in Piglets Fed a Diet Rich in N-3 PUFA. Animals 2019, 9, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eghbaliferiz, S.; Iranshahi, M. Prooxidant Activity of Polyphenols, Flavonoids, Anthocyanins and Carotenoids: Updated Review of Mechanisms and Catalyzing Metals. Phytother. Res. 2016, 30, 1379–1391. [Google Scholar] [CrossRef]
- León-González, A.J.; Auger, C.; Schini-Kerth, V.B. Pro-oxidant activity of polyphenols and its implication on cancer chemoprevention and chemotherapy. Biochem. Pharmacol. 2015, 98, 371–380. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Gupte, A.; Mumper, R.J. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 2009, 35, 32–46. [Google Scholar] [CrossRef]
- Weisburg, J.H.; Weissman, D.B.; Sedaghat, T.; Babich, H. In vitro Cytotoxicity of Epigallocatechin Gallate and Tea Extracts to Cancerous and Normal Cells from the Human Oral Cavity. Pharmacol. Toxicol. 2004, 95, 191–200. [Google Scholar] [CrossRef]
- Fu, D.-J.; Li, J.-H.; Yang, J.-J.; Li, P.; Zhang, Y.-B.; Liu, S.; Li, Z.-R.; Zhang, S.-Y. Discovery of novel chalcone-dithiocarbamates as ROS-mediated apoptosis inducers by inhibiting catalase. Bioorganic Chem. 2019, 86, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Kuete, V.; Nkuete, A.H.L.; Mbaveng, A.T.; Wiench, B.; Wabo, H.K.; Tane, P.; Efferth, T. Cytotoxicity and modes of action of 4′-hydroxy-2′,6′-dimethoxychalcone and other flavonoids toward drug-sensitive and multidrug-resistant cancer cell lines. Phytomedicine 2014, 21, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zheng, Q.; Chen, X.; Wang, Y.; Wang, D.; Wang, J. Isobavachalcone Induces ROS-Mediated Apoptosis via Targeting Thioredoxin Reductase 1 in Human Prostate Cancer PC-3 Cells. Oxidative Med. Cell. Longev. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.; Sun, T.; Du, J.; Zhang, B.; Xiang, D.; Li, W. Xanthohumol, a prenylated flavonoid from Hops, exerts anticancer effects against gastric cancer invitro. Oncol. Rep. 2018, 40, 3213–3222. [Google Scholar]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [Green Version]
- Radi, R. Peroxynitrite, a Stealthy Biological Oxidant. J. Biol. Chem. 2013, 288, 26464–26472. [Google Scholar] [CrossRef] [Green Version]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [Green Version]
- El-Aal, H.A.H.M.A. Lipid Peroxidation End-Products as a Key of Oxidative Stress: Effect of Antioxidant on Their Production and Transfer of Free Radicals. Lipid Peroxidation 2012, 10, 63–88. [Google Scholar]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Chang, Y.-T.; Huang, C.-Y.; Tang, J.-Y.; Liaw, C.-C.; Li, R.-N.; Liu, J.-R.; Sheu, J.-H.; Chang, H.-W. Reactive oxygen species mediate soft corals-derived sinuleptolide-induced antiproliferation and DNA damage in oral cancer cells. OncoTargets Ther. 2017, 10, 3289–3297. [Google Scholar] [CrossRef] [Green Version]
- Radak, Z.; Boldogh, I. 8-Oxo-7,8-dihydroguanine: Links to gene expression, aging, and defense against oxidative stress. Free. Radic. Biol. Med. 2010, 49, 587–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, L.J.; Yang, L.-X. Gamma-H2AX—A novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–309. [Google Scholar] [PubMed]
- Yazdi, P.T.; Wang, Y.; Zhao, S.; Patel, N.; Lee, E.Y.-H.; Qin, J. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genome Res. 2002, 16, 571–582. [Google Scholar] [CrossRef] [Green Version]
- Na Gil, H.; Jung, E.; Koh, D.; Lim, Y.; Lee, Y.; Shin, S.Y. A synthetic chalcone derivative, 2-hydroxy-3′,5,5′-trimethoxychalcone (DK-139), triggers reactive oxygen species-induced apoptosis independently of p53 in A549 lung cancer cells. Chem. Interact. 2019, 298, 72–79. [Google Scholar] [CrossRef]
- Halliwell, B. Free radicals and antioxidants—Quo vadis? Trends Pharmacol. Sci. 2011, 32, 125–130. [Google Scholar] [CrossRef]
- Henry-Mowatt, J.; Dive, C.; Martinou, J.-C.; James, D. Role of mitochondrial membrane permeabilization in apoptosis and cancer. Oncogene 2004, 23, 2850–2860. [Google Scholar] [CrossRef] [Green Version]
- Riedl, S.J.; Salvesen, G.S. The apoptosome: Signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 2007, 8, 405–413. [Google Scholar] [CrossRef]
- Riedl, S.J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004, 5, 897–907. [Google Scholar] [CrossRef]
- Yang, J. Prevention of Apoptosis by Bcl-2: Release of Cytochrome c from Mitochondria Blocked. Science 1997, 275, 1129–1132. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Ly, J.D.; Grubb, D.R.; Lawen, A. The mitochondrial membrane potential (Δψm) in apoptosis; an update. Apoptosis 2003, 8, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K. MAP Kinase Pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, D.; Hideshima, T.; Podar, K.; Mitsiades, C.; Munshi, N.C.; Kharbanda, S.; Anderson, K.C.; Li, G.; Mitsiades, N. JNK-dependent Release of Mitochondrial Protein, Smac, during Apoptosis in Multiple Myeloma (MM) Cells. J. Biol. Chem. 2003, 278, 17593–17596. [Google Scholar] [CrossRef] [Green Version]
- Porras, A.; Zuluaga, S.; Black, E.; Valladares, A.; Alvarez-Barrientos, A.; Ambrosino, C.; Benito, M.; Nebreda, A.R. p38α Mitogen-activated Protein Kinase Sensitizes Cells to Apoptosis Induced by Different Stimuli. Mol. Biol. Cell 2004, 15, 922–933. [Google Scholar] [CrossRef] [Green Version]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death—Apoptosis, autophagy and senescence. FEBS J. 2009, 277, 2–21. [Google Scholar] [CrossRef]
- Son, Y.; Kim, S.; Chung, H.-T.; Pae, H.-O. Reactive Oxygen Species in the Activation of MAP Kinases. Methods Enzymol. 2013, 528, 27–48. [Google Scholar]
- Wang, L.-H.; Li, H.-H.; Li, M.; Wang, S.; Jiang, X.-R.; Li, Y.; Ping, G.-F.; Cao, Q.; Liu, X.; Fang, W.-H.; et al. SL4, a chalcone-based compound, induces apoptosis in human cancer cells by activation of the ROS/MAPK signalling pathway. Cell Prolif. 2015, 48, 718–728. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analysis | Staining Solution | Manufacturer |
|---|---|---|
| ROS | DHR123 (Dihydrorhodamine 123), final concentration 200 nM | Sigma-Aldrich, St. Louis, MO, USA |
| RNS | DAF-FM (Diaminofluorescein-FM) diacetate, final concentration 2 mM | Sigma-Aldrich, St. Louis, MO, USA |
| Lipid peroxidation | BODIPY 581/591 C11, final concentration 1 mM | Sigma-Aldrich, St. Louis, MO, USA |
| Superoxide anion | MitoSox Red mitochondrial superoxide indicator, final concentration 5 µM | Sigma-Aldrich, St. Louis, MO, USA |
| Externalization of phosphatidylserine | Annexin V-FITC, 1:100 Propidium iodide, final concentration 25 µg/mL | BD Biosciences Pharmingen, San Diego, CA, USA |
| Caspase-3 activation | Cleaved caspase-3-PE, 1:200 | BD Biosciences Pharmingen, San Diego, CA, USA |
| Cytochrome c release | Cytochrome c antibody (6H2) FITC conjugate, 1:200 | Invitrogen, Carlsbad, CA, USA |
| Smac/DIABLO release | Smac/DIABLO rabbit mAb, 1:200 | Cell Signaling Technology, Danvers, MA, USA |
| Goat anti-rabbit IgG (H + L) secondary antibody, Alexa Fluor 488, 1:500 | Thermo Scientific, Rockford, IL, USA | |
| Mitochondrial membrane potential | TMRE (tetramethylrhodamine ethyl ester perchlorate), final concentration 0.1 µM | Sigma-Aldrich, St. Louis, MO, USA |
| Protein analysis | Phospho-Bcl-2 (Ser70) rabbit mAb Alexa Fluor 488 conjugate, 1:200 Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (E10) mouse mAb, 1:2000 Phospho-SAPK/JNK (Thr183/Tyr185) (G9) mouse mAb (PE conjugate), 1:200 Phospho-p38 MAPK (Thr180/Tyr182) (3D7) rabbit mAb PE conjugate, 1:200 | Cell Signaling Technology, Danvers, MA, USA |
| Goat anti-rabbit IgG (H+L) secondary antibody, Alexa Fluor 488, 1:500 | Thermo Scientific, Rockford, IL, USA | |
| DNA damage | Anti-pATM, PE conjugated antibody, 1:200 Anti-pHistone H2A.X, PerCP conjugated antibody, 1:200 Anti-pSMC1, Alexa Fluor 488 Antibody, 1:200 | Millipore Corporation, Temecula, CA, USA |
| Anti-oxoguanine 8 antibody | Abcam, Cambridge, United Kingdom | |
| Goat anti-mouse IgG (H + L) secondary antibody, Alexa Fluor 488 | Thermo Scientific, Rockford, IL, USA |
| Primary Antibodies | Mr (kDa) | Origin | Manufacturer |
| β-actin | 45 | Mouse | Cell Signaling Technology, Danvers, MA, USA |
| p38 MAPK | 43 | Rabbit | |
| Phospho-p38MAPK | 43 | Rabbit | |
| p44/42 MAPK (Erk1/2) | 42 + 44 | Rabbit | |
| Phospho-p44/42 MAPK (Erk 1/2) | 42 + 44 | Mouse | |
| JNK1 | 48 | Mouse | |
| Phospho-SAPK/JNK | 54 | Mouse | |
| Cleaved caspase-7 | 18 | Rabbit | |
| PARP | 116 + 89 | Rabbit | |
| Phospho-histone H2A.X | 15 | Rabbit | |
| Secondary Antibodies | Mr (kDa) | Origin | Manufacturer |
| Anti-rabbit IgG HRP | - | Goat | Cell Signalling Technology, Danvers, Massachusetts, USA |
| Anti-mouse IgG/HRP | - | Goat | Dako, Glostrup, Denmark |
| SubG0/G1 | G1 | S | G2 | |
|---|---|---|---|---|
| C_24h | 1.77 ± 0.74 | 44.65 ± 0.65 | 28.45 ± 2.55 | 25.25 ± 2.55 |
| NAC | 2.71 ± 1.36 | 41.30 ± 2.80 | 25.55 ± 0.45 | 30.60 ± 3.50 |
| 1C | 11.30 ± 2.31 b | 29.25 ± 3.15 b | 14.40 ± 2.10 b | 45.25 ± 1.45 c |
| NAC/1C | 4.25 ± 1.23 a** | 50.75 ± 3.45 *** | 15.85 ± 4.45 b | 29.35 ± 2.35 ** |
| C_48h | 0.95 ± 0.65 | 55.45 ± 6.45 | 19.10 ± 6.50 | 24.30 ± 0.90 |
| NAC | 1.92 ± 0.38 | 47.95 ± 0.15 | 23.60 ± 2.00 | 26.55 ± 2.55 |
| 1C | 11.85 ± 0.55 b | 31.80 ± 2.20 b | 17.75 ± 2.65 | 38.35 ± 1.25 b |
| NAC/1C | 5.55 ± 0.95 a*** | 52.95 ± 0.35 ** | 16.85 ± 3.85 | 24.50 ± 3.20 ** |
| C_72h | 0.71 ± 0.03 | 68.30 ± 6.00 | 13.25 ± 1.95 | 17.85 ± 4.45 |
| NAC | 1.53 ± 0.77 | 68.95 ± 6.35 | 14.75 ± 2.55 | 14.55 ± 3.15 |
| 1C | 15.63 ± 7.18 b | 26.25 ± 1.95 c | 18.80 ± 1.00 | 39.00 ± 4.20 b |
| NAC/1C | 3.85 ± 1.24 a* | 67.75 ± 0.65 *** | 10.65 ± 1.65 ** | 17.75 ± 0.25 ** |
| Live (Q1) | Early Apoptotic (Q2) | Late Apoptotic (Q3) | Death (Q4) | |
|---|---|---|---|---|
| C_24h | 97.80 ± 0.65 | 0.05 ± 0.04 | 0.18 ± 0.10 | 2.00 ± 0.49 |
| NAC | 90.95 ± 1.67 | 1.61 ± 1.14 b | 6.20 ± 3.43 b | 1.23 ± 0.633 b |
| 1C | 68.50 ± 0.98 c | 17.84 ± 2.25 c | 3.99 ± 0.99 c | 9.78 ± 0.34 c |
| NAC/1C | 86.65 ± 1.59 b*** | 10.92 ± 2.35 c*** | 0.71 ± 0.32 b** | 1.75 ± 0.45 *** |
| C_48h | 93.55 ± 0.86 | 1.37 ± 0.53 | 2.28 ± 0.62 | 2.75 ± 2.00 |
| NAC | 91.50 ± 0.98 | 0.16 ± 0.13 | 8.10 ± 0.65 b | 0.27 ± 0.19 b |
| 1C | 42.55 ± 5.76 c | 16.53 ± 2.96 c | 17.23 ± 5.69 c | 23.70 ± 3.02 c |
| NAC/1C | 62.15 ± 1.84 b** | 10.49 ± 0.35 c* | 15.61 ± 3.66 c | 11.75 ± 1.43 b** |
| C_72h | 94.30 ± 0.41 | 0.78 ± 0.21 | 1.38 ± 0.51 | 3.56 ± 0.29 |
| NAC | 94.95 ± 0.20 | 0.06 ± 0.05 c | 4.85 ± 0.05 | 0.17 ± 0.09 |
| 1C | 31.85 ± 5.59 c | 11.29 ± 0.58 c | 25.71 ± 4.07 c | 31.20 ± 0.89 c |
| NAC/1C | 61.10 ± 1.23 b** | 8.83 ± 1.48 c* | 11.83 ± 0.96 b* | 18.24 ± 0.70 b*** |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takac, P.; Kello, M.; Vilkova, M.; Vaskova, J.; Michalkova, R.; Mojzisova, G.; Mojzis, J. Antiproliferative Effect of Acridine Chalcone Is Mediated by Induction of Oxidative Stress. Biomolecules 2020, 10, 345. https://doi.org/10.3390/biom10020345
Takac P, Kello M, Vilkova M, Vaskova J, Michalkova R, Mojzisova G, Mojzis J. Antiproliferative Effect of Acridine Chalcone Is Mediated by Induction of Oxidative Stress. Biomolecules. 2020; 10(2):345. https://doi.org/10.3390/biom10020345
Chicago/Turabian StyleTakac, Peter, Martin Kello, Maria Vilkova, Janka Vaskova, Radka Michalkova, Gabriela Mojzisova, and Jan Mojzis. 2020. "Antiproliferative Effect of Acridine Chalcone Is Mediated by Induction of Oxidative Stress" Biomolecules 10, no. 2: 345. https://doi.org/10.3390/biom10020345