Discrimination of Naturally-Occurring 2-Arylbenzofurans as Cyclooxygenase-2 Inhibitors: Insights into the Binding Mode and Enzymatic Inhibitory Activity


Abstract
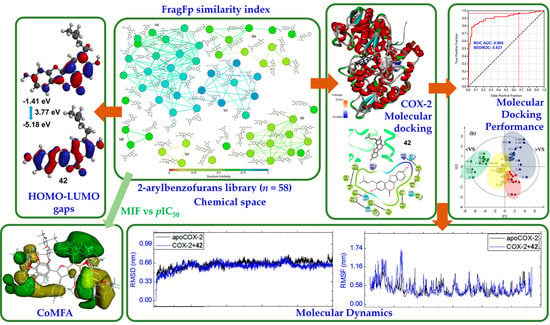
:
1. Introduction
2. Materials and Methods
2.1. Protein Preparation
2.2. Ligand Preparation
2.3. Molecular Docking
2.4. Statistical Analysis
2.5. Molecular Dynamics Simulations
2.6. Binding Free Energy Analyses
2.7. COX-2 Inhibition Assay
2.8. Comparative Molecular Field Analysis (CoMFA)
3. Results and Discussion
3.1. 2-Arylbenzofuran-Related Chemical Space
3.2. Molecular Docking Simulations
3.2.1. Performance and Trends of the Docking Results
3.2.2. Selection of the Best-Docked Compounds
3.2.3. Binding Mode and Residual Interactions of the Best-Docked Compounds
3.3. Stability and Chemical Reactivity of the Best-Docked 2-Arylbenzofurans
3.4. Molecular Dynamics Studies of the Best-Docked 2-Arylbenzofurans
3.5. Binding-Free Energy Calculations of the Best-Docked 2-Arylbenzofurans
3.6. In-Vitro COX-2 Activity of an in-House Collection of 2-Arylbenzofurans
Comparative Molecular Field Analysis (CoMFA)
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Common Name | Reference | No. | Common Name | Reference |
|---|---|---|---|---|---|
| 1 | moracin Y | [7] | 30 | moracin L | [7] |
| 2 | ocophyllal A | [8] | 31 | moracin U | [7] |
| 3 | ocophyllal B | [8] | 32 | moracin P | [7] |
| 4 | moracin A | [7] | 33 | moracin Q | [7] |
| 5 | moracin B | [7] | 34 | moracin V | [7] |
| 6 | moracin F | [7] | 35 | moracin O | [7] |
| 7 | moracin J | [7] | 36 | moracin X | [7] |
| 8 | moracin M | [7] | 37 | substituted benzofuran 2 b | [41] |
| 9 | (+)-obtusafuran | [42] | 38 | isoparvifuran | [43] |
| 10 | substituted benzofuran 1 a | [44] | 39 | moracin D | [7] |
| 11 | 9′-nor-7,8-dehydro-isolicarin-B | [8] | 40 | moracin K | [7] |
| 12 | egonol | [37] | 41 | kanzonol U | [45] |
| 13 | acetylegonol | [37] | 42 | psoralidin | [46] |
| 14 | homoegonol | [37] | 43 | liliflol B | [8] |
| 15 | zanthocapensol | [47] | 44 | (−)-licarin B | [8] |
| 16 | zanthocapensate | [47] | 45 | (+)-licarin B | [8] |
| 17 | acetylhomoegonol | [37] | 46 | (−)-licarin A | [8] |
| 18 | moracin C | [7] | 47 | (+)-licarin A | [8] |
| 19 | moracin I | [7] | 48 | (+)-acuminatin | [8] |
| 20 | moracin N | [7] | 49 | maximol | [48] |
| 21 | moracin S | [7] | 50 | picrasmalignan A | [49] |
| 22 | moracin T | [7] | 51 | salvianolic acid B | [50] |
| 23 | dinklagein A | [9] | 52 | substituted pterocarpan 1 c | [51] |
| 24 | dinklagein B | [9] | 53 | substituted pterocarpan 2 d | [51] |
| 25 | moracin R | [7] | 54 | (+)-glyceollin I | [52] |
| 26 | moracin Z | [7] | 55 | sulfuretin | [53] |
| 27 | moracin W | [7] | 56 | (+)-mirandin A | [8] |
| 28 | moracin G | [7] | 57 | kadsurenone | [8] |
| 29 | moracin H | [7] | 58 | (+)-denudatin B | [8] |
| No. | ES1 (1CVU) a | ES2 (1CX2) a | ES3 (4COX) a | ES4 (5COX) a | ES5 (1PXX) a | ES6 (3NT1) a | ES7 (3LN1) a | ES8 (4RS0) a | ES9 (4PH9) a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VS ± SD b | RMS c | VS ± SD b | RMS c | VS ± SD b | RMS c | VS ± SD b | RMS c | VS ± SD b | RMS c | VS± SD b | RMS c | VS ± SD b | RMS c | VS ± SD b | RMS c | VS ± SD b | RMS c | |
| 1 | −8.8 ± 0.1 | 0.435 | −9.1 ± 0.1 | 0.337 | −8.6 ± 0.1 | 0.808 | −8.4 ± 0.2 | 0.566 | −8.5 ± 0.2 | 0.361 | −8.3 ± 0.1 | 0.879 | −9.7 ± 0.2 | 0.699 | −8.1 ± 0.1 | 0.299 | −8.2 ± 0.1 | 0.303 |
| 2 | −9.6 ± 0.2 | 0.308 | −8.9 ± 0.2 | 0.225 | −10.0 ± 0.2 | 0.319 | −9.7 ± 0.1 | 0.269 | −9.7 ± 0.1 | 0.240 | −8.3 ± 0.2 | 0.282 | −10.2 ± 0.1 | 0.674 | −8.5 ± 0.1 | 0.183 | −7.8 ± 0.1 | 0.264 |
| 3 | −7.3 ± 0.2 | 0.239 | −9.0 ± 0.2 | 0.272 | −8.9 ± 0.2 | 0.255 | −8.6 ± 0.1 | 0.848 | −8.7 ± 0.1 | 0.441 | −7.6 ± 0.1 | 0.264 | −8.9 ± 0.1 | 0.178 | −7.6 ± 0.1 | 0.165 | −6.3 ± 0.2 | 0.210 |
| 4 | −8.7 ± 0.1 | 0.933 | −8.7 ± 0.1 | 0.877 | −8.5 ± 0.1 | 0.936 | −8.1 ± 0.4 | 0.492 | −8.3 ± 0.2 | 0.960 | −8.2 ± 0.2 | 0.358 | −9.5 ± 0.1 | 0.447 | −8.1 ± 0.2 | 0.334 | −7.8 ± 0.1 | 1.075 |
| 5 | −8.6 ± 0.2 | 0.973 | −9.1 ± 0.1 | 0.262 | −8.1 ± 0.1 | 0.699 | −8.4 ± 0.3 | 0.311 | −8.4 ± 0.1 | 0.355 | −8.2 ± 0.2 | 0.238 | −9.6 ± 0.1 | 0.128 | −8.1 ± 0.1 | 0.325 | −8.0 ± 0.1 | 0.344 |
| 6 | −8.5 ± 0.1 | 0.342 | −8.3 ± 0.1 | 0.344 | −8.2 ± 0.2 | 0.587 | −8.1 ± 0.1 | 0.288 | −8.0 ± 0.1 | 0.885 | −7.3 ± 0.1 | 0.927 | −9.8 ± 0.2 | 0.038 | −7.4 ± 0.1 | 0.351 | −7.7 ± 0.1 | 1.182 |
| 7 | −8.5 ± 0.1 | 0.346 | −9 ± 0.2 | 0.352 | −7.9 ± 0.2 | 0.279 | −8.4 ± 0.1 | 0.880 | −8.2 ± 0.1 | 0.334 | −8.4 ± 0.1 | 0.374 | −9.5 ± 0.1 | 0.673 | −8.0 ± 0.1 | 0.344 | −8.0 ± 0.1 | 1.201 |
| 8 | −8.5 ± 0.1 | 0.432 | −8.8 ± 0.1 | 0.854 | −8.4 ± 0.1 | 0.924 | −8.5 ± 0.1 | 0.869 | −8.4 ± 0.2 | 0.385 | −8.4 ± 0.1 | 0.400 | −9.3 ± 0.2 | 0.370 | −8.2 ± 0.1 | 0.650 | −7.9 ± 0.1 | 0.298 |
| 9 | −8.7 ± 0.2 | 0.869 | −8.8 ± 0.2 | 0.503 | −8.9 ± 0.2 | 0.986 | −9.8 ± 0.2 | 0.304 | −9.0 ± 0.1 | 0.308 | −8.6 ± 0.1 | 0.455 | −9.2 ± 0.2 | 0.734 | −8.6 ± 0.1 | 0.664 | −8.7 ± 0.1 | 0.619 |
| 10 | −8.4 ± 0.1 | 0.346 | −8.8 ± 0.1 | 0.314 | −8.7 ± 0.2 | 0.358 | −8.6 ± 0.1 | 0.707 | −8.8 ± 0.1 | 0.329 | −8.1 ± 0.1 | 0.490 | −9.3 ± 0.2 | 0.941 | −8.1 ± 0.1 | 0.362 | −7.8 ± 0.1 | 0.274 |
| 11 | −10.6 ± 0.2 | 0.276 | −10.5 ± 0.4 | 0.802 | −10.8 ± 0.2 | 0.299 | −10.9 ± 0.1 | 0.924 | −11.3 ± 0.1 | 0.178 | −10.8 ± 0.2 | 0.661 | −10.6 ± 0.1 | 0.935 | −10.4 ± 0.2 | 0.118 | −10.9 ± 0.2 | 0.220 |
| 12 | −9.2 ± 0.1 | 0.395 | −9.9 ± 0.4 | 0.797 | −9.7 ± 0.1 | 0.499 | −9.2 ± 0.1 | 0.789 | −9.2 ± 0.2 | 0.324 | −6.9 ± 0.1 | 0.252 | −9.9 ± 0.2 | 0.860 | −8.0 ± 0.1 | 0.213 | −8.9 ± 0.1 | 0.313 |
| 13 | −9.8 ± 0.1 | 0.652 | −10.6 ± 0.2 | 0.662 | −10.6 ± 0.1 | 0.287 | −9.5 ± 0.1 | 0.422 | −9.6 ± 0.1 | 0.273 | −7.6 ± 0.3 | 0.255 | −9.8 ± 0.2 | 0.594 | −7.4 ± 0.3 | 1.110 | −8.3 ± 0.2 | 0.947 |
| 14 | −8.7 ± 0.3 | 1.054 | −8.7 ± 0.2 | 0.577 | −9.4 ± 0.2 | 0.362 | −8.2 ± 0.1 | 0.716 | −8.8 ± 0.1 | 0.285 | −7.7 ± 0.1 | 0.307 | −9.0 ± 0.2 | 0.918 | −7.8 ± 0.1 | 0.275 | −8.9 ± 0.1 | 0.267 |
| 15 | −9.6 ± 0.1 | 1.134 | −10.3 ± 0.2 | 0.605 | −9.0 ± 0.1 | 0.327 | −9.3 ± 0.2 | 0.318 | −9.4 ± 0.1 | 0.295 | −6.9 ± 0.3 | 0.277 | −9.8 ± 0.1 | 0.829 | −7.1 ± 0.1 | 1.393 | −9.0 ± 0.2 | 0.244 |
| 16 | −9.6 ± 0.1 | 0.276 | −10.6 ± 0.2 | 0.767 | −9.6 ± 0.1 | 0.433 | −9.1 ± 0.3 | 0.258 | −9.6 ± 0.1 | 0.477 | −7.4 ± 0.2 | 0.252 | −9.6 ± 0.2 | 0.477 | −7.5 ± 0.2 | 0.107 | −8.8 ± 0.1 | 0.260 |
| 17 | −9.2 ± 0.1 | 0.587 | −9.2 ± 0.3 | 0.572 | −10.2 ± 0.2 | 0.424 | −8.9 ± 0.2 | 0.414 | −9.4 ± 0.1 | 0.253 | −7.6 ± 0.1 | 0.293 | −9.4 ± 0.1 | 0.573 | −7.7 ± 0.1 | 0.555 | −8.2 ± 0.1 | 0.974 |
| 18 | −9.0 ± 0.1 | 0.485 | −8.7 ± 0.3 | 0.350 | −9.5 ± 0.1 | 0.316 | −9.4 ± 0.1 | 0.373 | −9.2 ± 0.1 | 0.314 | −7.7 ± 0.1 | 0.351 | −11.0 ± 0.1 | 0.827 | −7.0 ± 0.5 | 0.391 | −7.4 ± 0.2 | 0.418 |
| 19 | −9.3 ± 0.2 | 0.385 | −8.5 ± 0.1 | 0.353 | −9.2 ± 0.2 | 0.768 | −8.9 ± 0.1 | 0.872 | −8.6 ± 0.1 | 0.386 | −8.5 ± 0.2 | 0.860 | −10.7 ± 0.1 | 0.144 | −9.1 ± 0.1 | 0.311 | −8.4 ± 0.1 | 0.360 |
| 20 | −9.5 ± 0.1 | 1.199 | −9.6 ± 0.2 | 0.900 | −9.6 ± 0.2 | 0.317 | −9.1 ± 0.2 | 0.333 | −9.8 ± 0.2 | 0.325 | −7.4 ± 0.1 | 0.924 | −11.1 ± 0.1 | 0.586 | −7.3 ± 0.1 | 0.393 | −8.6 ± 0.1 | 0.388 |
| 21 | −9.9 ± 0.1 | 0.361 | −9.6 ± 0.4 | 0.913 | −9.2 ± 0.1 | 0.673 | −10.3 ± 0.1 | 0.789 | −9.0 ± 0.1 | 0.344 | −8.1 ± 0.1 | 0.845 | −10.8 ± 0.2 | 0.532 | −8.6 ± 0.1 | 0.305 | −8.5 ± 0.1 | 0.403 |
| 22 | −9.1 ± 0.1 | 0.274 | −10.0 ± 0.2 | 0.489 | −8.8 ± 0.1 | 0.520 | −8.9 ± 0.1 | 0.394 | −9.1 ± 0.1 | 0.343 | −8.4 ± 0.1 | 0.331 | −10.7 ± 0.3 | 0.958 | −8.1 ± 0.1 | 1.139 | −8.3 ± 0.1 | 0.359 |
| 23 | −9.4 ± 0.2 | 0.345 | −9.6 ± 0.3 | 0.563 | −9 ± 0.1 | 0.569 | −10.4 ± 0.2 | 0.338 | −9.2 ± 0.1 | 0.784 | −8.5 ± 0.2 | 0.632 | −10.9 ± 0.1 | 0.358 | −8.9 ± 0.2 | 0.478 | −8.4 ± 0.2 | 0.412 |
| 24 | −9.3 ± 0.2 | 0.756 | −9.2 ± 0.3 | 0.478 | −9.2 ± 0.2 | 0.656 | −10.6 ± 0.2 | 0.589 | −9.4 ± 0.1 | 0.779 | −8.2 ± 0.1 | 0.456 | −10.5 ± 0.3 | 0.665 | −8.7 ± 0.2 | 0.851 | −8 ± 0.2 | 0.458 |
| 25 | −8.4 ± 0.1 | 0.372 | −8.7 ± 0.3 | 0.391 | −9.3 ± 0.1 | 0.401 | −8.7 ± 0.2 | 0.885 | −9.6 ± 0.1 | 0.953 | −6.7 ± 0.3 | 0.382 | −10.8 ± 0.1 | 0.249 | −7.6 ± 0.2 | 0.391 | −6.7 ± 0.1 | 0.412 |
| 26 | −9.0 ± 0.1 | 0.950 | −9.5 ± 0.2 | 0.919 | −9.4 ± 0.1 | 0.473 | −8.8 ± 0.3 | 0.776 | −9.3 ± 0.1 | 0.322 | −6.8 ± 0.7 | 0.395 | −10.7 ± 0.2 | 0.719 | −7.5 ± 0.1 | 0.554 | −8.0 ± 0.1 | 0.392 |
| 27 | −10.7 ± 0.1 | 0.535 | −9.7 ± 0.3 | 0.561 | −9.9 ± 0.1 | 0.977 | −10.0 ± 0.3 | 0.571 | −9.7 ± 0.5 | 0.823 | −10.0 ± 0.1 | 0.507 | −11.1 ± 0.3 | 0.413 | −9.8 ± 0.2 | 0.373 | −10.5 ± 0.2 | 0.403 |
| 28 | −10.7 ± 0.1 | 0.859 | −9.8 ± 0.1 | 0.344 | −10.4 ± 0.1 | 0.275 | −10.3 ± 0.1 | 0.336 | −9.9 ± 0.1 | 0.329 | −10.4 ± 0.5 | 0.303 | −11.3 ± 0.1 | 0.660 | −10.6 ± 0.1 | 0.290 | −10.8 ± 0.1 | 0.303 |
| 29 | −10.3 ± 0.2 | 0.351 | −10.9 ± 0.5 | 0.692 | −10.3 ± 0.1 | 0.328 | −10.0 ± 0.1 | 0.350 | −10.1 ± 0.1 | 0.336 | −10.5 ± 0.2 | 0.288 | −11.4 ± 0.1 | 0.883 | −10.6 ± 0.1 | 0.257 | −10.3 ± 0.5 | 3.621 |
| 30 | −10.2 ± 0.1 | 0.864 | −10.5 ± 0.2 | 0.329 | −10.2 ± 0.1 | 0.472 | −10.4 ± 0.1 | 0.366 | −9.3 ± 0.2 | 0.363 | −10.4 ± 0.4 | 0.346 | −11.3 ± 0.1 | 0.604 | −10.7 ± 0.1 | 0.354 | −10.8 ± 0.1 | 0.355 |
| 31 | −9.1 ± 0.1 | 0.340 | −9.9 ± 0.2 | 0.372 | −10.0 ± 0.4 | 0.396 | −9.7 ± 0.1 | 0.404 | −10.7 ± 0.1 | 0.932 | −7.2 ± 0.3 | 0.353 | −11.3 ± 0.1 | 0.622 | −8.7 ± 0.1 | 0.404 | −8.8 ± 0.1 | 0.419 |
| 32 | −8.8 ± 0.1 | 0.915 | −9.3 ± 0.1 | 0.316 | −10.3 ± 0.1 | 0.353 | −9.7 ± 0.1 | 0.918 | −9.5 ± 0.1 | 0.335 | −6.6 ± 1.8 | 0.547 | −11.3 ± 0.2 | 0.999 | −6.3 ± 0.2 | 0.300 | −7.8 ± 0.1 | 0.390 |
| 33 | −7.0 ± 0.1 | 0.311 | −9.2 ± 0.2 | 0.301 | −10.2 ± 0.2 | 0.302 | −9.0 ± 0.1 | 0.297 | −9.4 ± 0.1 | 0.263 | −7.1 ± 0.1 | 0.213 | −9.6 ± 0.2 | 0.662 | −6.4 ± 0.1 | 0.206 | −7.0 ± 0.1 | 0.288 |
| 34 | −8.6 ± 0.2 | 0.340 | −9.1 ± 0.1 | 0.359 | −10.3 ± 0.1 | 0.409 | −9.8 ± 0.3 | 0.931 | −9.0 ± 0.1 | 0.672 | −6.4 ± 1.7 | 0.380 | −11.2 ± 0.1 | 0.918 | −6.3 ± 0.1 | 0.364 | −7.0 ± 0.1 | 0.358 |
| 35 | −8.2 ± 0.2 | 0.481 | −9.6 ± 0.1 | 0.314 | −10.0 ± 0.1 | 0.353 | −9.2 ± 0.1 | 0.358 | −9.8 ± 0.1 | 0.893 | −7.2 ± 0.1 | 0.381 | −9.8 ± 0.1 | 0.683 | −6.9 ± 0.2 | 0.352 | −7.6 ± 0.1 | 1.113 |
| 36 | −9.1 ± 0.1 | 0.792 | −9.7 ± 0.3 | 0.321 | −9.0 ± 0.1 | 0.507 | −8.3 ± 0.3 | 0.898 | −9.1 ± 0.1 | 0.735 | −8.2 ± 0.1 | 0.296 | −10.7 ± 0.2 | 0.815 | −8.6 ± 0.1 | 0.302 | −8.5 ± 0.1 | 1.144 |
| 37 | −9.4 ± 0.1 | 0.592 | −9.4 ± 0.1 | 0.474 | −9.3 ± 0.1 | 0.630 | −9.1 ± 0.2 | 0.887 | −9.5 ± 0.1 | 0.773 | −7.9 ± 0.1 | 0.225 | −9.6 ± 0.2 | 0.865 | −8.4 ± 0.1 | 0.140 | −8.7 ± 0.2 | 0.216 |
| 38 | −9.0 ± 0.1 | 0.463 | −8.6 ± 0.2 | 0.839 | −8.8 ± 0.2 | 0.502 | −9.4 ± 0.1 | 0.301 | −8.5 ± 0.1 | 0.486 | −8.2 ± 0.1 | 0.327 | −9.8 ± 0.1 | 0.049 | −8.2 ± 0.1 | 0.538 | −8.1 ± 0.1 | 0.328 |
| 39 | −10.1 ± 0.1 | 0.306 | −10.3 ± 0.5 | 0.334 | −9.7 ± 0.1 | 0.922 | −9.2 ± 0.1 | 0.299 | −10.2 ± 0.1 | 0.312 | −10.2 ± 0.3 | 0.339 | −11.8 ± 0.2 | 0.826 | −10.2 ± 0.1 | 0.316 | −10.1 ± 0.2 | 0.331 |
| 40 | −10.0 ± 0.1 | 0.562 | −10.5 ± 0.1 | 0.283 | −10.1 ± 0.1 | 0.954 | −10.1 ± 0.1 | 0.492 | −9.8 ± 0.2 | 0.335 | −9.8 ± 0.5 | 0.886 | −11.7 ± 0.1 | 0.712 | −10.6 ± 0.1 | 0.305 | −10.1 ± 0.2 | 0.398 |
| 41 | −10.3 ± 0.1 | 0.322 | −10.3 ± 0.1 | 0.258 | −10.3 ± 0.2 | 0.307 | −9.6 ± 0.1 | 0.793 | −9.3 ± 0.1 | 0.780 | −8.7 ± 0.1 | 0.242 | −10.7 ± 0.1 | 0.747 | −8.9 ± 0.2 | 0.222 | −9.2 ± 0.1 | 0.352 |
| 42 | −10.5 ± 0.1 | 0.302 | −11.6 ± 0.1 | 0.328 | −10.9 ± 0.1 | 0.324 | −10.9 ± 0.1 | 0.315 | −10.6 ± 0.1 | 0.303 | −11.0 ± 0.1 | 0.258 | −12.0 ± 0.2 | 0.683 | −11.9 ± 0.1 | 0.264 | −11.9 ± 0.1 | 0.241 |
| 43 | −9.2 ± 0.1 | 0.221 | −9.6 ± 0.2 | 0.324 | −10.2 ± 0.1 | 0.301 | −10.0 ± 0.1 | 0.310 | −9.9 ± 0.2 | 0.283 | −8.0 ± 0.1 | 0.296 | −9.7 ± 0.1 | 0.665 | −8.6 ± 0.1 | 0.226 | −8.7 ± 0.1 | 0.275 |
| 44 | −9.1 ± 0.3 | 0.255 | −9.7 ± 0.2 | 0.249 | −9.8 ± 0.2 | 0.289 | −10.2 ± 0.2 | 0.239 | −9.6 ± 0.1 | 0.243 | −7.1 ± 0.1 | 0.278 | −9.8 ± 0.2 | 0.915 | −8.3 ± 0.1 | 0.172 | −8.6 ± 0.1 | 0.239 |
| 45 | −7.4 ± 0.2 | 0.246 | −9.9 ± 0.4 | 0.236 | −10.1 ± 0.3 | 0.234 | −9.3 ± 0.2 | 0.303 | −9.9 ± 0.2 | 0.287 | −8.5 ± 0.2 | 0.411 | −10.2 ± 0.2 | 0.904 | −8.7 ± 0.1 | 0.142 | −8.9 ± 0.1 | 0.253 |
| 46 | −9.0 ± 0.2 | 0.286 | −9.2 ± 0.1 | 0.908 | −9.7 ± 0.1 | 0.264 | −9.5 ± 0.2 | 0.295 | −9.7 ± 0.2 | 0.290 | −6.8 ± 0.1 | 0.738 | −9.8 ± 0.2 | 0.943 | −7.9 ± 0.1 | 0.292 | −7.5 ± 0.2 | 0.262 |
| 47 | −7.3 ± 0.1 | 0.267 | −9.4 ± 0.2 | 0.300 | −9.6 ± 0.1 | 0.295 | −9.4 ± 0.1 | 0.282 | −9.5 ± 0.2 | 0.225 | −7.3 ± 0.1 | 0.261 | −10.1 ± 0.2 | 0.891 | −7.9 ± 0.1 | 0.254 | −7.5 ± 0.1 | 0.330 |
| 48 | −7.1 ± 0.2 | 0.274 | −9.8 ± 0.3 | 0.298 | −9.5 ± 0.1 | 0.405 | −9.5 ± 0.2 | 0.245 | −9.7 ± 0.1 | 0.229 | −7.9 ± 0.1 | 0.264 | −10.1 ± 0.1 | 0.912 | −8.1 ± 0.1 | 0.187 | −7.4 ± 0.1 | 0.179 |
| 49 | −10.2 ± 0.1 | 0.582 | −11.4 ± 0.1 | 0.329 | −11.6 ± 0.1 | 0.513 | −12.2 ± 0.2 | 0.319 | −10.8 ± 0.2 | 0.671 | −11.1 ± 0.6 | 0.512 | −12.7 ± 0.2 | 0.878 | −10.6 ± 0.1 | 0.352 | −10.7 ± 0.1 | 0.308 |
| 50 | −8.3 ± 0.1 | 0.281 | −9.1 ± 0.1 | 0.515 | −11.3 ± 0.2 | 0.712 | −9.2 ± 0.2 | 0.846 | −6.5 ± 0.2 | 0.428 | −7.1 ± 0.3 | 0.258 | −10.1 ± 0.1 | 0.910 | −7.1 ± 0.1 | 0.240 | −7.6 ± 0.1 | 0.488 |
| 51 | −9.2 ± 0.1 | 0.510 | −10.5 ± 0.5 | 0.459 | −10.6 ± 0.2 | 0.808 | −9.9 ± 0.2 | 0.643 | −10.2 ± 0.1 | 0.372 | −10.8 ± 0.1 | 0.726 | −11.2 ± 0.2 | 0.978 | −10.4 ± 0.2 | 0.568 | −10.5 ± 0.2 | 0.525 |
| 52 | −8.4 ± 0.1 | 0.494 | −8.6 ± 0.2 | 0.334 | −9.0 ± 0.1 | 0.352 | −8.7 ± 0.2 | 0.336 | −8.8 ± 0.2 | 0.793 | −6.9 ± 0.2 | 0.297 | −9.1 ± 0.3 | 0.916 | −7.4 ± 0.1 | 0.256 | −6.7 ± 0.1 | 0.293 |
| 53 | −9.5 ± 0.1 | 0.358 | −9.4 ± 0.1 | 0.375 | −9.6 ± 0.1 | 0.556 | −9.1 ± 0.2 | 0.337 | −9.8 ± 0.1 | 0.653 | −8.4 ± 0.1 | 0.389 | −9.4 ± 0.1 | 0.889 | −8.9 ± 0.1 | 0.281 | −7.5 ± 0.2 | 0.368 |
| 54 | −10.5 ± 0.1 | 0.365 | −10.5 ± 0.1 | 0.312 | −10.7 ± 0.1 | 0.348 | −10.7 ± 0.1 | 0.354 | −8.9 ± 0.1 | 0.307 | −10.7 ± 0.6 | 0.324 | −10.3 ± 0.2 | 0.453 | −10.4 ± 0.1 | 0.242 | −10.8 ± 0.1 | 0.372 |
| 55 | −8.6 ± 0.1 | 0.376 | −9.1 ± 0.1 | 0.361 | −8.7 ± 0.2 | 0.297 | −8.9 ± 0.1 | 0.374 | −8.4 ± 0.2 | 0.338 | −8.3 ± 0.1 | 0.364 | −9.8 ± 0.1 | 0.998 | −8.4 ± 0.1 | 0.374 | −8.3 ± 0.2 | 0.412 |
| 56 | −7.8 ± 0.1 | 0.256 | −8.8 ± 0.2 | 0.252 | −9.0 ± 0.1 | 0.307 | −9.0 ± 0.1 | 0.300 | −8.4 ± 0.2 | 0.641 | −8.2 ± 0.1 | 0.257 | −8.8 ± 0.3 | 0.856 | −8.5 ± 0.1 | 0.193 | −7.9 ± 0.2 | 0.250 |
| 57 | −6.4 ± 0.1 | 0.275 | −8.5 ± 0.4 | 0.436 | −8.8 ± 0.2 | 0.281 | −8.2 ± 0.4 | 0.232 | −9.0 ± 0.1 | 0.260 | −6.7 ± 0.1 | 0.240 | −9.8 ± 0.2 | 0.643 | −5.9 ± 0.1 | 0.172 | −6.6 ± 0.2 | 0.228 |
| 58 | −7.6 ± 0.1 | 0.247 | −8.4 ± 0.1 | 0.471 | −9.6 ± 0.1 | 0.230 | −9.3 ± 0.1 | 0.246 | −7.8 ± 0.2 | 0.508 | −8.3 ± 0.1 | 0.173 | −8.8 ± 0.2 | 0.995 | −8.4 ± 0.1 | 0.186 | −7.9 ± 0.2 | 0.249 |
| Natural Ligand d and Known Inhibitors e | ||||||||||||||||||
| AA | −8.6 ± 0.2 | 1.381 | −8.4 ± 0.3 | 1.259 | −7.8 ± 0.3 | 1.331 | −7.5 ± 0.6 | 1.236 | −8.1 ± 0.4 | 1.562 | −7.7 ± 0.2 | 0.417 | −8.2 ± 0.3 | 0.533 | −7.8 ± 0.4 | 1.252 | −7.7 ± 0.3 | 2.993 |
| CEL | −10.7 ± 0.1 | 0.493 | −11.6 ± 0.1 | 0.421 | −10.9 ± 0.1 | 0.551 | −10.8 ± 0.1 | 0.313 | −10.9 ± 0.1 | 0.434 | −11.0 ± 0.1 | 0.411 | −12.7 ± 0.2 | 0.885 | −11.1 ± 0.1 | 0.500 | −11.4 ± 0.1 | 0.384 |
| DCL | −8.7 ± 0.3 | 0.289 | −8.2 ± 0.2 | 0.270 | −8.3 ± 0.4 | 0.256 | −8.1 ± 0.1 | 0.370 | −8.7 ± 0.1 | 0.174 | −8.5 ± 0.2 | 0.264 | −8.6 ± 0.1 | 0.838 | −8.0 ± 0.1 | 0.152 | −7.9 ± 0.1 | 0.248 |
| IMD | −8.2 ± 0.1 | 0.530 | −10.3 ± 0.3 | 0.446 | −10.7 ± 0.3 | 0.825 | −10.5 ± 0.1 | 0.298 | −9.2 ± 0.1 | 0.457 | −9.5 ± 0.1 | 0.289 | −9.9 ± 0.4 | 0.821 | −9.6 ± 0.1 | 0.169 | −9.1 ± 0.1 | 0.205 |
| NPX | −8.6 ± 0.1 | 0.854 | −8.5 ± 0.2 | 0.339 | −8.7 ± 0.1 | 0.275 | −8.4 ± 0.2 | 0.266 | −8.3 ± 0.1 | 0.309 | −9.3 ± 0.1 | 0.276 | −8.6 ± 0.1 | 0.765 | −8.7 ± 0.2 | 0.075 | −8.8 ± 0.1 | 0.221 |
| SC58 | −10.7 ± 0.1 | 0.310 | −12 ± 0.2 | 0.313 | −10.9 ± 0.1 | 0.613 | −10.6 ± 0.2 | 0.349 | −11.2 ± 0.1 | 0.2631 | −10.6 ± 0.1 | 0.449 | −12.0 ± 0.2 | 0.786 | −10.6 ± 0.1 | 0.268 | −11.2 ± 0.1 | 0.356 |
| S−IBP | −7.5 ± 0.1 | 0.222 | −7.6 ± 0.1 | 0.650 | −8.2 ± 0.1 | 0.199 | −8.2 ± 0.2 | 0.235 | −8.5 ± 0.1 | 0.592 | −8.2 ± 0.1 | 0.180 | −7.5 ± 0.1 | 0.528 | −7.7 ± 0.1 | 0.589 | −7.8 ± 0.2 | 2.549 |
| R−IBP | −6.5 ± 0.1 | 0.377 | −6.0 ± 0.1 | 1.065 | −6.7 ± 0.1 | 0.550 | −6.1 ± 0.1 | 1.376 | −6.5 ± 0.2 | 0.267 | −6.9 ± 0.1 | 1.434 | −6.7 ± 0.1 | 0.665 | −5.5 ± 0.1 | 0.574 | −5.8 ± 0.1 | 0.751 |
| Descriptors | 11 | 29 | 42 | 49 |
|---|---|---|---|---|
| MW | 316.39 | 348.43 | 348.43 | 431.54 |
| logP | 2.0404 | 1.0172 | 0.9567 | 2.0612 |
| logD | 3.2 | 1.05 | 1.47 | 3.72 |
| logSw | −2.82 | −2.51 | −2.41 | −3.48 |
| tPSA | 36.92 | 68.15 | 79.15 | 69.92 |
| RotatableB | 4 | 2 | 3 | 5 |
| RigidB | 20 | 22 | 20 | 28 |
| Flexibility | 0.17 | 0.08 | 0.13 | 0.15 |
| HBD | 0 | 2 | 3 | 3 |
| HBA | 4 | 5 | 5 | 4 |
| HBD_HBA | 4 | 7 | 8 | 7 |
| Rings | 2 | 2 | 1 | 4 |
| MaxSizeRing | 9 | 14 | 17 | 9 |
| NumCharges | 0 | 0 | 0 | 0 |
| TotalCharge | 0 | 0 | 0 | 0 |
| HeavyAtoms | 23 | 25 | 25 | 32 |
| CarbonAtoms | 19 | 20 | 20 | 28 |
| HeteroAtoms | 4 | 5 | 5 | 4 |
| ratioH/C | 0.21 | 0.25 | 0.25 | 0.14 |
| Lipinski_Violation | 0 | 0 | 0 | 0 |
| Sol (mg/L) | 18,800.83 | 28,341.42 | 31,451.27 | 13,240.01 |
| Sol Forecast Index | Good Solubility | Good Solubility | Good Solubility | Good Solubility |
| OB (VEBER) | Good | Good | Good | Good |
| OB (EGAN) | Good | Good | Good | Good |
| TrafficLights | 0 | 0 | 0 | 1 |
| Phospholipidosis | Non Inducer | Non Inducer | Non Inducer | Non Inducer |
| Overall Result | Accepted | Accepted | Accepted | Accepted |


References
- Clària, J. Cyclooxygenase-2 biology. Curr. Pharm. Des. 2003, 9, 2177–2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Praticò, D.; Dogné, J.-M. Selective cyclooxygenase-2 inhibitors development in cardiovascular medicine. Circulation 2005, 112, 1073–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayar, A.M.; Lagarde, N.; Ferroud, C.; Zagury, J.F.; Montes, M.; Sylla-Iyarreta Veitia, M. Update on COX-2 selective inhibitors: Chemical classification, side effects and their use in cancers and neuronal diseases. Curr. Top. Med. Chem. 2017, 17, 2935–2956. [Google Scholar] [CrossRef] [PubMed]
- White, W.B.; Faich, G.; Borer, J.S.; Makuch, R.W. Cardiovascular thrombotic events in arthritis trials of the cyclooxygenase-2 inhibitor celecoxib. ACC Curr. J. Rev. 2003, 12, 14–15. [Google Scholar] [CrossRef]
- Khanam, H. Shamsuzzaman Bioactive Benzofuran derivatives: A review. Eur. J. Med. Chem. 2015, 97, 483–504. [Google Scholar] [CrossRef] [PubMed]
- Ragab, F.A.E.-F.; Eid, N.M.; Hassan, G.S.; Nissan, Y.M. Synthesis and anti-inflammatory activity of some benzofuran and benzopyran-4-one derivatives. Chem. Pharm. Bull. (Tokyo) 2012, 60, 110–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, R.; Harmalkar, D.S.; Xu, X.; Jang, K.; Lee, K. Bioactive benzofuran derivatives: Moracins A–Z in medicinal chemistry. Eur. J. Med. Chem. 2015, 90, 379–393. [Google Scholar] [CrossRef]
- Coy, E.D.; Cuca, L.E.; Sefkow, M. COX, LOX and platelet aggregation inhibitory properties of Lauraceae neolignans. Bioorg. Med. Chem. Lett. 2009, 19, 6922–6925. [Google Scholar] [CrossRef]
- Kapche, D.W.F.G.; Lekane, N.M.; Kulabas, S.S.; Ipek, H.; Tok, T.T.; Ngadjui, B.T.; Demirtas, I.; Tumer, T.B. Aryl benzofuran derivatives from the stem bark of Calpocalyx dinklagei attenuate inflammation. Phytochemistry 2017, 141, 70–79. [Google Scholar] [CrossRef]
- Kiefer, J.R.; Pawlitz, J.L.; Moreland, K.T.; Stegeman, R.A.; Hood, W.F.; Gierse, J.K.; Stevens, A.M.; Goodwin, D.C.; Rowlinson, S.W.; Marnett, L.J.; et al. Structural insights into the stereochemistry of the cyclooxygenase reaction. Nature 2000, 405, 97–101. [Google Scholar] [CrossRef]
- Kurumbail, R.G.; Stevens, A.M.; Gierse, J.K.; McDonald, J.J.; Stegeman, R.A.; Pak, J.Y.; Gildehaus, D.; iyashiro, J.M.; Penning, T.D.; Seibert, K.; et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996, 384, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Rowlinson, S.W.; Kiefer, J.R.; Prusakiewicz, J.J.; Pawlitz, J.L.; Kozak, K.R.; Kalgutkar, A.S.; Stallings, W.C.; Kurumbail, R.G.; Marnett, L.J. A novel mechanism of cyclooxygenase-2 inhibition involving interactions with Ser-530 and Tyr-385. J. Biol. Chem. 2003, 278, 45763–45769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duggan, K.C.; Walters, M.J.; Musee, J.; Harp, J.M.; Kiefer, J.R.; Oates, J.A.; Marnett, L.J. Molecular basis for cyclooxygenase inhibition by the non-steroidal anti-inflammatory drug naproxen. J. Biol. Chem. 2010, 285, 34950–34959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.L.; Limburg, D.; Graneto, M.J.; Springer, J.; Hamper, J.R.B.; Liao, S.; Pawlitz, J.L.; Kurumbail, R.G.; Maziasz, T.; Talley, J.J.; et al. The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: The second clinical candidate having a shorter and favorable human half-life. Bioorg. Med. Chem. Lett. 2010, 20, 7159–7163. [Google Scholar] [CrossRef]
- Blobaum, A.L.; Xu, S.; Rowlinson, S.W.; Duggan, K.C.; Banerjee, S.; Kudalkar, S.N.; Birmingham, W.R.; Ghebreselasie, K.; Marnett, L.J. Action at a distance: Mutations of peripheral residues transform rapid reversible inhibitors to slow, tight binders of cyclooxygenase-2. J. Biol. Chem. 2015, 290, 12793–12803. [Google Scholar] [CrossRef] [Green Version]
- Orlando, B.J.; Lucido, M.J.; Malkowski, M.G. The structure of ibuprofen bound to cyclooxygenase-2. J. Struct. Biol. 2015, 189, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Empereur-Mot, C.; Zagury, J.-F.; Montes, M. Screening explorer–An interactive tool for the analysis of screening results. J. Chem. Inf. Model. 2016, 56, 2281–2286. [Google Scholar] [CrossRef]
- Guerrero-Perilla, C.; Bernal, F.A.; Coy-Barrera, E. Insights into the interaction and binding mode of a set of antifungal azoles as inhibitors of potential fungal enzyme-based targets. Mol. Divers. 2018, 22, 929–942. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, Y.; Tian, S.; Xu, L.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 2014, 16, 16719–16729. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, Y.; Shen, M.; Tian, S.; Xu, L.; Pan, P.; Guan, Y.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys. Chem. Chem. Phys. 2014, 16, 22035–22045. [Google Scholar] [CrossRef]
- Tosco, P.; Balle, T. Open3DQSAR: A new open-source software aimed at high-throughput chemometric analysis of molecular interaction fields. J. Mol. Model. 2010, 17, 201–208. [Google Scholar] [CrossRef]
- Morris, G.M.; Lim-Wilby, M. Molecular docking BT. In Molecular Modeling of Proteins; Kukol, A., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 365–382. ISBN 978-1-59745-177-2. [Google Scholar]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2013, 66, 334–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Shah, S. Structure-based virtual screening BT. In Protein Bioinformatics: From Protein Modifications and Networks to Proteomics; Wu, C.H., Arighi, C.N., Ross, K.E., Eds.; Springer: New York, NY, USA, 2017; pp. 111–124. ISBN 978-1-4939-6783-4. [Google Scholar]
- Westermaier, Y.; Barril, X.; Scapozza, L. Virtual screening: An in silico tool for interlacing the chemical universe with the proteome. Methods 2015, 71, 44–57. [Google Scholar] [CrossRef]
- Chen, Y.-C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef]
- Ma, H.; Bandos, A.I.; Gur, D. On the use of partial area under the ROC curve for comparison of two diagnostic tests. Biom. J. 2015, 57, 304–320. [Google Scholar] [CrossRef]
- Yang, H.J.; Youn, H.; Seong, K.M.; Yun, Y.J.; Kim, W.; Kim, Y.H.; Lee, J.Y.; Kim, C.S.; Jin, Y.-W.; Youn, B. Psoralidin, a dual inhibitor of COX-2 and 5-LOX, regulates ionizing radiation (IR)-induced pulmonary inflammation. Biochem. Pharmacol. 2011, 82, 524–534. [Google Scholar] [CrossRef]
- Pang, X.; Zhou, L.; Zhang, L.; Xu, L.; Zhang, X. Two rules on the protein-ligand interaction. Nat. Preced. 2008. [Google Scholar] [CrossRef]
- Chaudhary, N.; Aparoy, P. Deciphering the mechanism behind the varied binding activities of COXIBs through Molecular Dynamic Simulations, MM-PBSA binding energy calculations and per-residue energy decomposition studies. J. Biomol. Struct. Dyn. 2017, 35, 868–882. [Google Scholar] [CrossRef] [PubMed]
- Bouaziz-Terrachet, S.; Toumi-Maouche, A.; Maouche, B.; Taïri-Kellou, S. Modeling the binding modes of stilbene analogs to cyclooxygenase-2: A molecular docking study. J. Mol. Model. 2010, 16, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Bouslama, L.; Becot, J.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs4: Free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics 2017, 33, 3658–3660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertanha, C.S.; Braguine, C.G.; Moraes, A.C.G.; Gimenez, V.M.M.; Groppo, M.; Silva, M.L.A.; Cunha, W.R.; Januário, A.H.; Pauletti, P.M. Cyclooxygenase inhibitory properties of nor-neolignans from Styrax pohlii. Nat. Prod. Res. 2012, 26, 2323–2329. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Kar, S.; Das, R.N. Introduction to 3D-QSAR. In Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment; Elsevier: London, UK, 2015; pp. 291–317. ISBN 9780128015056. [Google Scholar]
- Doweyko, A.M. Three-dimensional quantitative structure–activity relationship: The state of the art. In Comprehensive Medicinal Chemistry II; Elsevier: London, UK, 2007; pp. 575–595. ISBN 9780080450445. [Google Scholar]
- Clark, R.D.; Fox, P.C. Statistical variation in progressive scrambling. J. Comput. Aided Mol. Des. 2004, 18, 563–576. [Google Scholar] [CrossRef]
- Yadav, P.; Singh, P.; Tewari, A.K. Design, synthesis, docking and anti-inflammatory evaluation of novel series of benzofuran based prodrugs. Bioorg. Med. Chem. Lett. 2014, 24, 2251–2255. [Google Scholar] [CrossRef]
- Gregson, M.; Ollis, W.D.; Redman, B.T.; Sutherland, I.O.; Dietrichs, H.H.; Gottlieb, O.R. Obtusastyrene and obtustyrene, cinnamylphenols from Dalbergia retusa. Phytochemistry 1978, 17, 1395–1400. [Google Scholar] [CrossRef]
- Muangnoicharoen, N.; Frahm, A.W. Arylbenzofurans from Dalbergia parviflora. Phytochemistry 1981, 20, 291–293. [Google Scholar] [CrossRef]
- Jurd, L.; Wong, R.Y. Diarylheptanoid and other phenolic constituents of Centrolobium species. Aust. J. Chem. 1984, 37, 1127. [Google Scholar] [CrossRef]
- Fukai, T.; Sheng, C.-B.; Horikoshi, T.; Nomura, T. Isoprenylated flavonoids from underground parts of Glycyrrhiza glabra. Phytochemistry 1996, 43, 1119–1124. [Google Scholar] [CrossRef]
- O’Neill, M.J.; Adesanya, S.A.; Roberts, M.F.; Pantry, I.R. Inducible isoflavonoids from the lima bean, Phaseolus lunatus. Phytochemistry 1986, 25, 1315–1322. [Google Scholar] [CrossRef]
- Luo, X.; Pedro, L.; Milic, V.; Mulhovo, S.; Duarte, A.; Duarte, N.; Ferreira, M.-J. Antibacterial Benzofuran Neolignans and Benzophenanthridine Alkaloids from the Roots ofZanthoxylum capense. Planta Med. 2011, 78, 148–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zawawi, N.K.N.A.; Ahmat, N.; Mazatulikhma, M.Z.; Shafiq, R.M.; Wahid, N.H.; Sufian, A.S. Bioactive oligostilbenoids from Shorea maxwelliana King and their chemotaxonomic significance. Nat. Prod. Res. 2012, 27, 1589–1593. [Google Scholar] [CrossRef]
- Zhao, F.; Chen, L.; Bi, C.; Zhang, M.; Jiao, W.; Yao, X. In vitro anti-inflammatory effect of picrasmalignan A by the inhibition of iNOS and COX-2 expression in LPS-activated macrophage RAW 264.7 cells. Mol. Med. Rep. 2013, 8, 1575–1579. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.-M.; Liang, X.-T.; Li, L.-N. Prionitisides A and B, two phenolic glycosides from Salvia prionitis. Phytochemistry 1996, 42, 899–901. [Google Scholar] [CrossRef]
- Máximo, P.; Lourenço, A. A pterocarpan from Ulex parviflorus. Phytochemistry 1998, 48, 359–362. [Google Scholar] [CrossRef]
- Burden, R.S.; Bailey, J.A. Structure of the phytoalexin from soybean. Phytochemistry 1975, 14, 1389–1390. [Google Scholar] [CrossRef]
- Shu, L.; Shen, Y.; Yang, L.; Gao, X.; Hu, Q.-F. Flavonoids derivatives from arundina graminifolia and their cytotoxicity. Asian J. Chem. 2013, 25, 8358–8360. [Google Scholar] [CrossRef]








| Code a | PDB ID b | Resolution [Å] | Bound | Source | Reference c |
|---|---|---|---|---|---|
| ES1 | 1CVU | 2.40 | arachidonic acid | Mus musculus | [10] |
| ES2 | 5COX | 3.00 | unbounded | M. musculus | [11] |
| ES3 | 4COX | 2.90 | indomethacin | M. musculus | [11] |
| ES4 | 1CX2 | 3.00 | SC-558 | M. musculus | [11] |
| ES5 | 1PXX | 2.90 | diclofenac | M. musculus | [12] |
| ES6 | 3NT1 | 1.73 | naproxen | M. musculus | [13] |
| ES7 | 3LN1 | 2.40 | celecoxib | M. musculus | [14] |
| ES8 | 4RS0 | 2.81 | S-ibuprofen | M. musculus | [15] |
| ES9 | 4PH9 | 1.81 | rac-ibuprofen | M. musculus | [16] |
| No. | GMVS a | %RSD b | RMSD c | No. | GMVS a | %RSD b | RMSD c | No. | GMVS a | %RSD b | RMSD c |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −8.63 ± 0.50 | 5.85 | 0.521 | 23 | −9.37 ± 0.83 | 8.88 | 0.513 | 45 | −9.21 ± 0.93 | 10.06 | 0.335 |
| 2 | −9.19 ± 0.84 | 9.14 | 0.307 | 24 | −9.23 ± 0.89 | 9.67 | 0.626 | 46 | −8.79 ± 1.11 | 12.60 | 0.475 |
| 3 | −8.10 ± 0.94 | 11.61 | 0.319 | 25 | −8.50 ± 1.35 | 15.85 | 0.493 | 47 | −8.67 ± 1.14 | 13.14 | 0.345 |
| 4 | −8.43 ± 0.50 | 5.90 | 0.712 | 26 | −8.78 ± 1.18 | 4.88 | 0.574 | 48 | −8.79 ± 1.15 | 13.11 | 0.333 |
| 5 | −8.50 ± 0.53 | 6.25 | 0.404 | 27 | −10.16 ± 0.50 | 13.40 | 0.611 | 49 | −11.26 ± 0.81 | 5.48 | 0.496 |
| 6 | −8.14 ± 0.74 | 9.09 | 0.549 | 28 | −10.47 ± 0.46 | 9.38 | 0.711 | 50 | −8.48 ± 1.58 | 18.66 | 0.520 |
| 7 | −8.43 ± 0.52 | 6.19 | 0.531 | 29 | −10.49 ± 0.43 | 4.14 | 0.450 | 51 | −10.37 ± 0.57 | 7.16 | 0.621 |
| 8 | −8.49 ± 0.39 | 4.58 | 0.576 | 30 | −10.42 ± 0.54 | 12.75 | 0.790 | 52 | −8.18 ± 0.92 | 11.30 | 0.452 |
| 9 | −8.92 ± 0.38 | 4.30 | 0.605 | 31 | −9.49 ± 1.21 | 5.21 | 0.471 | 53 | −9.07 ± 0.72 | 5.60 | 0.467 |
| 10 | −8.51 ± 0.46 | 9.03 | 0.490 | 32 | −8.84 ± 1.66 | 18.80 | 0.564 | 54 | −10.39 ± 0.58 | 7.95 | 0.842 |
| 11 | −10.76 ± 0.27 | 5.40 | 0.458 | 33 | −8.32 ± 1.42 | 17.12 | 0.316 | 55 | −8.72 ± 0.49 | 5.61 | 0.433 |
| 12 | −8.99 ± 0.98 | 10.88 | 0.494 | 34 | −8.63 ± 1.74 | 20.14 | 0.526 | 56 | −8.49 ± 0.45 | 5.32 | 0.368 |
| 13 | −9.24 ± 1.20 | 12.96 | 0.578 | 35 | −8.70 ± 1.23 | 14.15 | 0.548 | 57 | −7.77 ± 1.38 | 17.81 | 0.307 |
| 14 | −8.58 ± 0.57 | 6.59 | 0.529 | 36 | −9.02 ± 0.79 | 8.71 | 0.646 | 58 | −8.46 ± 0.67 | 7.98 | 0.367 |
| 15 | −8.93 ± 1.17 | 13.08 | 0.602 | 37 | −9.03 ± 0.58 | 6.41 | 0.534 | AA | −7.98 ± 0.37 | 4.60 | 1.329 |
| 16 | −9.09 ± 1.05 | 11.53 | 0.367 | 38 | −8.73 ± 0.58 | 6.65 | 0.426 | CEL | −11.23 ± 0.62 | 5.52 | 0.488 |
| 17 | −8.87 ± 0.86 | 9.75 | 0.516 | 39 | −10.2 ± 0.69 | 6.79 | 0.443 | DCL | −8.33 ± 0.30 | 3.65 | 0.318 |
| 18 | −8.77 ± 1.24 | 14.14 | 0.425 | 40 | −10.3 ± 0.59 | 7.42 | 0.547 | IMD | −9.67 ± 0.79 | 8.13 | 0.449 |
| 19 | −9.02 ± 0.71 | 7.89 | 0.493 | 41 | −9.70 ± 0.72 | 5.74 | 0.447 | NPX | −8.66 ± 0.29 | 3.32 | 0.376 |
| 20 | −9.11 ± 1.20 | 13.18 | 0.596 | 42 | −11.25 ± 0.59 | 5.28 | 0.335 | SC558 | −11.09 ± 0.57 | 5.13 | 0.412 |
| 21 | −9.33 ± 0.89 | 9.55 | 0.574 | 43 | −9.32 ± 0.75 | 8.01 | 0.322 | S−IBP | −7.71 ± 0.33 | 4.22 | 0.638 |
| 22 | −9.04 ± 0.84 | 9.25 | 0.534 | 44 | −9.13 ± 0.98 | 10.73 | 0.320 | R−IBP | −7.71 ± 0.27 | 3.52 | 0.784 |
| Ligands | ΔEvdW a | ΔEele b | ΔGsol c | ΔGsasa d | ΔGbind e |
|---|---|---|---|---|---|
| 11 | −211.7 ± 2.5 | −96.3 ± 1.6 | 185.6 ± 7.5 | −19.0 ± 0.2 | −141.5 ± 5.8 |
| 29 | −221.2 ± 5.4 | −109.2 ± 3.9 | 175.6 ± 2.7 | −20.2 ± 0.3 | −175.0 ± 4.3 |
| 42 | −205.6 ± 3.5 | −102.5 ± 3.6 | 132.2 ± 4.8 | −19.8 ± 0.2 | −195.7 ± 4.5 |
| 49 | −226.9 ± 4.5 | −120.8 ± 4.6 | 189.2 ± 3.7 | −24.4 ± 0.4 | −182.9 ± 2.0 |
| celecoxib | −225.5 ± 8.9 | −138.2 ± 5.7 | 184.5 ± 8.5 | −18.6 ± 0.3 | −197.8 ± 7.5 |
| Residue | 11 | 29 | 42 | 49 | Cel |
|---|---|---|---|---|---|
| Val74 | −0.22 ± 0.42 | −0.15 ± 0.07 | −0.08 ± 0.05 | −4.12 ± 0.76 | −0.37 ± 0.08 |
| His75 | −0.57 ± 0.40 | −2.12 ± 0.58 | −0.24 ± 0.82 | −2.26 ± 0.73 | −1.93 ± 0.37 |
| Leu103 | −1.07 ± 0.15 | −1.02 ± 0.26 | −3.63 ± 0.83 | −0.77 ± 0.05 | −0.54 ± 0.04 |
| Arg106 | −9.64 ± 3.76 | −0.35 ± 0.51 | −6.67 ± 0.99 | −0.36 ± 2.84 | −8.94 ± 1.33 |
| Gln178 | −1.77 ± 0.30 | −1.68 ± 0.32 | −1.48 ± 0.21 | −0.53 ± 0.29 | −4.97 ± 0.38 |
| Val330 | −2.08 ± 0.06 | −0.08 ± 0.08 | −2.13 ± 0.38 | −0.02 ± 0.02 | −0.28 ± 0.15 |
| Val335 | −5.43 ± 0.46 | −9.27 ± 0.34 | −11.49 ± 0.72 | −2.17 ± 0.61 | −4.65 ± 0.81 |
| Leu338 | −1.41 ± 0.90 | −3.29 ± 1.09 | −4.13 ± 0.74 | −5.39 ± 0.77 | −8.87 ± 1.19 |
| Ser339 | −2.59 ± 0.81 | −4.39 ± 0.56 | −4.86 ± 0.39 | −4.98 ± 0.91 | −6.37 ± 1.01 |
| Tyr341 | −0.07 ± 0.63 | −4.58 ± 0.55 | −1.56 ± 0.54 | −1.18 ± 0.75 | −3.01 ± 0.52 |
| Leu345 | −2.29 ± 0.41 | −1.59 ± 0.26 | −1.42 ± 0.43 | −0.85 ± 0.15 | −1.80 ± 0.08 |
| Leu370 | −0.16 ± 0.04 | −0.13 ± 0.03 | −0.17 ± 0.03 | −1.91 ± 0.15 | −1.32 ± 0.38 |
| Tyr371 | −0.08 ± 0.11 | −0.55 ± 0.16 | −0.58 ± 0.32 | −0.46 ± 0.26 | −1.49 ± 0.92 |
| Arg499 | −5.32 ± 1.33 | −3.83 ± 1.39 | −1.35 ± 1.41 | −6.24 ± 1.26 | −7.90 ± 1.17 |
| Phe504 | −3.12 ± 0.44 | −2.93 ± 0.91 | −2.37 ± 0.51 | −2.41 ± 0.48 | −6.23 ± 1.03 |
| Val509 | −6.17 ± 0.58 | −6.41 ± 1.03 | −8.11 ± 0.33 | −7.36 ± 0.91 | −9.91 ± 0.43 |
| Glu510 | −3.01 ± 1.03 | −0.61 ± 0.99 | −1.27 ± 0.55 | −13.3 ± 1.76 | −6.89 ± 0.58 |
| Gly512 | −0.61 ± 0.17 | −1.32 ± 0.25 | −1.07 ± 0.07 | −3.68 ± 0.56 | −3.29 ± 0.66 |
| Ala513 | −3.84 ± 1.03 | −5.55 ± 0.71 | −9.86 ± 0.24 | −7.57 ± 0.37 | −5.09 ± 0.83 |
| Ser516 | −2.17 ± 0.91 | −0.52 ± 0.61 | −2.48 ± 0.58 | −2.17 ± 0.71 | −2.78 ± 0.49 |
| Leu517 | −1.50 ± 1.24 | −1.36 ± 0.58 | −5.96 ± 0.59 | −1.54 ± 0.28 | −1.89 ± 0.32 |
| Ligand | −60.9 ± 1.5 | −61.5 ± 1.97 | −83.4 ± 2.44 | −69.6 ± 2.24 | −92.6 ± 3.70 |
| No | COX-2 IC50 | pIC50 a | pIC50 b | No | COX-2 IC50 | pIC50 a | pIC50 b | No | COX-2 IC50 | pIC50 a | pIC50 b |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (µM) | (exp) | (pred) | (µM) | (exp) | (pred) | (µM) | (exp) | (pred) | |||
| 2 | 16.7 c | 3.78 | 4.04 | 29 | 3.75 | 4.43 | 3.94 | 49 | 1.25 | 4.90 | 4.71 |
| 3 | 13.2 c | 3.49 | 3.35 | 38 | 158 | 2.80 | 2.86 | 50 | 45.6 | 3.34 | 3.21 |
| 8 | 28.7 | 3.54 | 3.72 | 42 | 0.752 | 5.12 | 5.18 | 51 | 10.5 | 3.98 | 4.14 |
| 9 | 119 | 2.92 | 3.32 | 43 c | 102 | 2.99 | 3.45 | 54 | 10.9 | 3.96 | 3.84 |
| 11 | 3.27 c | 4.49 | 4.09 | 44 c | 26.2 | 3.58 | 3.12 | 55 | 12.6 | 3.90 | 4.17 |
| 12 | 4.33 c | 4.36 | 3.98 | 45 c | 52.7 | 3.28 | 2.95 | 56 c | 125 | 2.90 | 2.51 |
| 13 | 8.56 c | 4.07 | 4.46 | 46 c | 32.9 | 3.48 | 3.59 | 57 c | 438 | 2.36 | 2.21 |
| 14 | 7.37 c | 4.13 | 4.55 | 47 c | 71.5 | 3.15 | 3.55 | 58 c | 240 | 2.62 | 2.72 |
| 17 | 12.3 c | 3.91 | 4.22 | 48 c | 29.3 | 3.53 | 3.78 | cel | 0.102 | - | - |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coy-Barrera, E. Discrimination of Naturally-Occurring 2-Arylbenzofurans as Cyclooxygenase-2 Inhibitors: Insights into the Binding Mode and Enzymatic Inhibitory Activity. Biomolecules 2020, 10, 176. https://doi.org/10.3390/biom10020176
Coy-Barrera E. Discrimination of Naturally-Occurring 2-Arylbenzofurans as Cyclooxygenase-2 Inhibitors: Insights into the Binding Mode and Enzymatic Inhibitory Activity. Biomolecules. 2020; 10(2):176. https://doi.org/10.3390/biom10020176
Chicago/Turabian StyleCoy-Barrera, Ericsson. 2020. "Discrimination of Naturally-Occurring 2-Arylbenzofurans as Cyclooxygenase-2 Inhibitors: Insights into the Binding Mode and Enzymatic Inhibitory Activity" Biomolecules 10, no. 2: 176. https://doi.org/10.3390/biom10020176