R-MetaboList 2: A Flexible Tool for Metabolite Annotation from High-Resolution Data-Independent Acquisition Mass Spectrometry Analysis

 and
and
Abstract
:1. Introduction
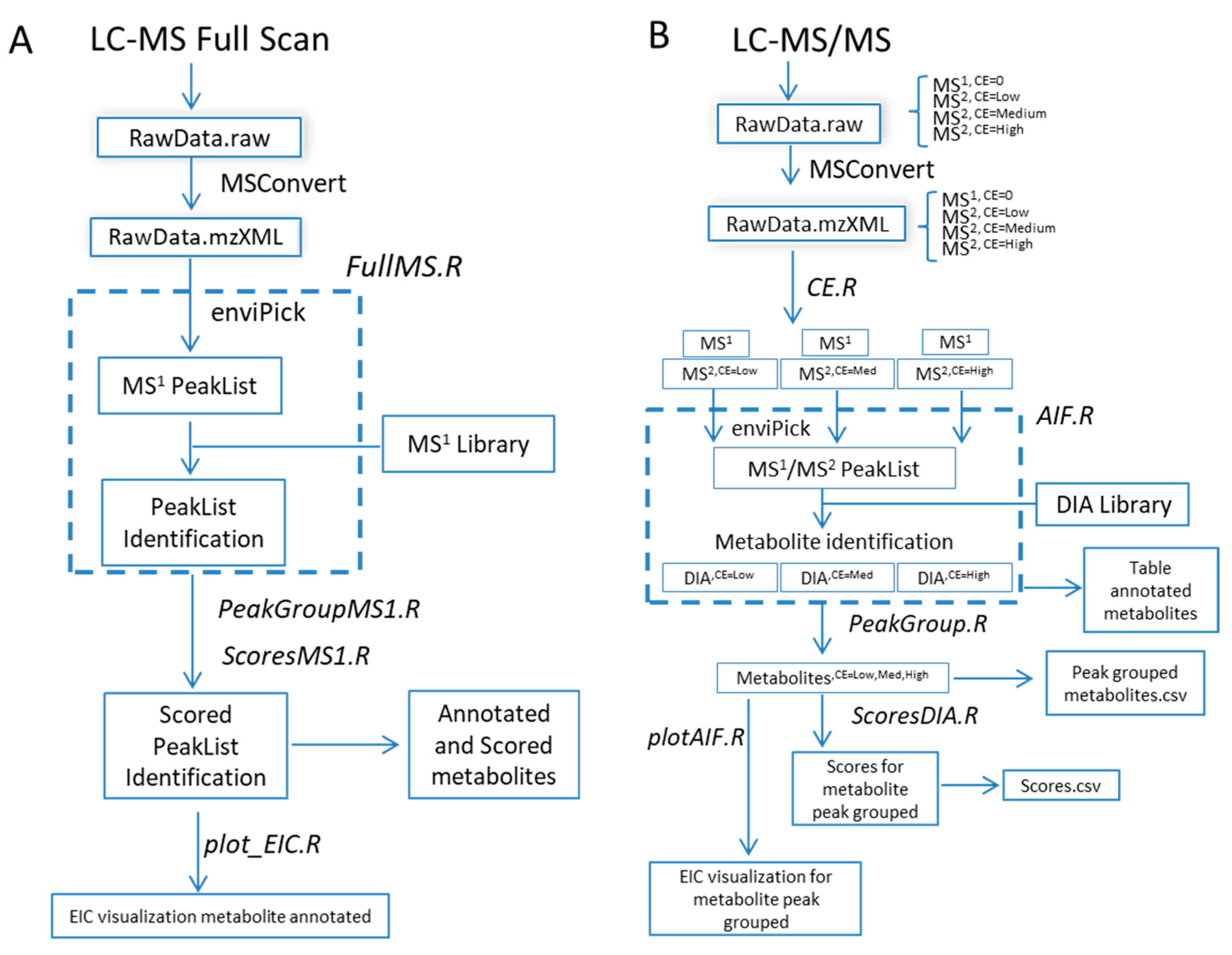
2. Results and Discussion
- (1)
- Processing workflow for full-scan MS1 analysis (Figure 1A) independent of full-scan MS/MS analysis. The previous version did not include the processing of full-scan MS1 data outside the scope of the associated MS/MS data.
- (2)
- Simultaneous processing of full-scan MS/MS data generated under different instrumental conditions (Figure 1B).
- (3)
- Incorporation of scoring functions to evaluate metabolite annotation of both full-scan MS1 and MS/MS approaches (Figure 1B).
- (4)
- Improved graphical representation of the results.
- (5)
- Incorporation of a batch job function for compilation of full-scan MSn reports from multiple samples for high-throughput applications.
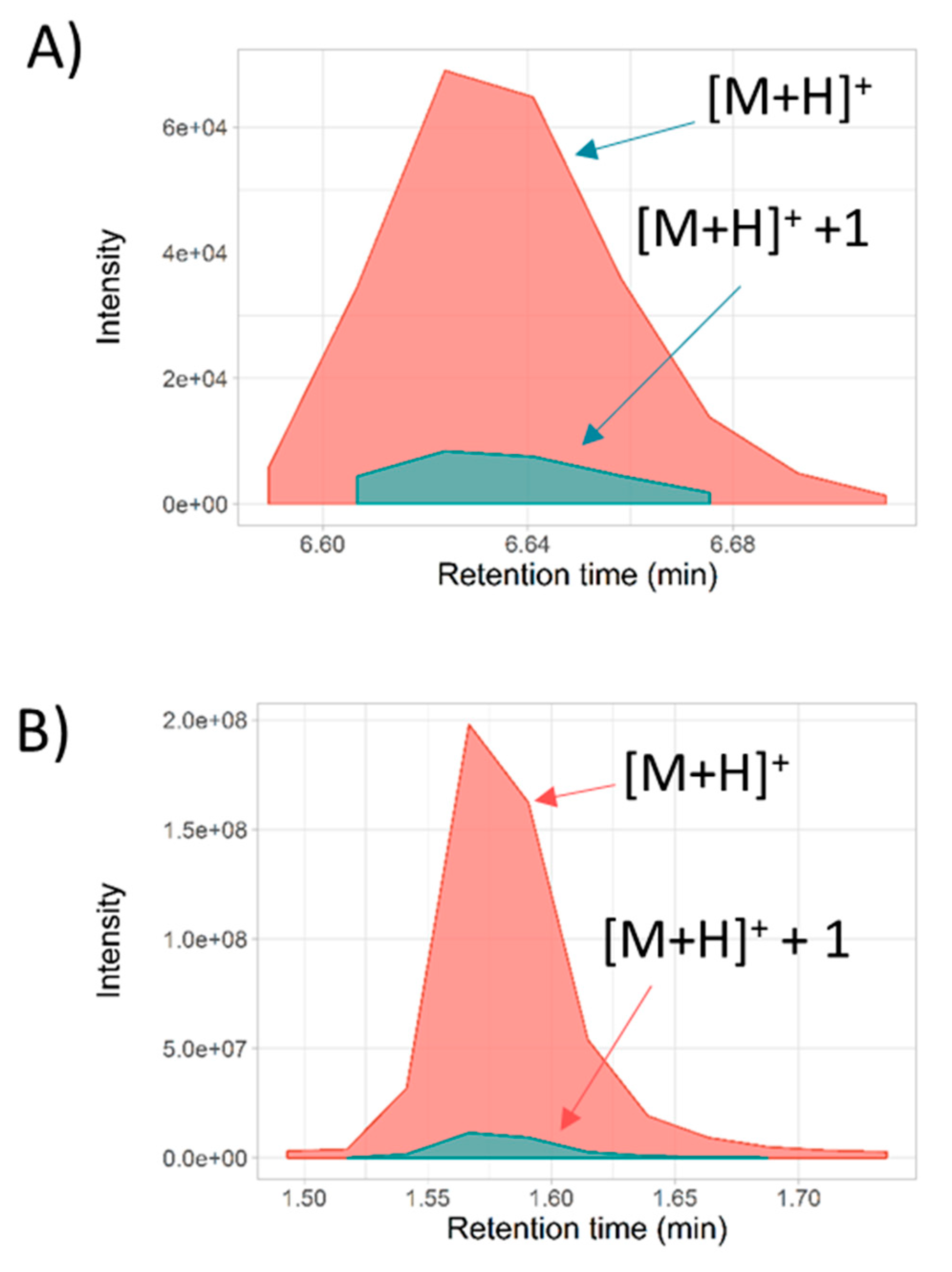
2.1. Metabolite Profiling of Samples from Full-Scan MS1 Analysis
2.2. Metabolites Annotation by Full-Scan MS/MS Approach
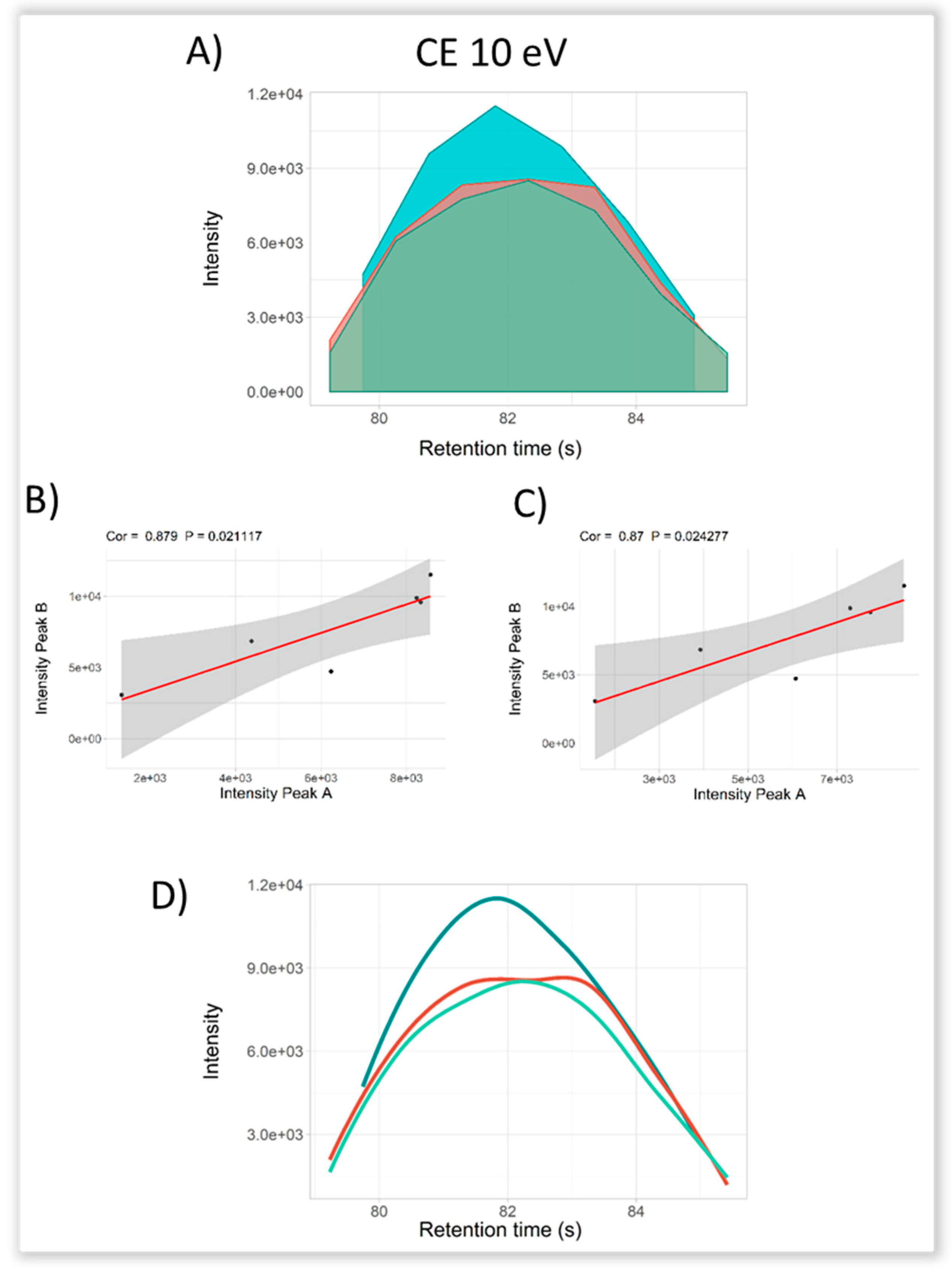
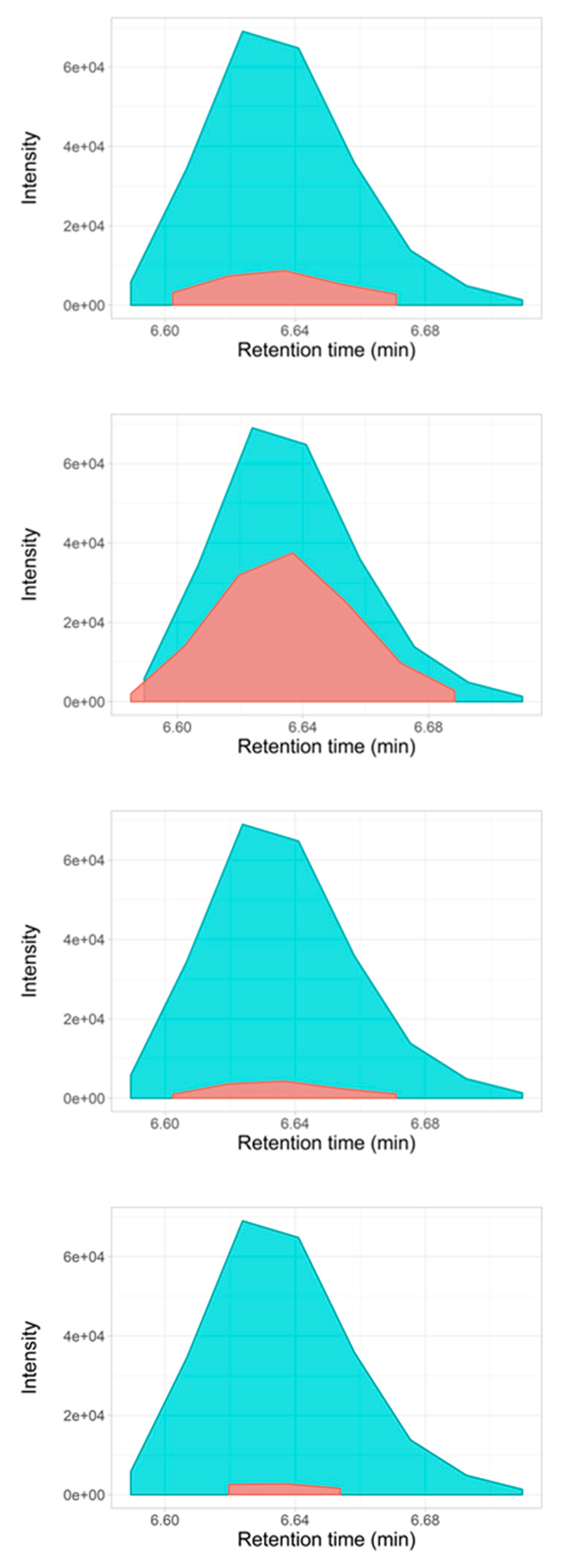
2.3. Selectivity for Metabolite Annotation by LC-DIA-MS: Quality Control and Scores Test
3. Materials and Methods
3.1. Chemicals and Sample Preparation
3.2. LC-MS/MS Analysis
3.3. Automated Data Processing by R-MetaboList 2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Financial Disclosure Statement
References
- Rochat, B. Quantitative and Qualitative LC-High-Resolution MS: The Technological and Biological Reasons for a Shift of Paradigm. In Recent Advances in Analytical Chemistry; IntechOpen: London, UK, 2018. [Google Scholar]
- Bouatra, S.; Aziat, F.; Mandal, R.; Guo, A.C.; Wilson, M.R.; Knox, C.; Bjorndahl, T.C.; Krishnamurthy, R.; Saleem, F.; Liu, P.; et al. The Human Urine Metabolome. PLoS ONE 2013, 8, e73076. [Google Scholar] [CrossRef] [PubMed]
- Psychogios, N.; Hau, D.D.; Peng, J.; Guo, A.C.; Mandal, R.; Bouatra, S.; Sinelnikov, I.; Krishnamurthy, R.; Eisner, R.; Gautam, B.; et al. The Human Serum Metabolome. PLoS ONE 2011, 6, e16957. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.F.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0-The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Boil. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Metabolomics: The apogee of the omic triology. Nat. Rev. Mol. Cell Boil. 2012, 13, 263–269. [Google Scholar] [CrossRef]
- Forcisi, S.; Moritz, F.; Kanawati, B.; Tziotis, D.; Lehmann, R.; Schmitt-Kopplin, P. Liquid chromatography–mass spectrometry in metabolomics research: Mass analyzers in ultra high pressure liquid chromatography coupling. J. Chromatogr. A 2013, 1292, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Eliuk, S.; Makarov, A. Evolution of Orbitrap Mass Spectrometry Instrumentation. In Annual Review of Analytical Chemistry; Cooks, R.G., Pemberton, J.E., Eds.; Annual Reviews: Palo Alto, CA, USA, 2015; Volume 8, pp. 61–80. [Google Scholar]
- Bateman, K.P.; Kellmann, M.; Muenster, H.; Papp, R.; Taylor, L. Quantitative-qualitative data acquisition using a benchtop orbitrap mass spectrometer. J. Am. Soc. Mass Spectrom. 2009, 20, 1441–1450. [Google Scholar] [CrossRef] [Green Version]
- Sentandreu, E.; Peris-Díaz, M.D.; Sweeney, S.R.; Chiou, J.; Muñoz, N.; Tiziani, S. A Survey of Orbitrap All Ion Fragmentation Analysis Assessed by an R-MetaboList Package to Study Small-Molecule Metabolites. Chromatographia 2018. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteom. 2012, 11. [Google Scholar] [CrossRef] [Green Version]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef]
- Shulaev, V. Metabolomics technology and bioinformatics. Briefings Bioinform. 2006, 7, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data Independent MS/MS Deconvolution for Comprehensive Metabolome Analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhu, Z.-J.; Li, H.; Guo, Y.; Chen, F. MetDIA: Targeted Metabolite Extraction of Multiplexed MS/MS Spectra Generated by Data-Independent Acquisition. Anal. Chem. 2016, 88, 8757–8764. [Google Scholar]
- Zhou, J.; Li, Y.; Chen, X.; Zhong, L.; Yin, Y. Development of data-independent acquisition workflows for metabolomic analysis on a quadrupole-orbitrap platform. Talanta 2017, 164, 128–136. [Google Scholar] [CrossRef]
- Peris-Díaz, M.D.; Rodak, O.; Sweeney, S.R.; Krężel, A.; Sentandreu, E. Chemometrics-assisted optimization of liquid chromatography-quadrupole-time-of-flight mass spectrometry analysis for targeted metabolomics. Talanta 2019, 199, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Naz, S.; Gallart-Ayala, H.; Reinke, S.N.; Mathon, C.; Blankley, R.; Chaleckis, R.; Wheelock, C.E. Development of a Liquid Chromatography–High Resolution Mass Spectrometry Metabolomics Method with High Specificity for Metabolite Identification Using All Ion Fragmentation Acquisition. Anal. Chem. 2017, 89, 7933–7942. [Google Scholar] [CrossRef]
- Ipsen, A.; Want, E.J.; Lindon, J.C.; Ebbels, T.M.D. A Statistically Rigorous Test for the Identification of Parent−Fragment Pairs in LC-MS Datasets. Anal. Chem. 2010, 82, 1766–1778. [Google Scholar] [CrossRef]
- Loos, M.; Singer, H. Nontargeted homologue series extraction from hyphenated high resolution mass spectrometry data. J. Cheminformatics 2017, 9, 12. [Google Scholar] [CrossRef]
- Senan, O.; Aguilar-Mogas, A.; Navarro, M.; Capellades, J.; Noon, L.; Burks, D.; Yanes, O.; Guimerà, R.; Sales-Pardo, M. CliqueMS: A computational tool for annotating in-source metabolite ions from LC-MS untargeted metabolomics data based on a coelution similarity network. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Kuhl, C.; Tautenhahn, R.; Böttcher, C.; Larson, T.R.; Neumann, S. CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Bruderer, T.; Varesio, E.; Hidasi, A.O.; Duchoslav, E.; Burton, L.; Bonner, R.; Hopfgartner, G. Metabolomic spectral libraries for data-independent SWATH liquid chromatography mass spectrometry acquisition. Anal. Bioanal. Chem. 2018, 410, 1873–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Fragment [M+H]+(m/z) | CE (eV) | PPC | PPS | a Product/Precursor Ion Ratio | |
|---|---|---|---|---|---|
| Glutamine | |||||
| 84.0444 | 5 | 0.93 | 0.22 | 0.23 | |
| 130.0499 | 5 | 0.97 | 1.00 | 0.72 | |
| 84.0444 | 10 | 0.87 | 0.67 | 0.74 | |
| 130.0499 | 10 | 0.87 | 0.67 | 0.74 | |
| 84.0444 | 20 | 0.87 | 0.67 | 0.99 | |
| Phenylacetylglutamine | |||||
| 84.0444 | 5 | 0.39 | 0.80 | 0.04 | |
| 130.0499 | 5 | 0.76 | 0.60 | 0.20 | |
| 84.0444 | 10 | 0.72 | 0.40 | 0.07 | |
| 130.0499 | 10 | 0.91 | 0.80 | 0.54 | |
| 136.0756 | 10 | 0.67 | 0.40 | 0.06 | |
| 147.0762 | 10 | 0.80 | 0.40 | 0.06 | |
| 84.0444 | 20 | 0.89 | 0.40 | 0.13 | |
| 130.0499 | 20 | 0.95 | 0.40 | 0.54 | |
| 136.0757 | 20 | 0.89 | 0.40 | 0.06 | |
| 147.0762 | 20 | 0.97 | 0.40 | 0.04 | |
| Phenylalanine | |||||
| 120.0809 | 5 | 0.60 | 3.00 | 0.77 | |
| 120.0809 | 10 | 0.87 | 1.50 | 1.39 | |
| 103.0543 | 20 | 0.76 | 2.00 | 0.43 | |
| 120.0809 | 20 | 0.93 | 1.50 | 1.05 |
| Experimental Fragment [M+H]+ (m/z) | CE (eV) | PPC | PPS | a Product/Precursor Ion Ratio | |
|---|---|---|---|---|---|
| Glutathione | |||||
| 76.0214 | 30 | 0.99 | 0.79 | 0.44 | |
| 116.0163 | 30 | 0.99 | 0.30 | 0.08 | |
| 144.0112 | 30 | 0.99 | 0.35 | 0.08 | |
| 162.0217 | 30 | 0.99 | 0.40 | 0.17 | |
| 179.0482 | 30 | 0.99 | 0.25 | 0.04 | |
| 233.0585 | 30 | 0.99 | 0.20 | 0.02 | |
| 130.0497 | 30 | 0.99 | 0.60 | 0.08 | |
| 84.0443 | 30 | 0.99 | 0.60 | 0.15 | |
| Methionine | |||||
| 133.0315 | 30 | 0.98 | 0.17 | 0.02 | |
| 104.0526 | 30 | 0.96 | 0.49 | 0.03 | |
| 61.0107 | 30 | 0.99 | 1.25 | 0.30 | |
| 56.0497 | 30 | 0.99 | 0.49 | 0.22 | |
| Tyrosine | |||||
| 147.0438 | 30 | 0.99 | 0.50 | 0.015 | |
| 136.0754 | 30 | 0.99 | 0.49 | 0.16 | |
| 123.0439 | 30 | 0.99 | 0.99 | 0.40 | |
| 119.0490 | 30 | 0.99 | 1.25 | 0.22 | |
| 95.0490 | 30 | 0.99 | 0.99 | 0.19 | |
| 91.0541 | 30 | 0.99 | 0.99 | 0.40 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peris-Díaz, M.D.; Sweeney, S.R.; Rodak, O.; Sentandreu, E.; Tiziani, S. R-MetaboList 2: A Flexible Tool for Metabolite Annotation from High-Resolution Data-Independent Acquisition Mass Spectrometry Analysis. Metabolites 2019, 9, 187. https://doi.org/10.3390/metabo9090187
Peris-Díaz MD, Sweeney SR, Rodak O, Sentandreu E, Tiziani S. R-MetaboList 2: A Flexible Tool for Metabolite Annotation from High-Resolution Data-Independent Acquisition Mass Spectrometry Analysis. Metabolites. 2019; 9(9):187. https://doi.org/10.3390/metabo9090187
Chicago/Turabian StylePeris-Díaz, Manuel D., Shannon R. Sweeney, Olga Rodak, Enrique Sentandreu, and Stefano Tiziani. 2019. "R-MetaboList 2: A Flexible Tool for Metabolite Annotation from High-Resolution Data-Independent Acquisition Mass Spectrometry Analysis" Metabolites 9, no. 9: 187. https://doi.org/10.3390/metabo9090187