Structure Elucidation of Unknown Metabolites in Metabolomics by Combined NMR and MS/MS Prediction

 ,
, 
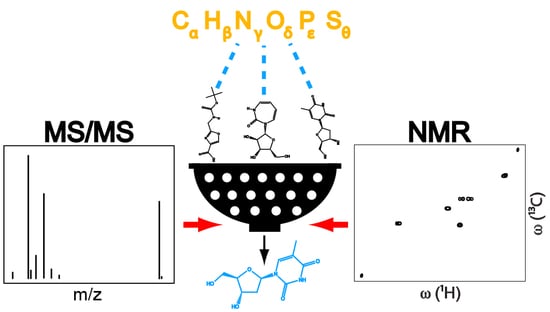
Abstract
:
1. Introduction
2. Results
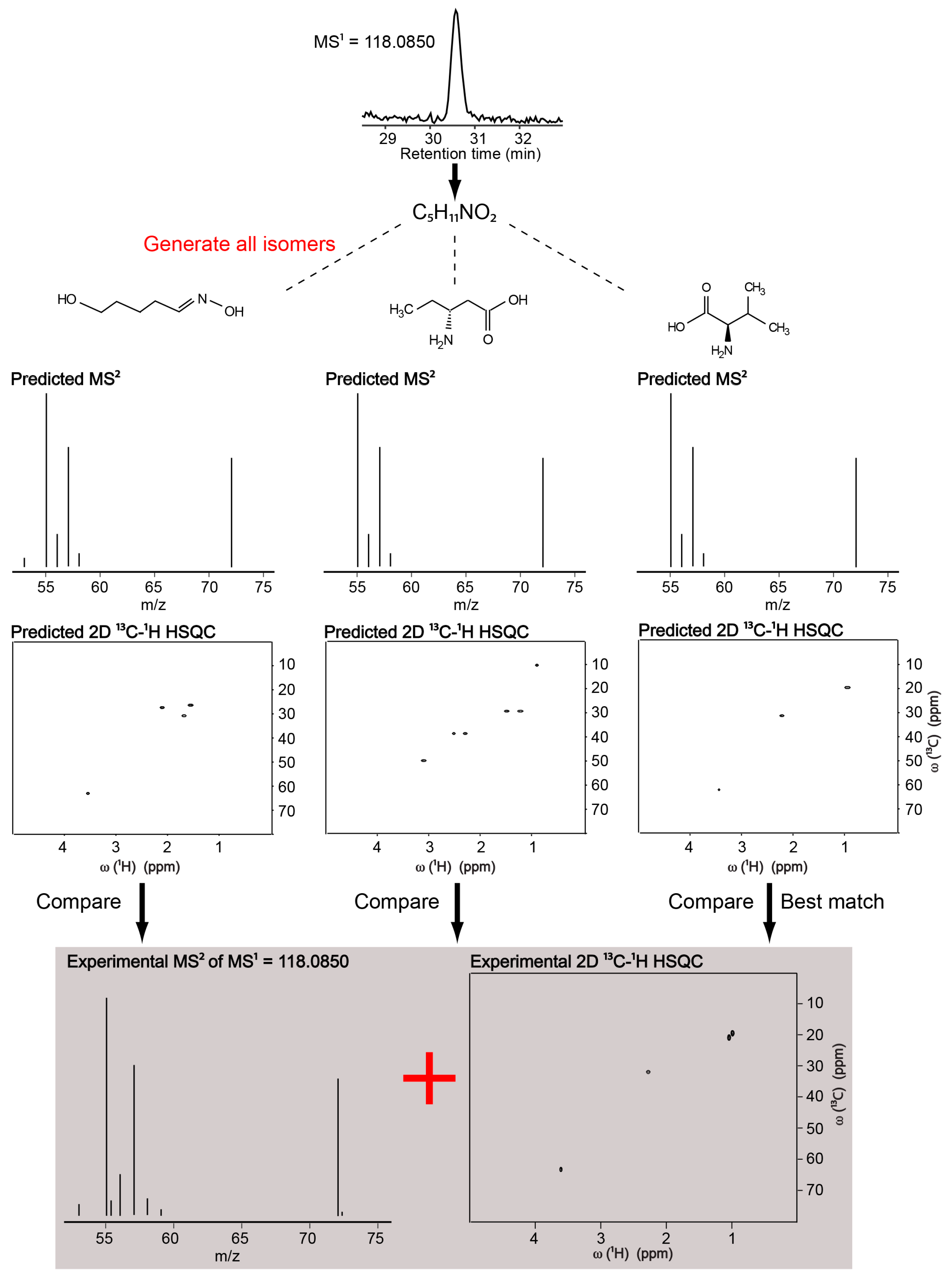
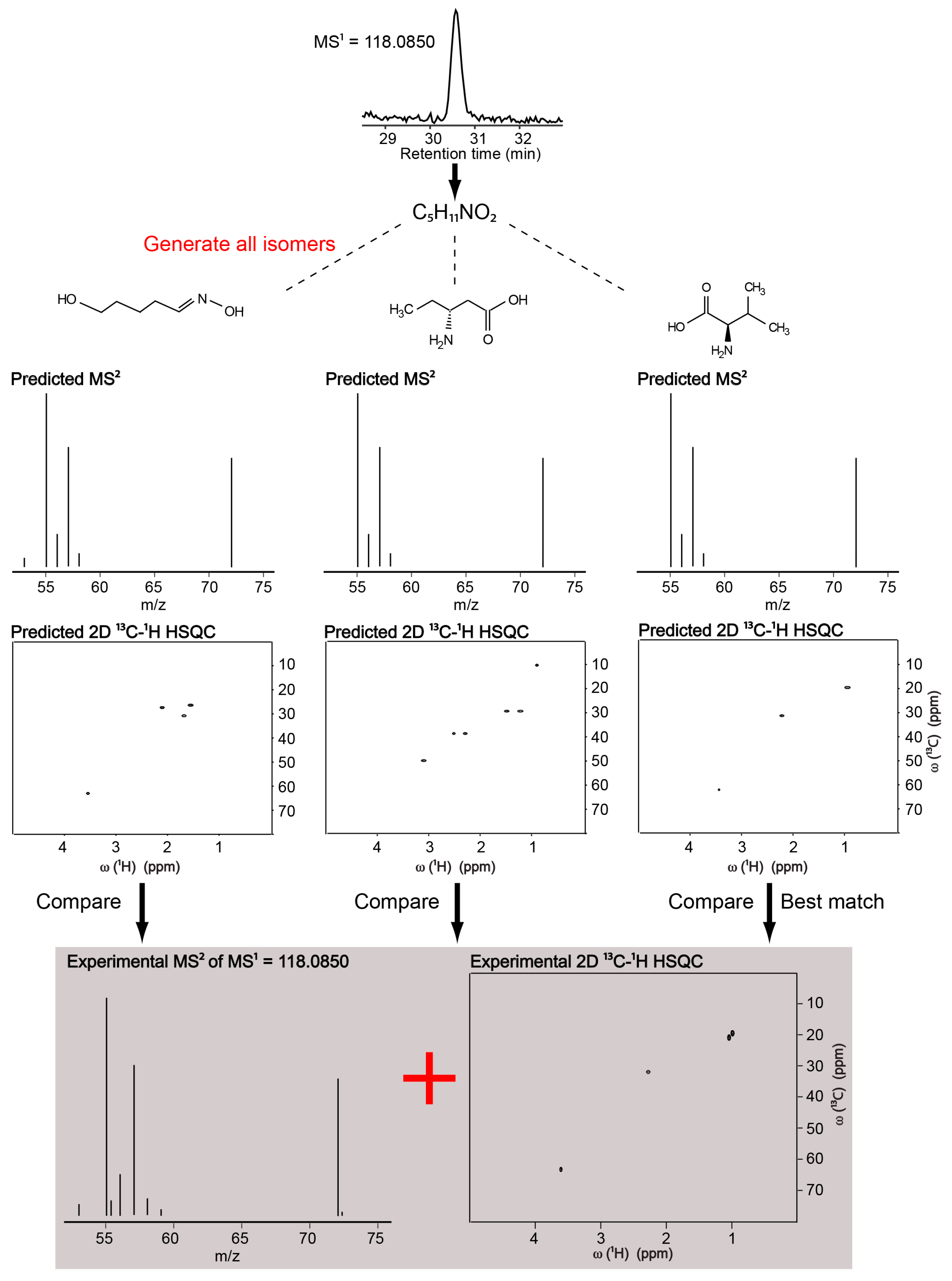
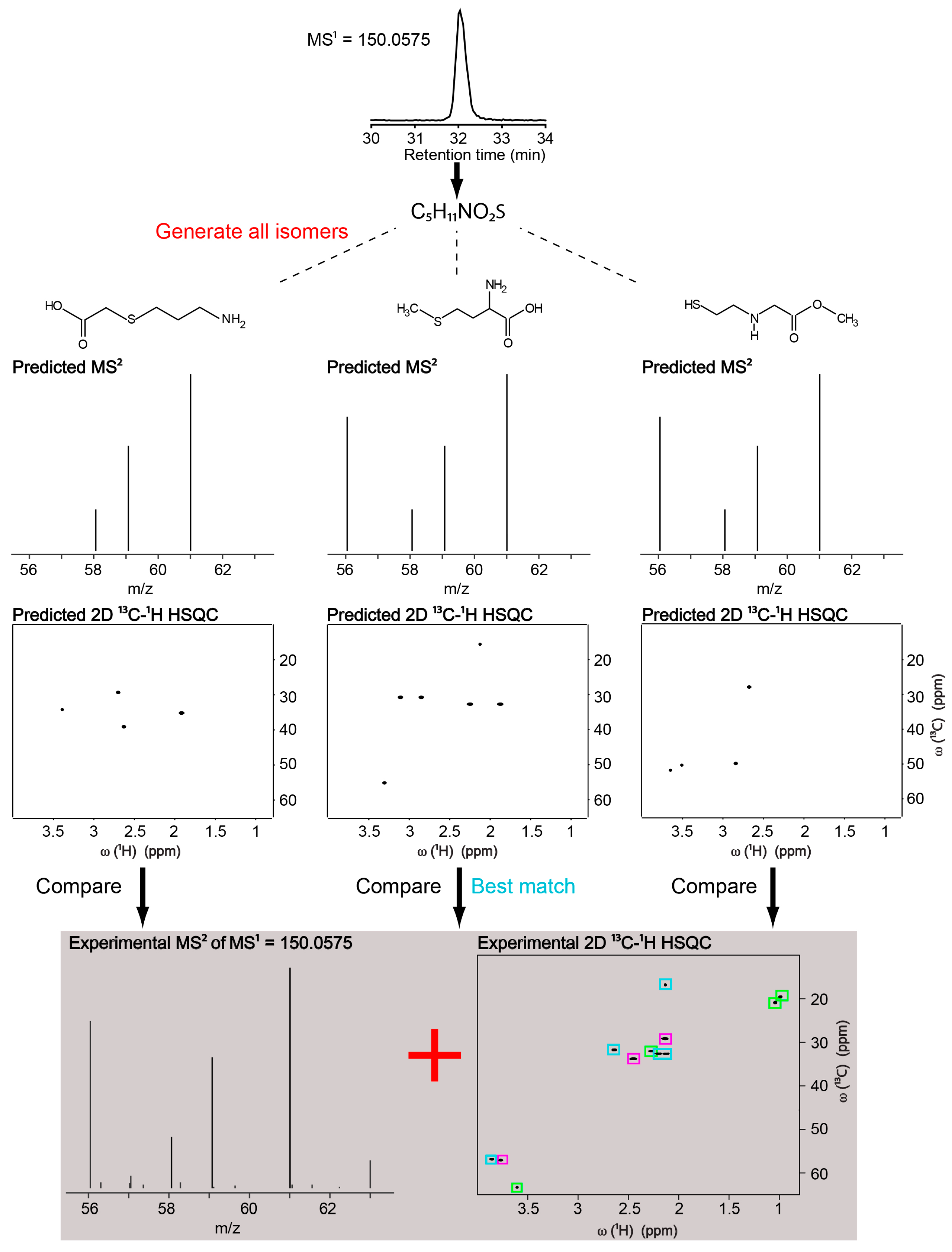
2.1. Identification of a Known Metabolite without the Use of a Database for Matching
2.2. Identification of Known Metabolites in a Test Mixture without the Use of a Database for Matching
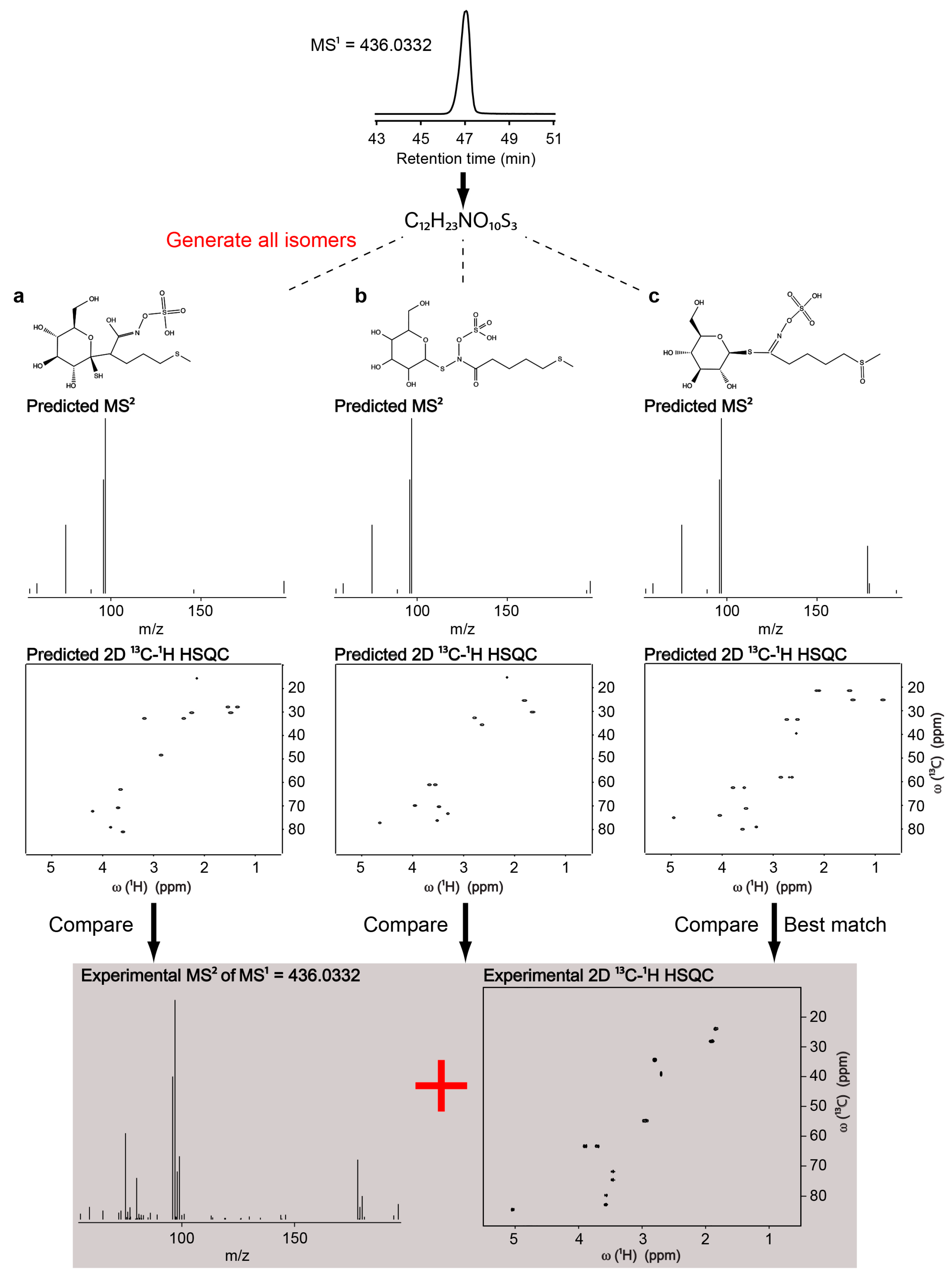
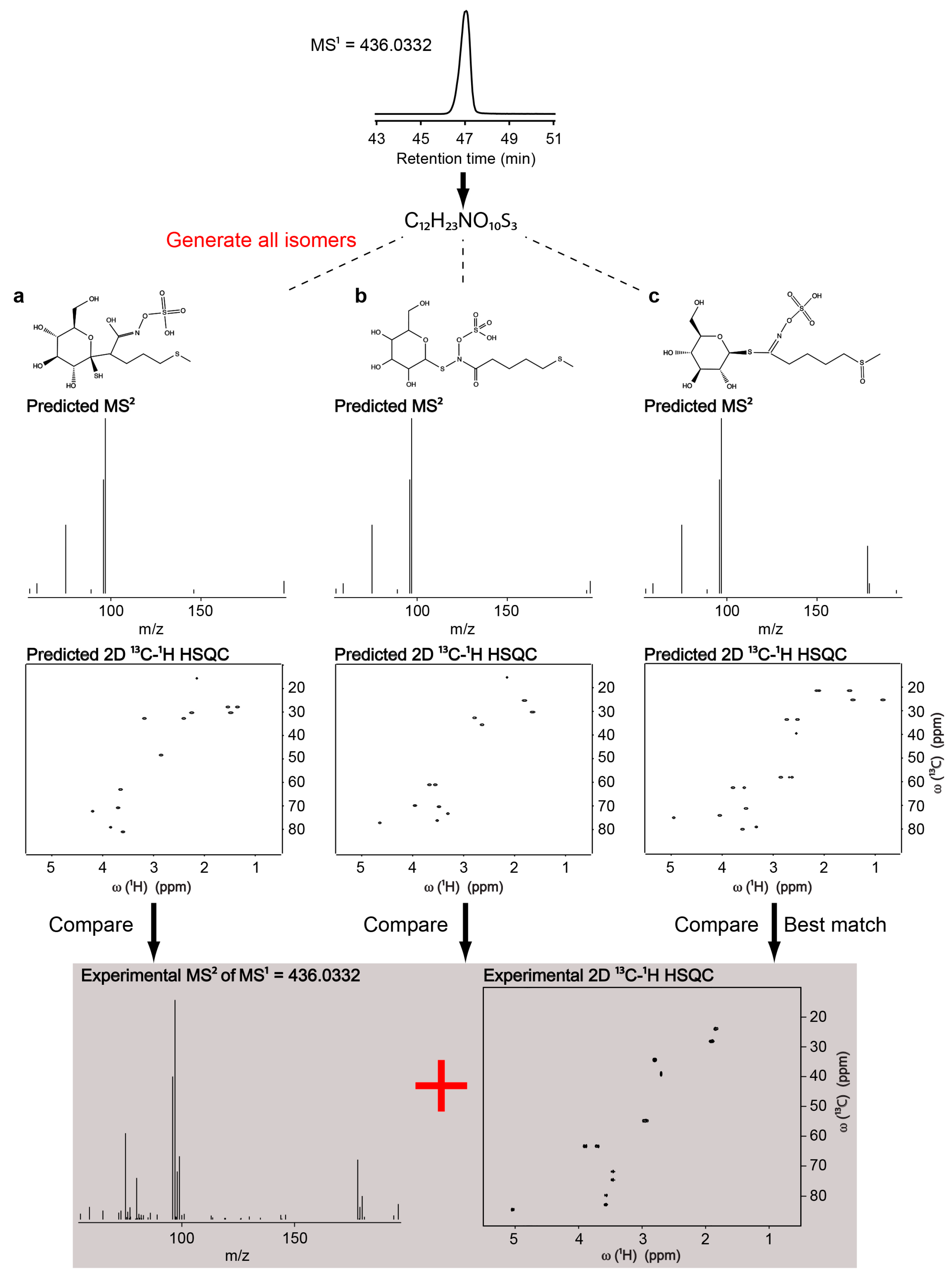
2.3. Identification of Uncatalogued Metabolite in Arabidopsis thaliana
3. Discussion
4. Materials and Methods
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nicholson, J.K.; Holmes, E.; Kinross, J.M.; Darzi, A.W.; Takats, Z.; Lindon, J.C. Metabolic phenotyping in clinical and surgical environments. Nature 2012, 491, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Bundy, J.G.; Davey, M.P.; Viant, M.R. Environmental metabolomics: A critical review and future perspectives. Metabolomics 2009, 5, 3–21. [Google Scholar] [CrossRef]
- Martien, J.I.; Amador-Noguez, D. Recent applications of metabolomics to advance microbial biofuel production. Curr. Opin. Biotechnol. 2017, 43, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Scalbert, A.; Brennan, L.; Manach, C.; Andres-Lacueva, C.; Dragsted, L.O.; Draper, J.; Rappaport, S.M.; van der Hooft, J.J.; Wishart, D.S. The food metabolome: A window over dietary exposure. Am. J. Clin. Nutr. 2014, 99, 1286–1308. [Google Scholar] [CrossRef] [PubMed]
- Ellis, D.I.; Goodacre, R. Metabolomics-assisted synthetic biology. Curr. Opin. Biotechnol. 2012, 23, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Markley, J.L.; Brüschweiler, R.; Edison, A.S.; Eghbalnia, H.R.; Powers, R.; Raftery, D.; Wishart, D.S. The future of NMR-based metabolomics. Curr. Opin. Biotechnol. 2017, 43, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Su, X.; Klein, M.S.; Lewis, I.A.; Fiehn, O.; Rabinowitz, J.D. Metabolite measurement: Pitfalls to avoid and practices to follow. Annu. Rev. Biochem. 2017, 86, 277–304. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, E.L.; Akutsu, H.; Doreleijers, J.F.; Harano, Y.; Ioannidis, Y.E.; Lin, J.; Livny, M.; Mading, S.; Maziuk, D.; Miller, Z.; et al. BioMagResBank. Nucleic Acids Res. 2008, 36, D402–D408. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The human metabolome database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.J.; Schultz, A.W.; Wang, J.; Johnson, C.H.; Yannone, S.M.; Patti, G.J.; Siuzdak, G. Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nat. Protocols 2013, 8, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Li, D.W.; Zhang, B.; Brüschweiler, R. Comprehensive metabolite identification strategy using multiple two-dimensional nmr spectra of a complex mixture implemented in the COLMARm web server. Anal. Chem. 2016, 88, 12411–12418. [Google Scholar] [CrossRef] [PubMed]
- Koehn, F.E.; Carter, G.T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 2005, 4, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kora, G.; Bowen, B.P.; Pan, C. MIDAS: A database-searching algorithm for metabolite identification in metabolomics. Anal. Chem. 2014, 86, 9496–9503. [Google Scholar] [CrossRef] [PubMed]
- Duhrkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Bocker, S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef] [PubMed]
- Ruttkies, C.; Schymanski, E.L.; Wolf, S.; Hollender, J.; Neumann, S. Metfrag relaunched: Incorporating strategies beyond in silico fragmentation. J. Cheminform. 2016, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Zhang, F.; Bruschweiler-Li, L.; Brüschweiler, R. Carbon backbone topology of the metabolome of a cell. J. Am. Chem. Soc. 2012, 134, 9006–9011. [Google Scholar] [CrossRef] [PubMed]
- Clendinen, C.S.; Pasquel, C.; Ajredini, R.; Edison, A.S. 13C NMR metabolomics: Inadequate network analysis. Anal. Chem. 2015, 87, 5698–5706. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Ohishi, R.; Shino, A.; Kikuchi, J. Structure and metabolic-flow analysis of molecular complexity in a 13C-labeled tree by 2D and 3D NMR. Angew. Chem. 2016, 128, 6104–6107. [Google Scholar] [CrossRef]
- Bingol, K.; Bruschweiler-Li, L.; Yu, C.; Somogyi, A.; Zhang, F.; Brüschweiler, R. Metabolomics beyond spectroscopic databases: A combined MS/NMR strategy for the rapid identification of new metabolites in complex mixtures. Anal. Chem. 2015, 87, 3864–3870. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; He, L.; Li, D.W.; Bruschweiler-Li, L.; Marshall, A.G.; Brüschweiler, R. Accurate identification of unknown and known metabolic mixture components by combining 3D NMR with fourier transform ion cyclotron resonance tandem mass spectrometry. J. Proteome Res. 2017, 16, 3774–3786. [Google Scholar] [CrossRef] [PubMed]
- Pence, H.E.; Williams, A. ChemSpider: An online chemical information resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Bodenhausen, G.; Ruben, D.J. Natural abundance N15 nmr by enhanced heteronuclear spectroscopy. Chem. Phys. Lett. 1980, 69, 185–189. [Google Scholar] [CrossRef] [Green Version]
- Braunschweiler, L.; Ernst, R.R. Coherence transfer by isotropic mixing—Application to proton correlation spectroscopy. J. Magn. Reson. 1983, 53, 521–528. [Google Scholar] [CrossRef]
- Bax, A.; Summers, M.F. H-1 and C-13 assignments from sensitivity-enhanced detection of heteronuclear multiple-bond connectivity by 2D multiple quantum NMR. J. Am. Chem. Soc. 1986, 108, 2093–2094. [Google Scholar] [CrossRef]
- Bingol, K.; Li, D.W.; Bruschweiler-Li, L.; Cabrera, O.A.; Megraw, T.; Zhang, F.; Brüschweiler, R. Unified and isomer-specific NMR metabolomics database for the accurate analysis of 13C-1H HSQC spectra. ACS Chem. Biol. 2015, 10, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Fiehn, O. Metabolomic database annotations via query of elemental compositions: Mass accuracy is insufficient even at less than 1 ppm. BMC Bioinform. 2006, 7, 234. [Google Scholar] [CrossRef] [PubMed]
- Benecke, C.; Grund, R.; Hohberger, R.; Kerber, A.; Laue, R.; Wieland, T. Molgen+, a generator of connectivity isomers and stereoisomers for molecular-structure elucidation. Anal. Chim. Acta 1995, 314, 141–147. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Greiner, R.; Wishart, D. Competitive fragmentation modeling of esi-ms/ms spectra for putative metabolite identification. Metabolomics 2015, 11, 98–110. [Google Scholar] [CrossRef]
- Metz, T.O.; Baker, E.S.; Schymanski, E.L.; Renslow, R.S.; Thomas, D.G.; Causon, T.J.; Webb, I.K.; Hann, S.; Smith, R.D.; Teeguarden, J.G. Integrating ion mobility spectrometry into mass spectrometry-based exposome measurements: What can it add and how far can it go? Bioanalysis 2017, 9, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Chikayama, E.; Shimbo, Y.; Komatsu, K.; Kikuchi, J. The effect of molecular conformation on the accuracy of theoretical 1H and 13C chemical shifts calculated by ab initio methods for metabolic mixture analysis. J. Phys. Chem. B 2016, 120, 3479–3487. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, F.; Li, D.W.; Sebastiani, D.; Brüschweiler, R. Improved quantum chemical nmr chemical shift prediction of metabolites in aqueous solution toward the validation of unknowns. J. Phys. Chem. A 2017, 121, 3071–3078. [Google Scholar] [CrossRef] [PubMed]
- Lerner, L.; Bax, A. Sensitivity-enhanced two-dimensional heteronuclear relayed coherence transfer nmr-spectroscopy. J. Magn. Reson. 1986, 69, 375–380. [Google Scholar] [CrossRef]
- Tolstikov, V.; Costisella, B.; Weckwerth, W.; Zhang, B.; Fiehn, O. Accurate mass QTOF and MSn Ion trap measurements require additional NMR data for plant metabolites de-novo identification. In Proceedings of the 50th ASMS Conference on Mass Spectrometry and Allied Topics, Orlando, FL, USA, 2–6 June 2002; pp. 417–418. [Google Scholar]
- Kliebenstein, D.J.; Kroymann, J.; Brown, P.; Figuth, A.; Pedersen, D.; Gershenzon, J.; Mitchell-Olds, T. Genetic control of natural variation in arabidopsis glucosinolate accumulation. Plant Physiol. 2001, 126, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Beekwilder, J.; van Leeuwen, W.; van Dam, N.M.; Bertossi, M.; Grandi, V.; Mizzi, L.; Soloviev, M.; Szabados, L.; Molthoff, J.W.; Schipper, B.; et al. The impact of the absence of aliphatic glucosinolates on insect herbivory in arabidopsis. PLoS ONE 2008, 3, e2068. [Google Scholar] [CrossRef] [PubMed]
- Higdon, J.V.; Delage, B.; Williams, D.E.; Dashwood, R.H. Cruciferous vegetables and human cancer risk: Epidemiologic evidence and mechanistic basis. Pharmacol. Res. 2007, 55, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Brüschweiler, R. Knowns and unknowns in metabolomics identified by multidimensional nmr and hybrid ms/nmr methods. Curr. Opin. Biotechnol. 2017, 43, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.R.; Hoyt, D.W.; Walker, S.M., II; Ward, J.K.; Nicora, C.D.; Bingol, K. Unambiguous metabolite identification in high-throughput metabolomics by hybrid 1D 1H NMR/ESI MS1 approach. Magn. Reson. Chem. 2016, 54, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. Nmrpipe: A multidimensional spectral processing system based on unix pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. Mzmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | m/z a | ppm b | Formula | Size c | Score d | Rank e |
|---|---|---|---|---|---|---|
| Phenylalanine | 166.0859 | 2 | C9H11NO2 | 1881 | 1.0000 | 1 |
| Valine | 118.0850 | 10 | C5H11NO2 | 453 | 0.9240 | 6 |
| Nicotinate | 124.0392 | 0 | C6H5NO2 | 100 | 0.8900 | 7 |
| Pantothenate | 220.1174 | 2 | C9H17NO5 | 563 | 0.8963 | 8 |
| Glutamine | 147.0746 | 12 | C5H10N2O3 | 283 | 0.8404 | 10 |
| Methionine | 150.0575 | 5 | C5H11NO2S | 212 | 0.8952 | 13 |
| Isoleucine | 132.1016 | 2 | C6H13NO2 | 962 | 0.8723 | 22 |
| Proline | 116.0703 | 2 | C5H9NO2 | 528 | 0.8515 | 26 |
| Thymidine | 243.0971 | 1 | C10H14N2O5 | 948 | 0.9110 | 40 |
| Leucine | 132.1018 | 0 | C6H13NO2 | 962 | 0.7002 | 148 |
| Metabolite | Size a | 1H b | 13C c | Rank d | Rank e |
|---|---|---|---|---|---|
| Phenylalanine | 1881 | 0.1224 | 2.8505 | 2 | 2 |
| Valine | 453 | 0.0773 | 0.6653 | 1 | 1 |
| Nicotinate | 100 | 0.1187 | 3.1317 | 1 | 1 |
| Pantothenate | 563 | 0.1320 | 0.8340 | 4 | 16 |
| Glutamine | 283 | 0.0852 | 1.4473 | 1 | 1 |
| Methionine | 212 | 0.2094 | 0.9670 | 1 | 1 |
| Isoleucine | 962 | 0.0923 | 2.2827 | 1 | 1 |
| Proline | 528 | 0.3077 | 1.0908 | 2 | 2 |
| Thymidine | 948 | 0.1959 | 2.0899 | 2 | 2 |
| Leucine | 962 | 0.1220 | 1.5720 | 1 | 3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boiteau, R.M.; Hoyt, D.W.; Nicora, C.D.; Kinmonth-Schultz, H.A.; Ward, J.K.; Bingol, K. Structure Elucidation of Unknown Metabolites in Metabolomics by Combined NMR and MS/MS Prediction. Metabolites 2018, 8, 8. https://doi.org/10.3390/metabo8010008
Boiteau RM, Hoyt DW, Nicora CD, Kinmonth-Schultz HA, Ward JK, Bingol K. Structure Elucidation of Unknown Metabolites in Metabolomics by Combined NMR and MS/MS Prediction. Metabolites. 2018; 8(1):8. https://doi.org/10.3390/metabo8010008
Chicago/Turabian StyleBoiteau, Rene M., David W. Hoyt, Carrie D. Nicora, Hannah A. Kinmonth-Schultz, Joy K. Ward, and Kerem Bingol. 2018. "Structure Elucidation of Unknown Metabolites in Metabolomics by Combined NMR and MS/MS Prediction" Metabolites 8, no. 1: 8. https://doi.org/10.3390/metabo8010008