Metabolic Effects of Known and Novel HDAC and SIRT Inhibitors in Glioblastomas Independently or Combined with Temozolomide

 ,
,
Abstract
:1. Introduction
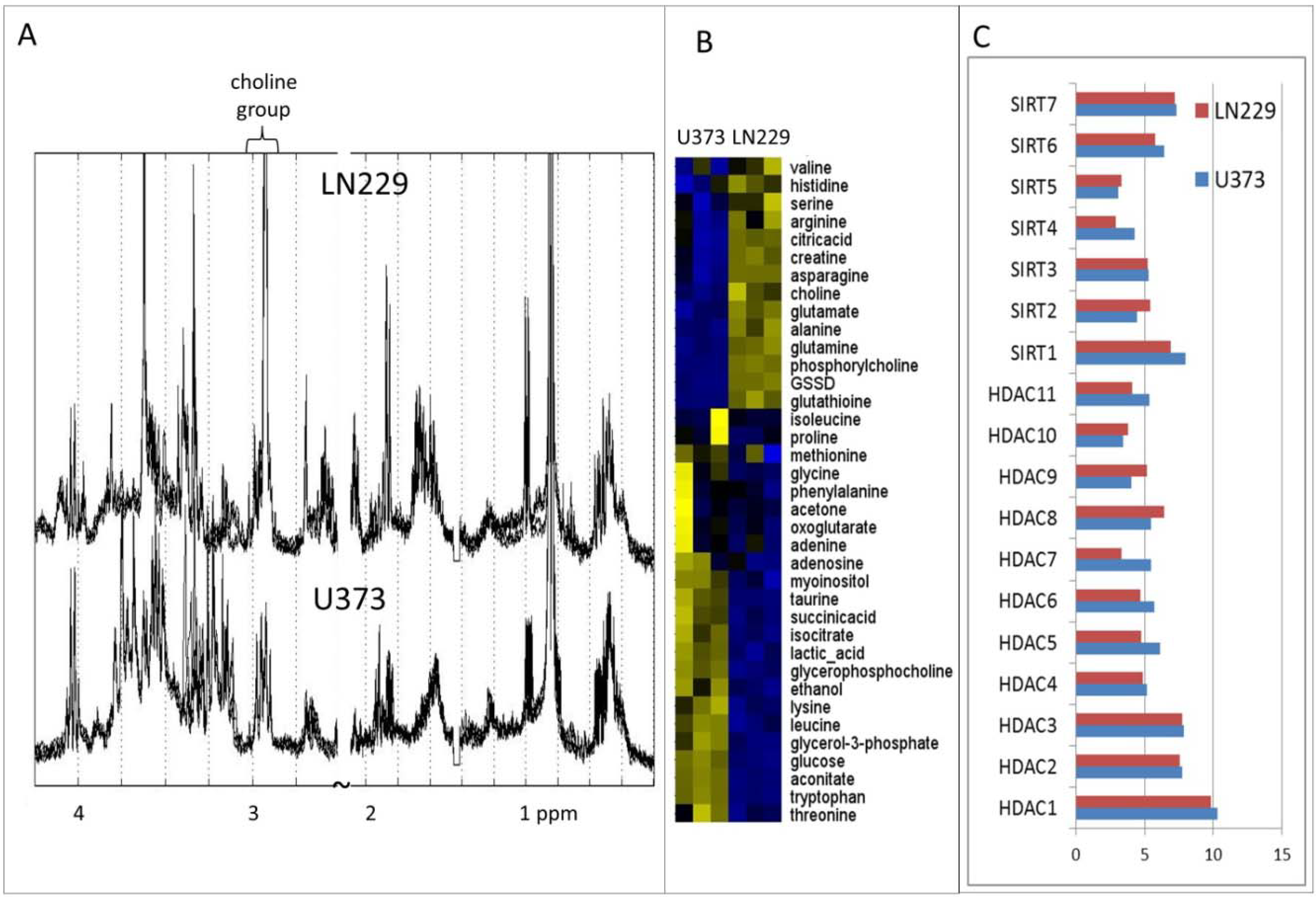
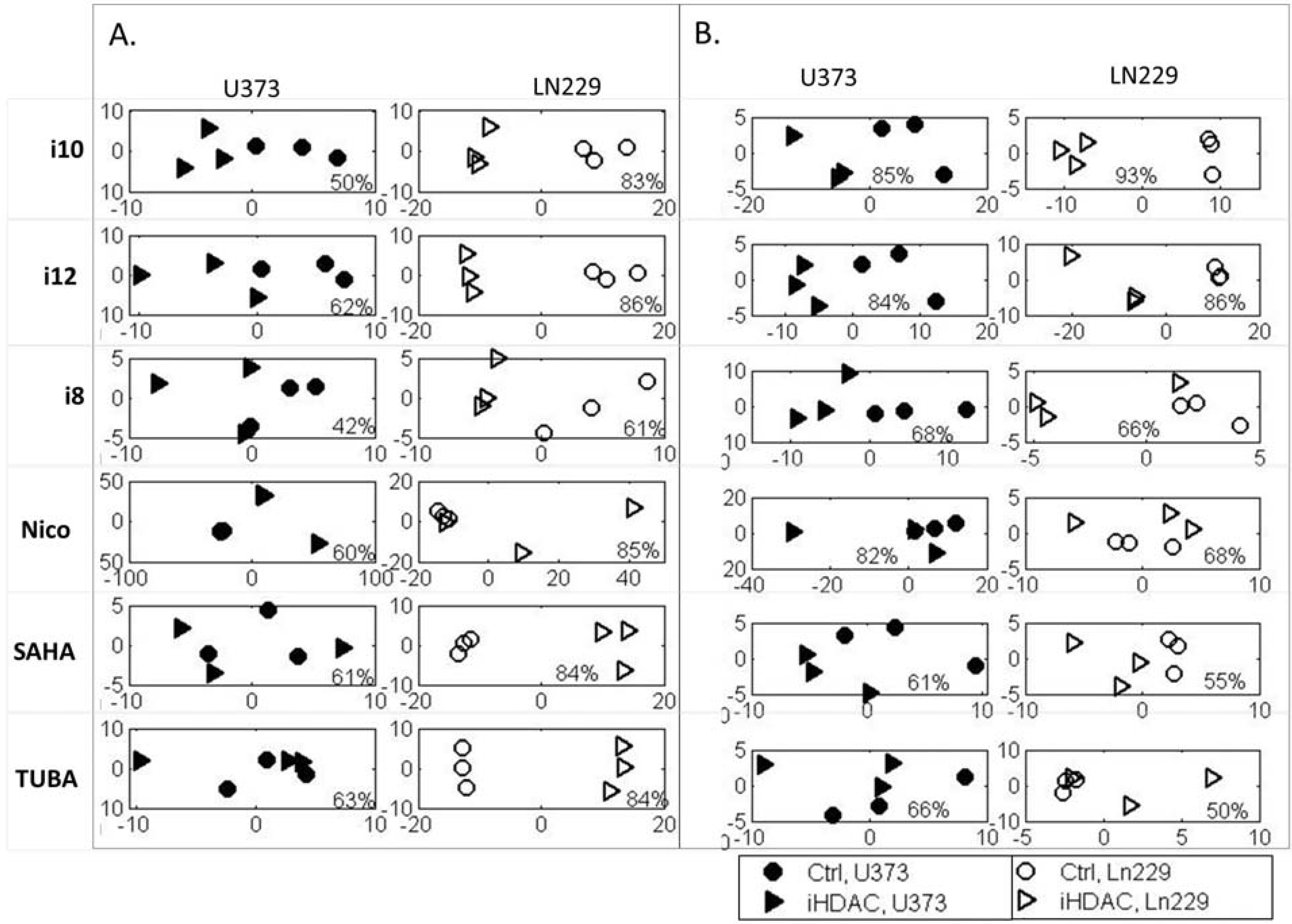
2. Results and Discussion
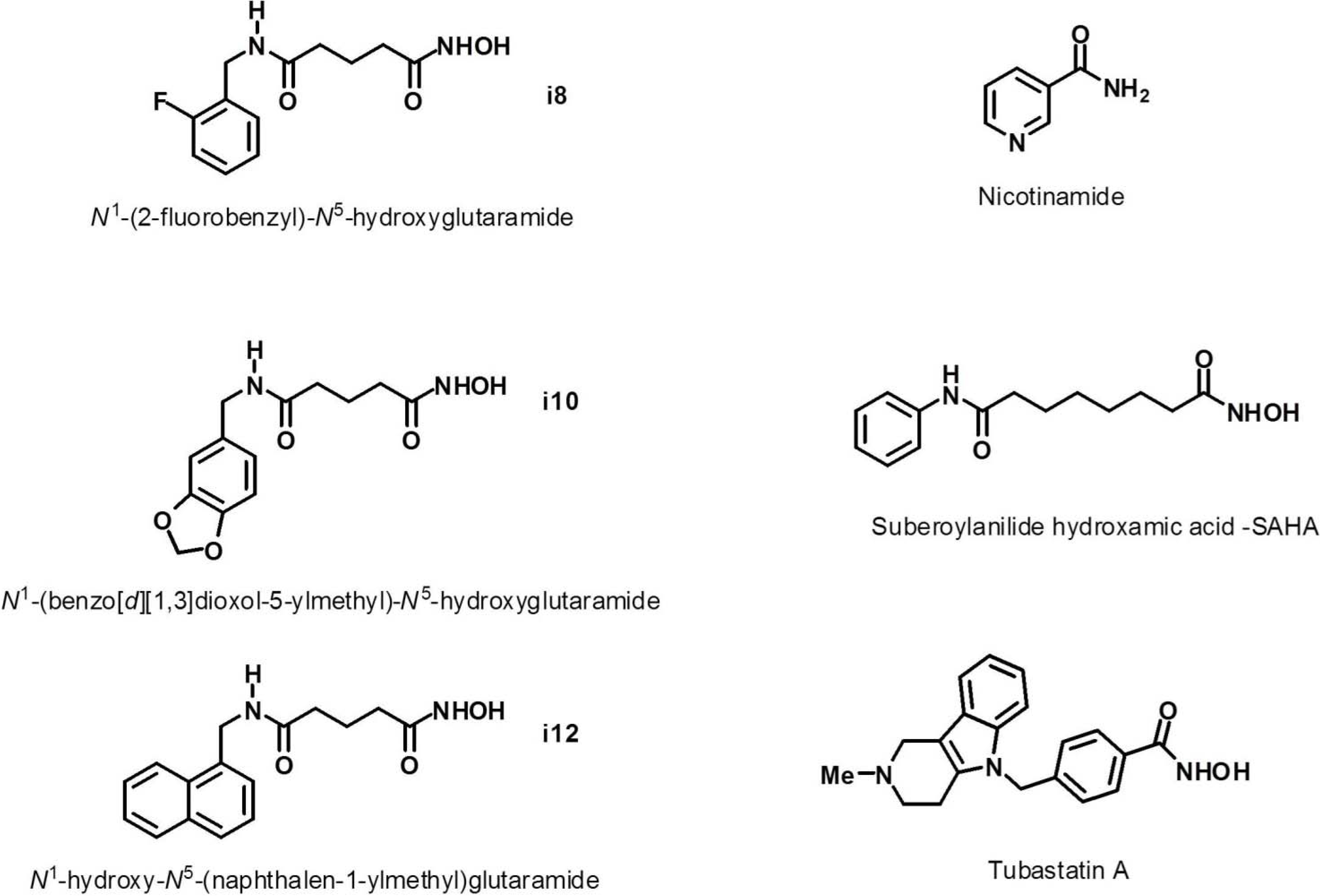

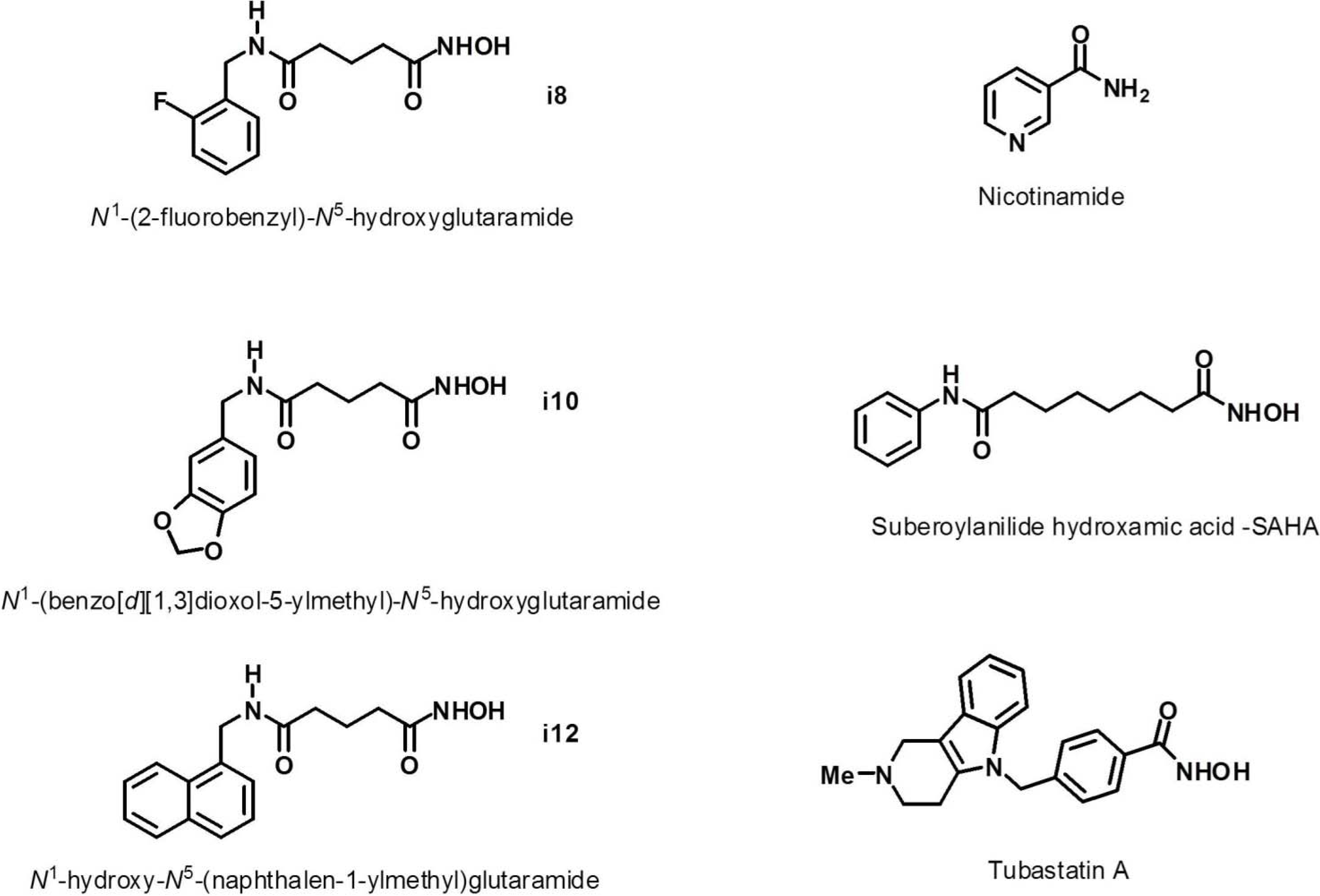{kind=link}
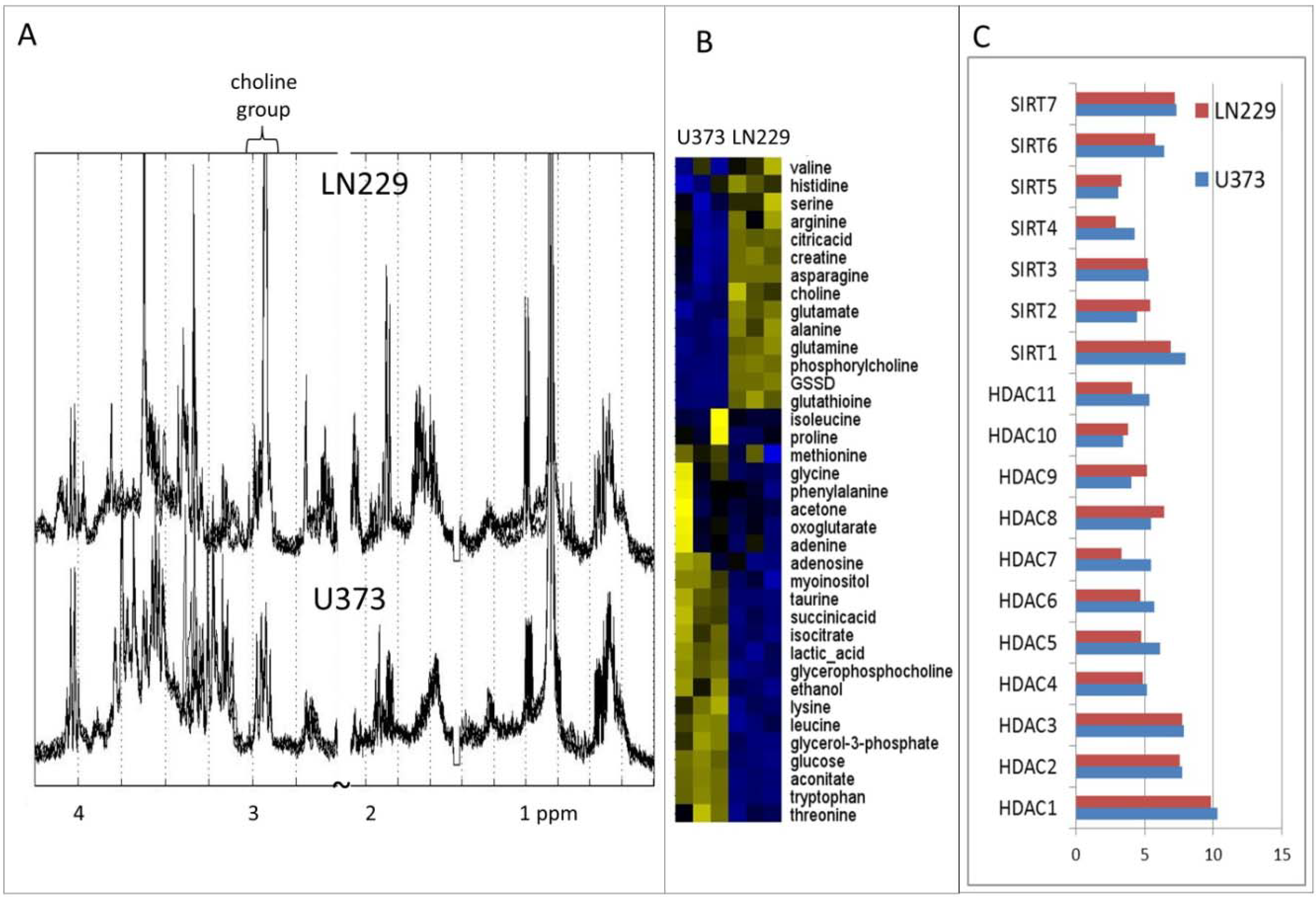{kind=link}
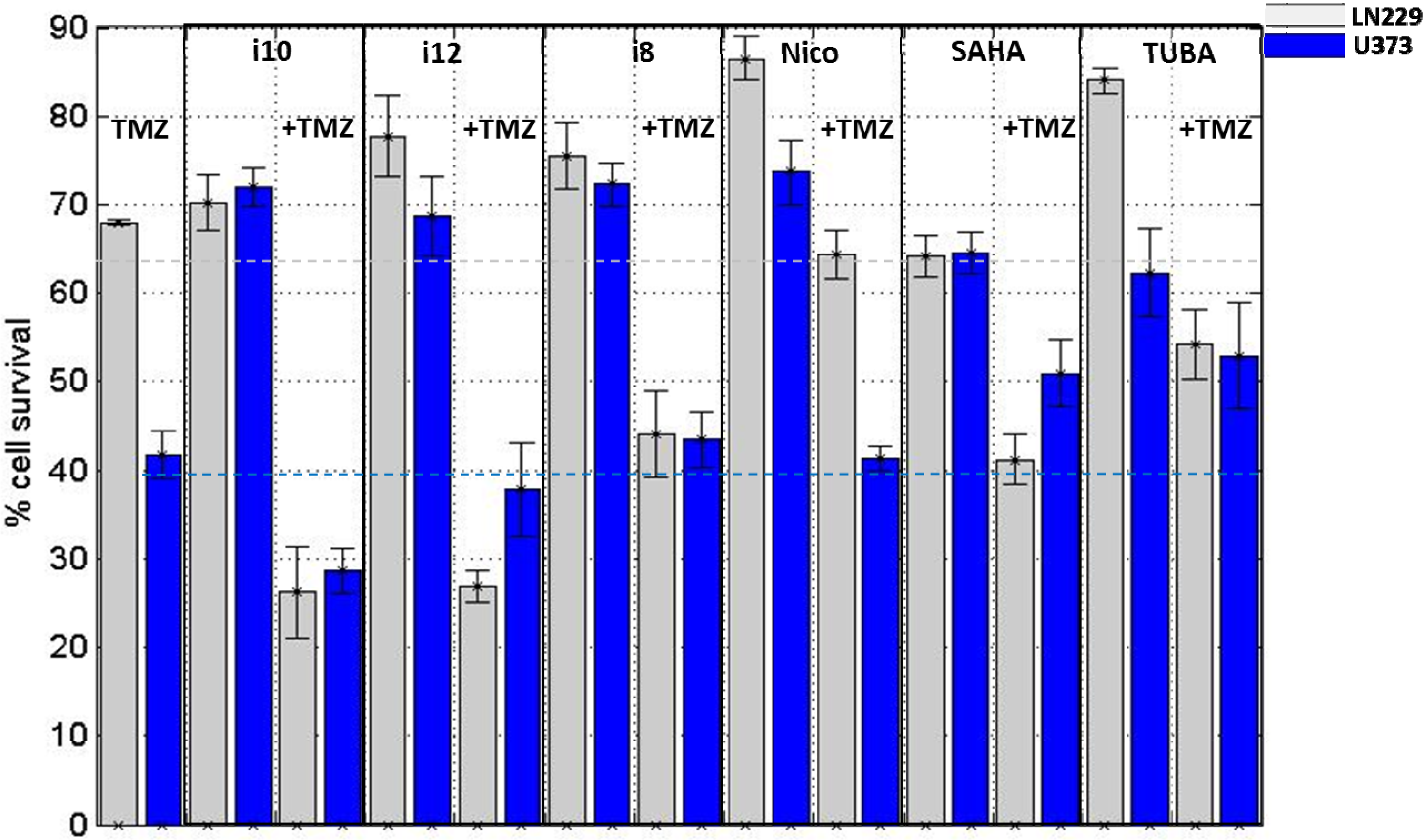{kind=link}
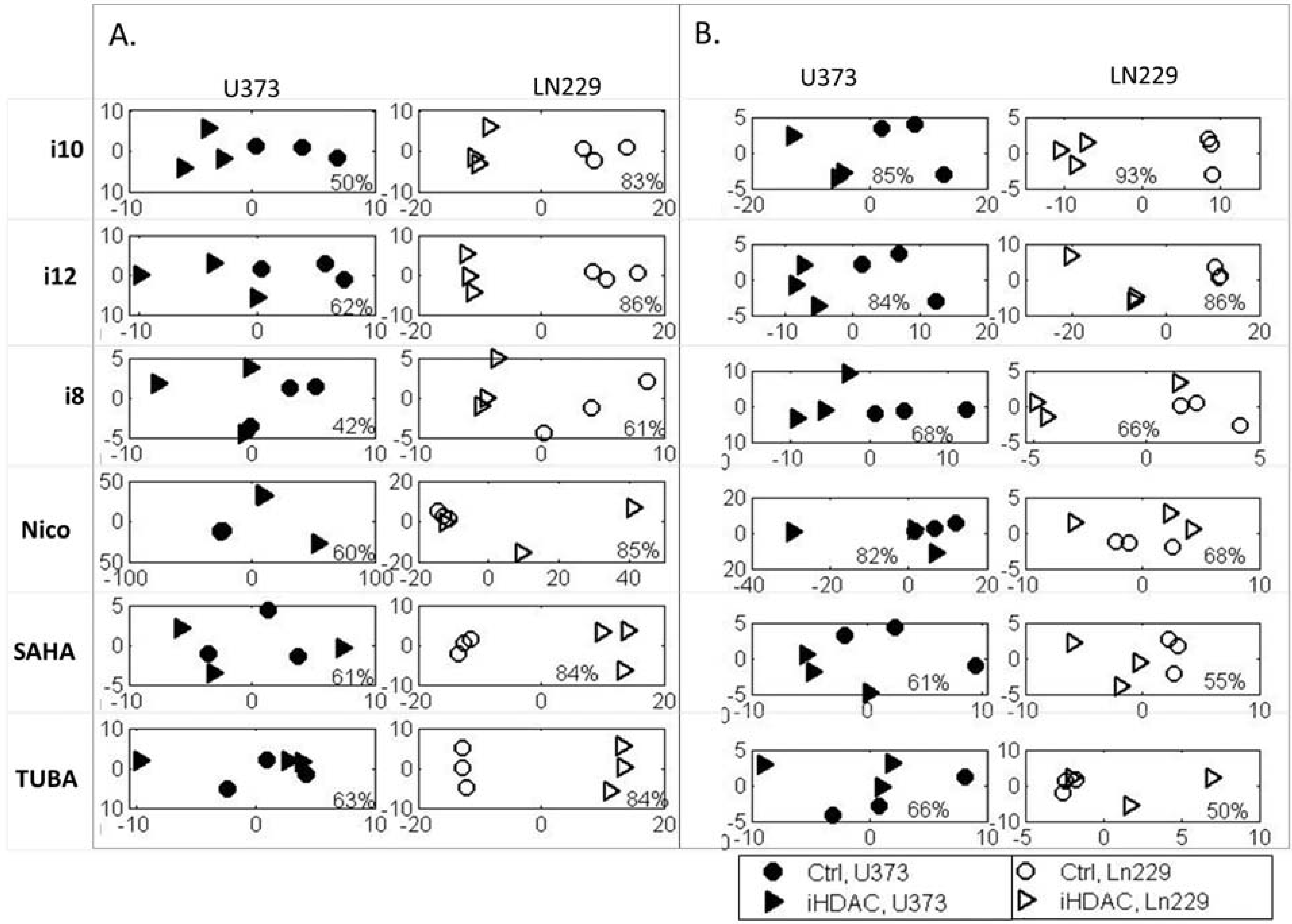{kind=link}
{kind=link}
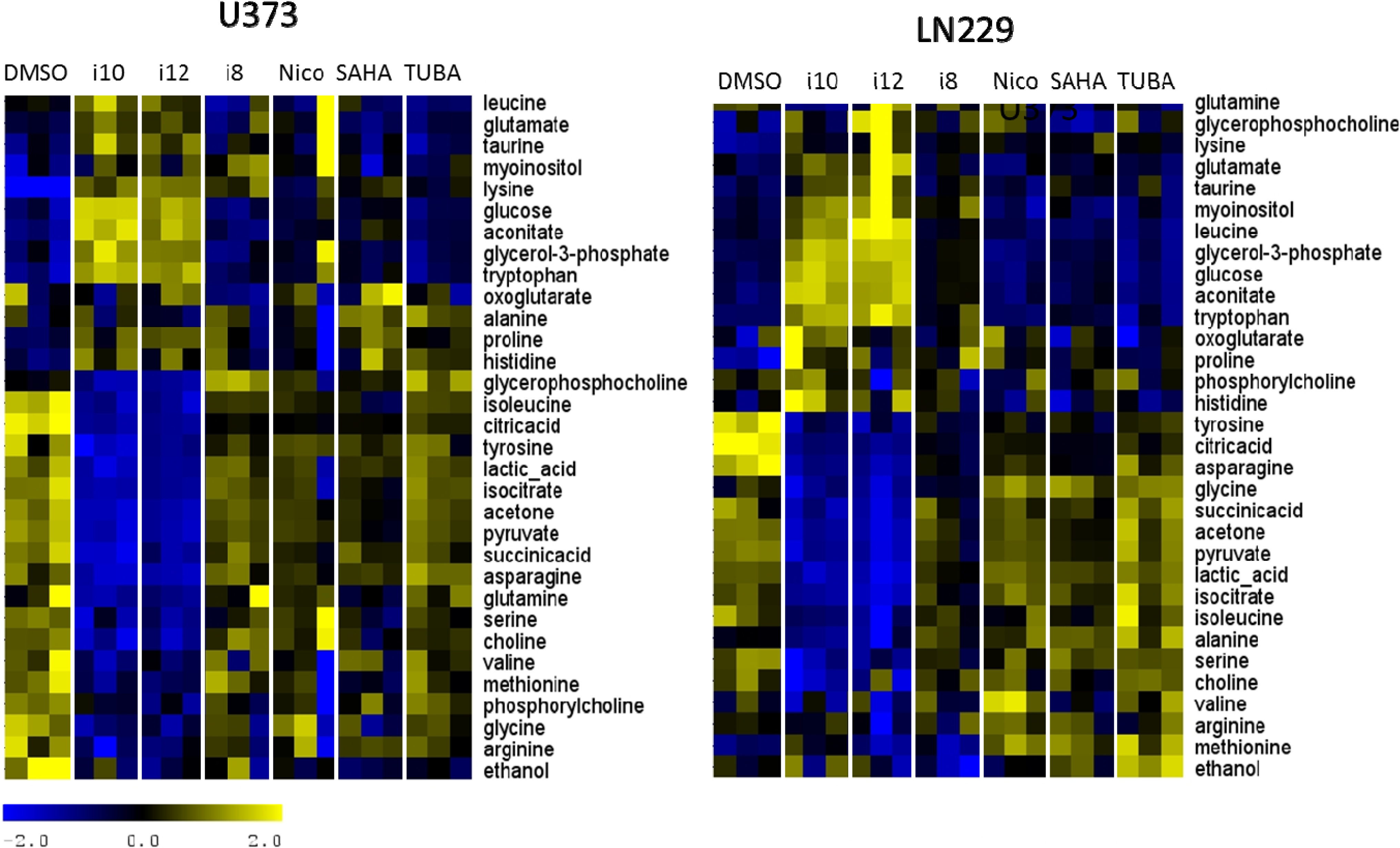{kind=link}
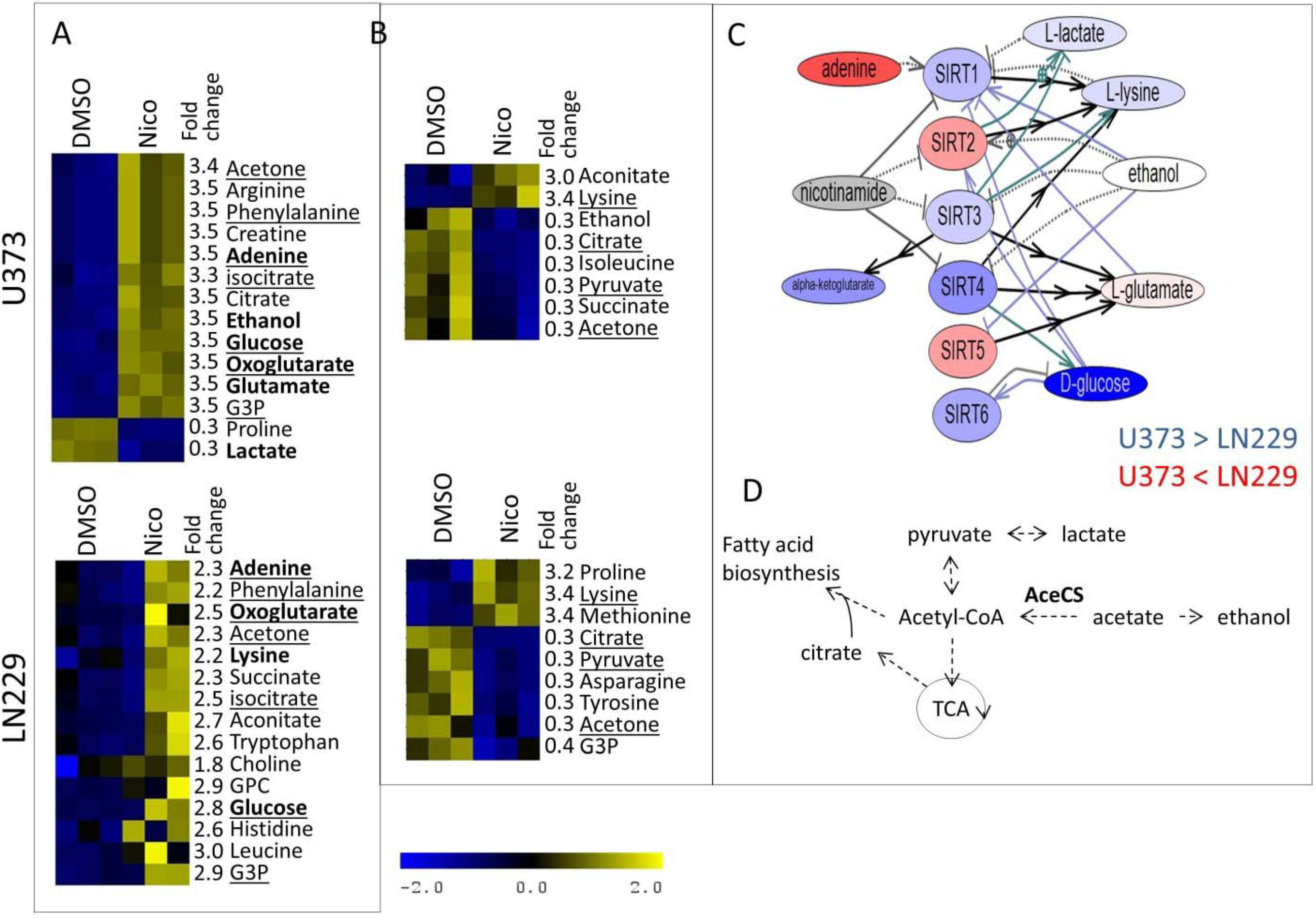{kind=link}
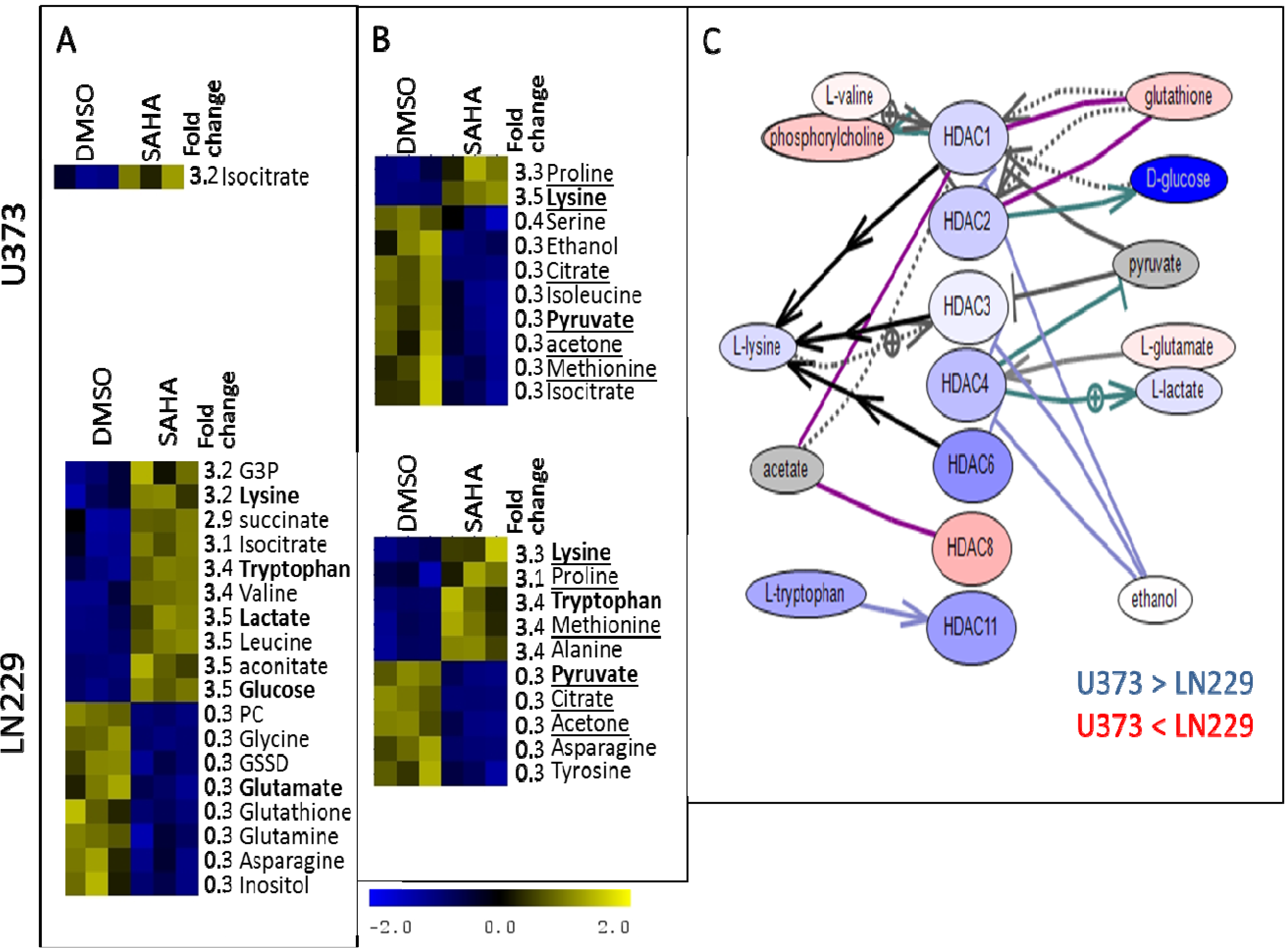{kind=link}
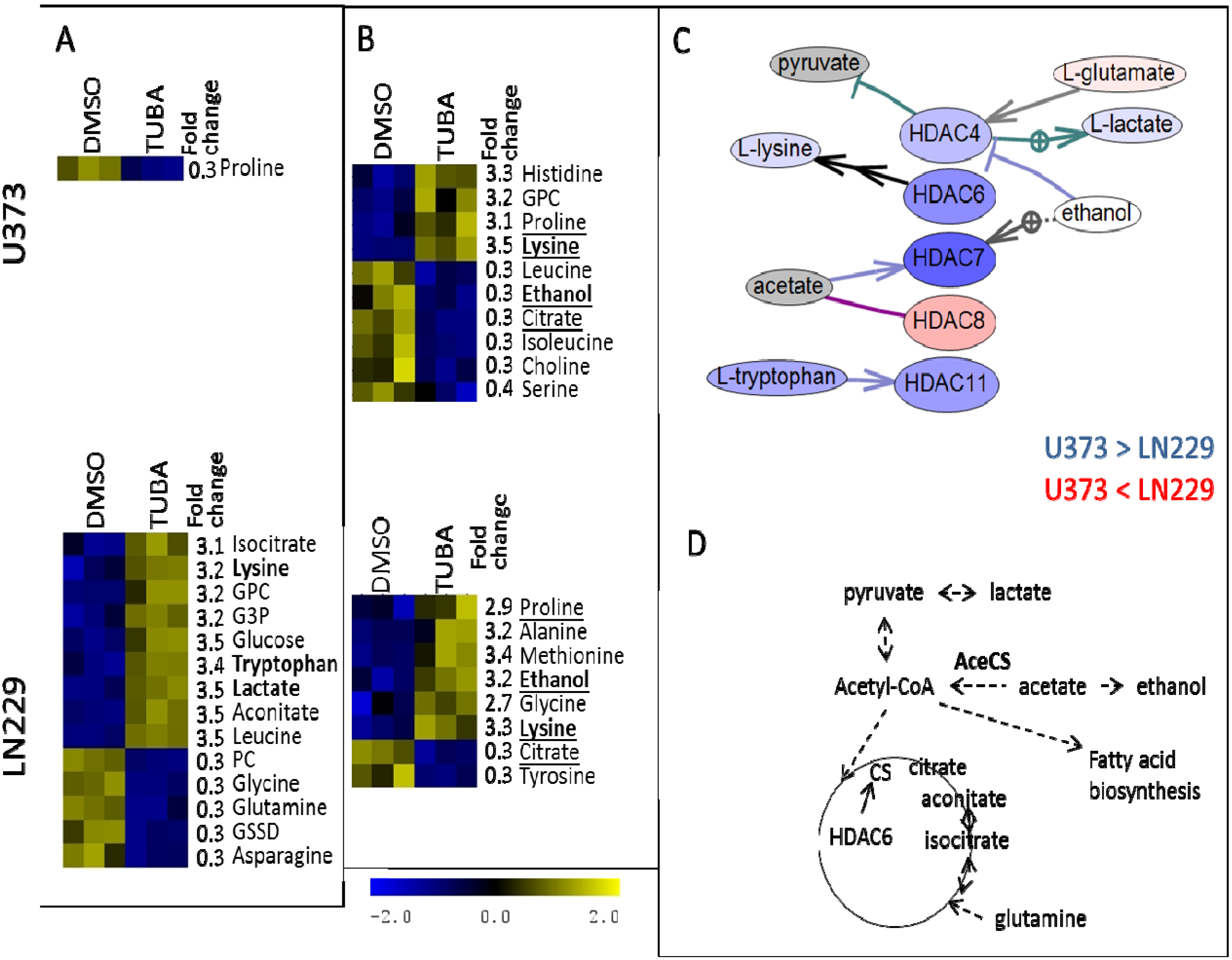{kind=link}
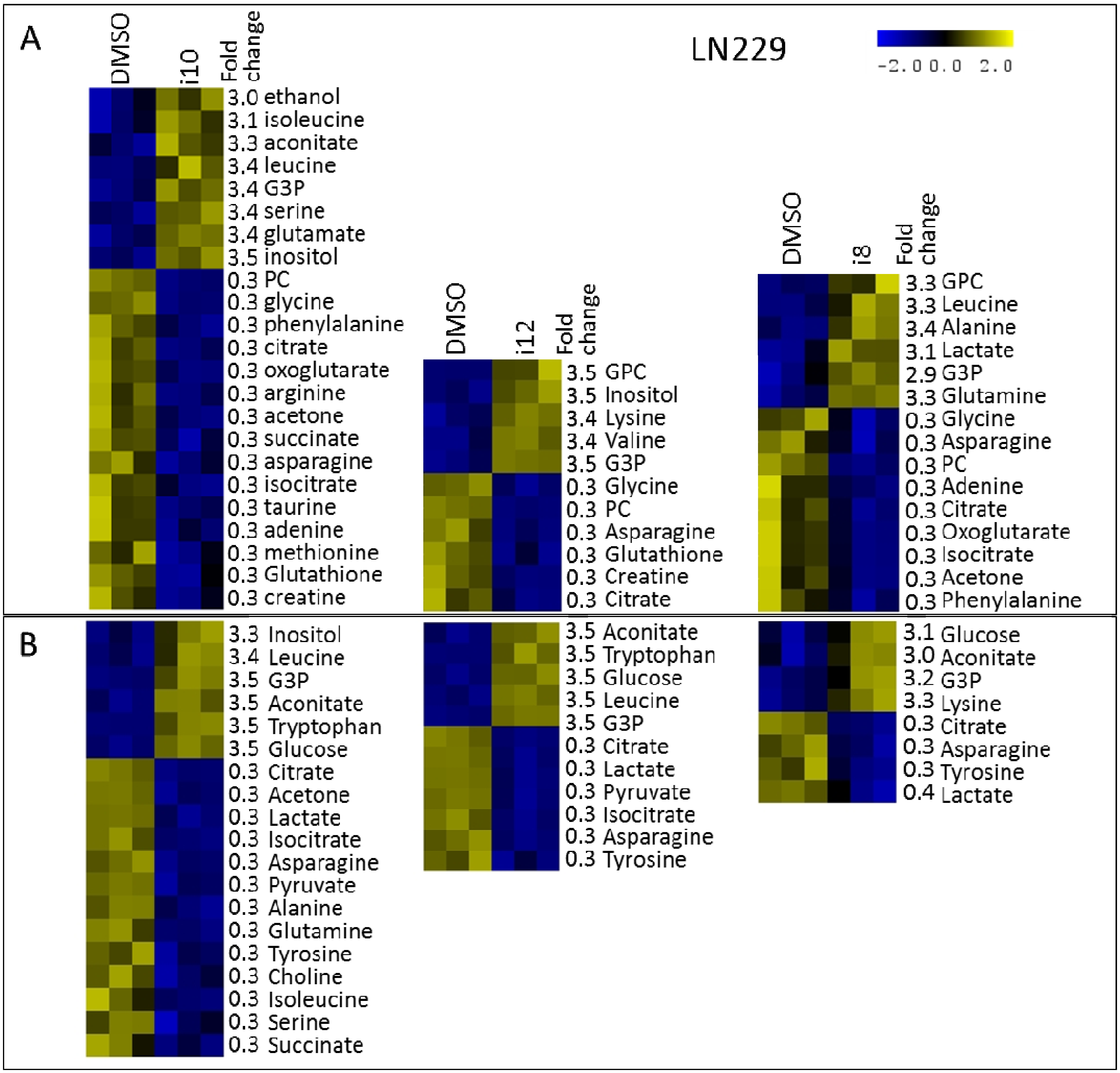{kind=link}
| Compound | cLogP (≤5)* (0.16–6.59)# | MW (≤500)* (151–655)# | H-Bond Donors (∑OHs + ∑NHs) (≤5)* (0–3)# | H-Bond Acceptors (∑Ns + ∑Os + ƩFs) (≤10)* (1–10)# | No. Rotatable Bonds (≤10)* (0–5)# |
|---|---|---|---|---|---|
| TMZ | −0.81 | 194.15 | 2 | 8 | 3 |
| i8 | 0.28 | 254.26 | 3 | 6 | 8 |
| i10 | 0.10 | 280.28 | 3 | 7 | 8 |
| i12 | 1.31 | 286.33 | 3 | 5 | 8 |
| Nicotinamide | −0.21 | 122.12 | 2 | 3 | 2 |
| SAHA | 0.99 | 264.32 | 3 | 5 | 10 |
| TUBA | 2.38 | 335.4 | 2 | 5 | 6 |
| Compound | Concentration (µM) |
|---|---|
| TMZ | 250 |
| i8 | 20 |
| i10 | 20 |
| i12 | 20 |
| Nicotinamide | 5 mM |
| SAHA | 0.5 |
| TUBA | 0.5 |

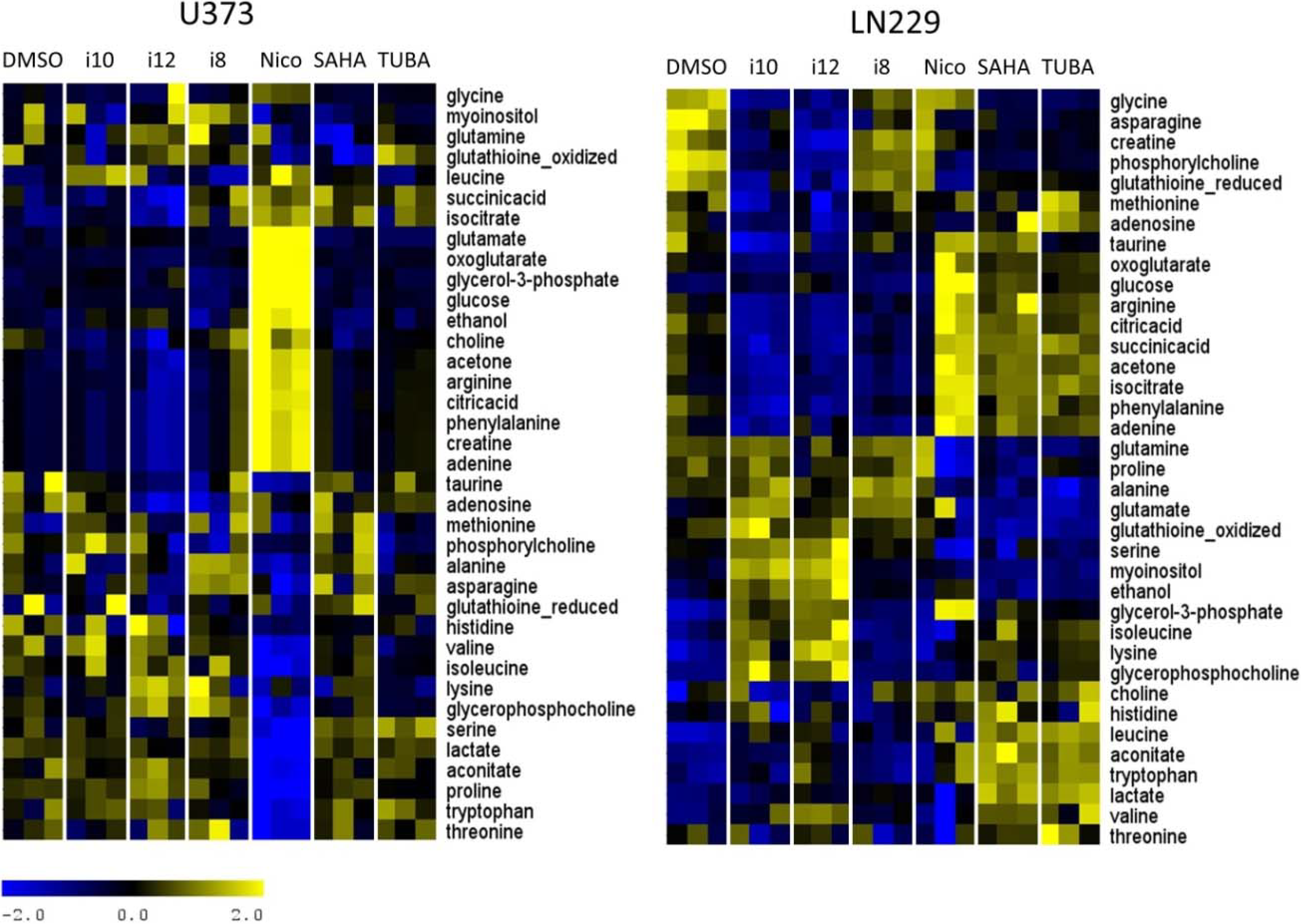
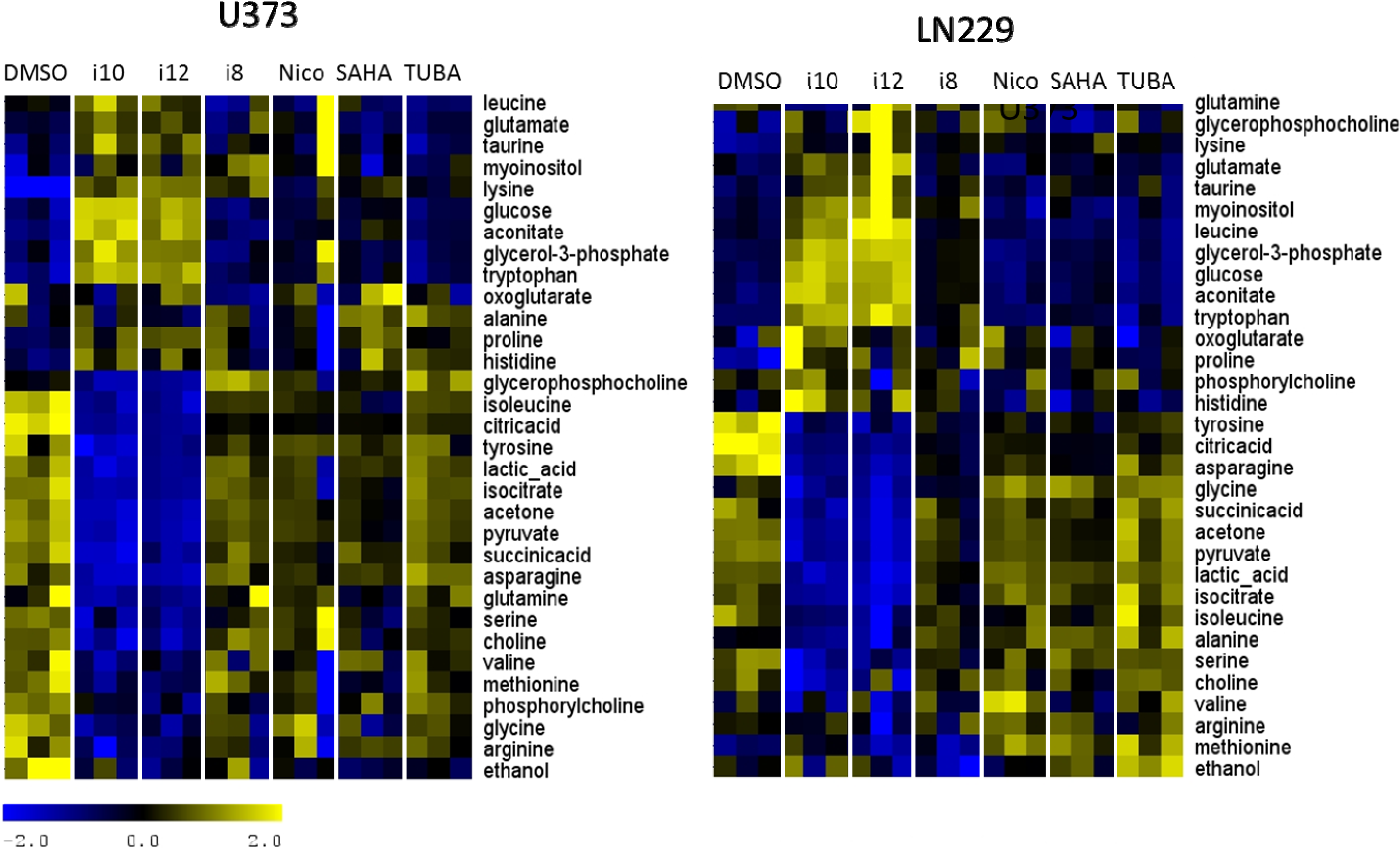
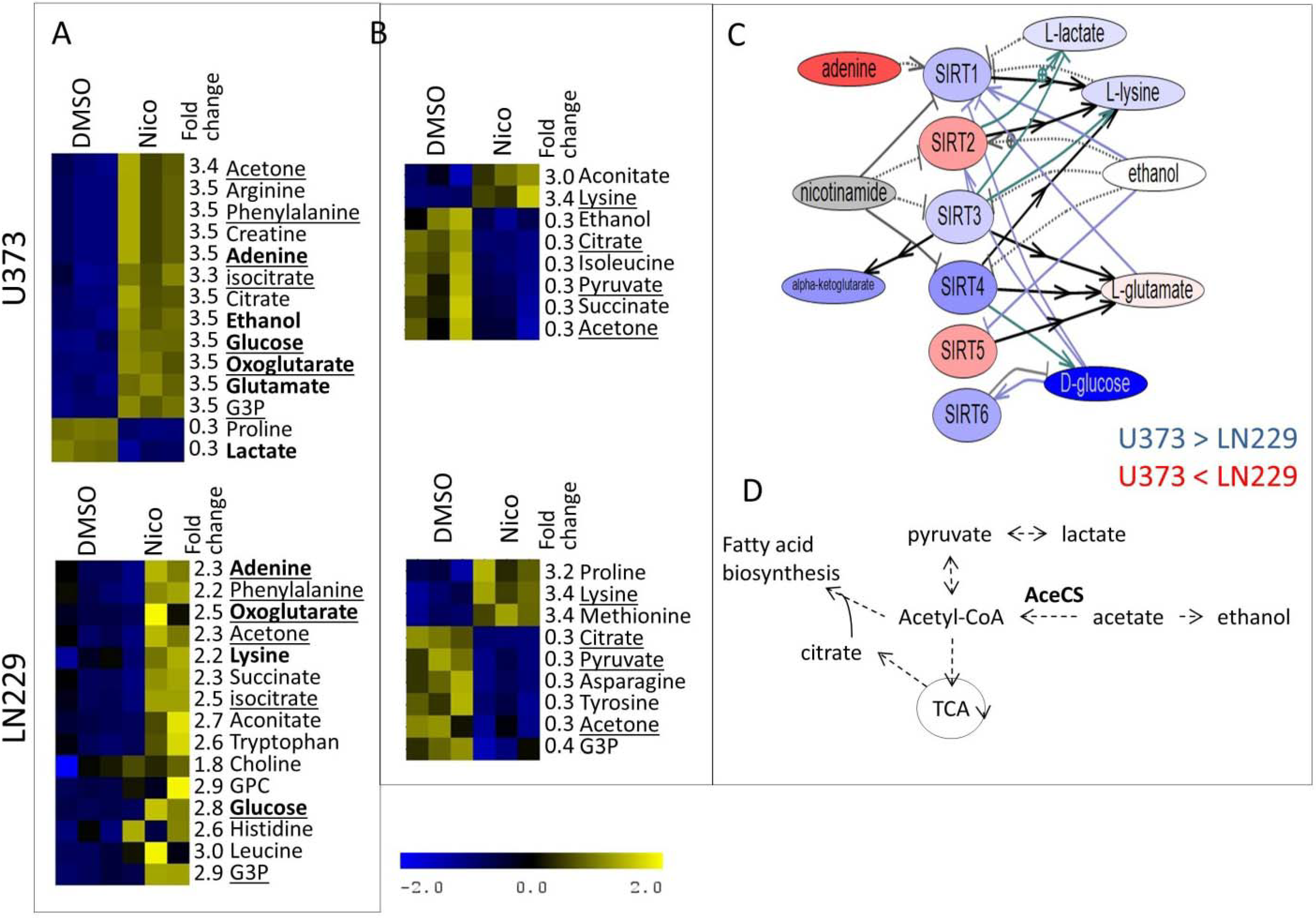
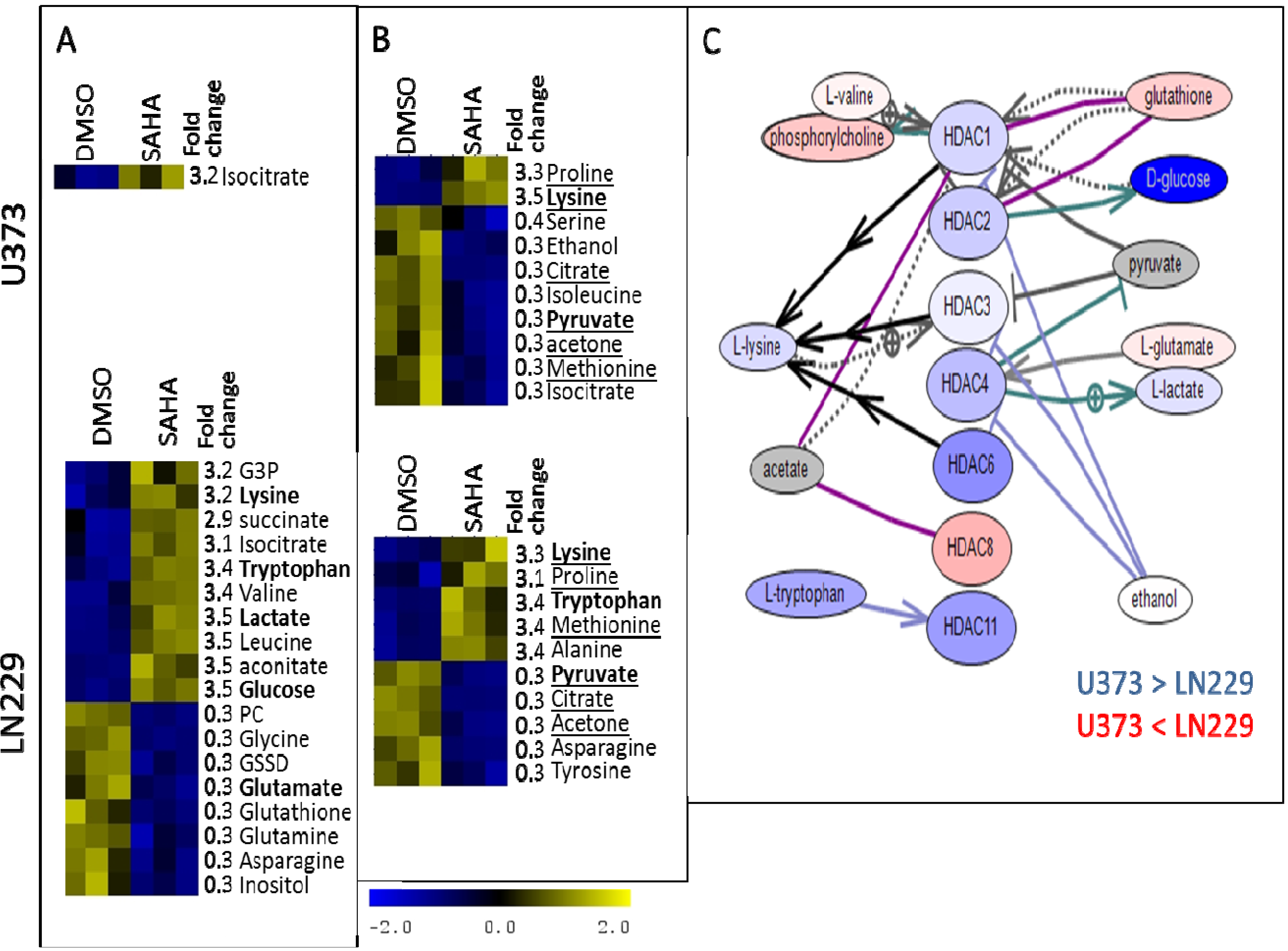
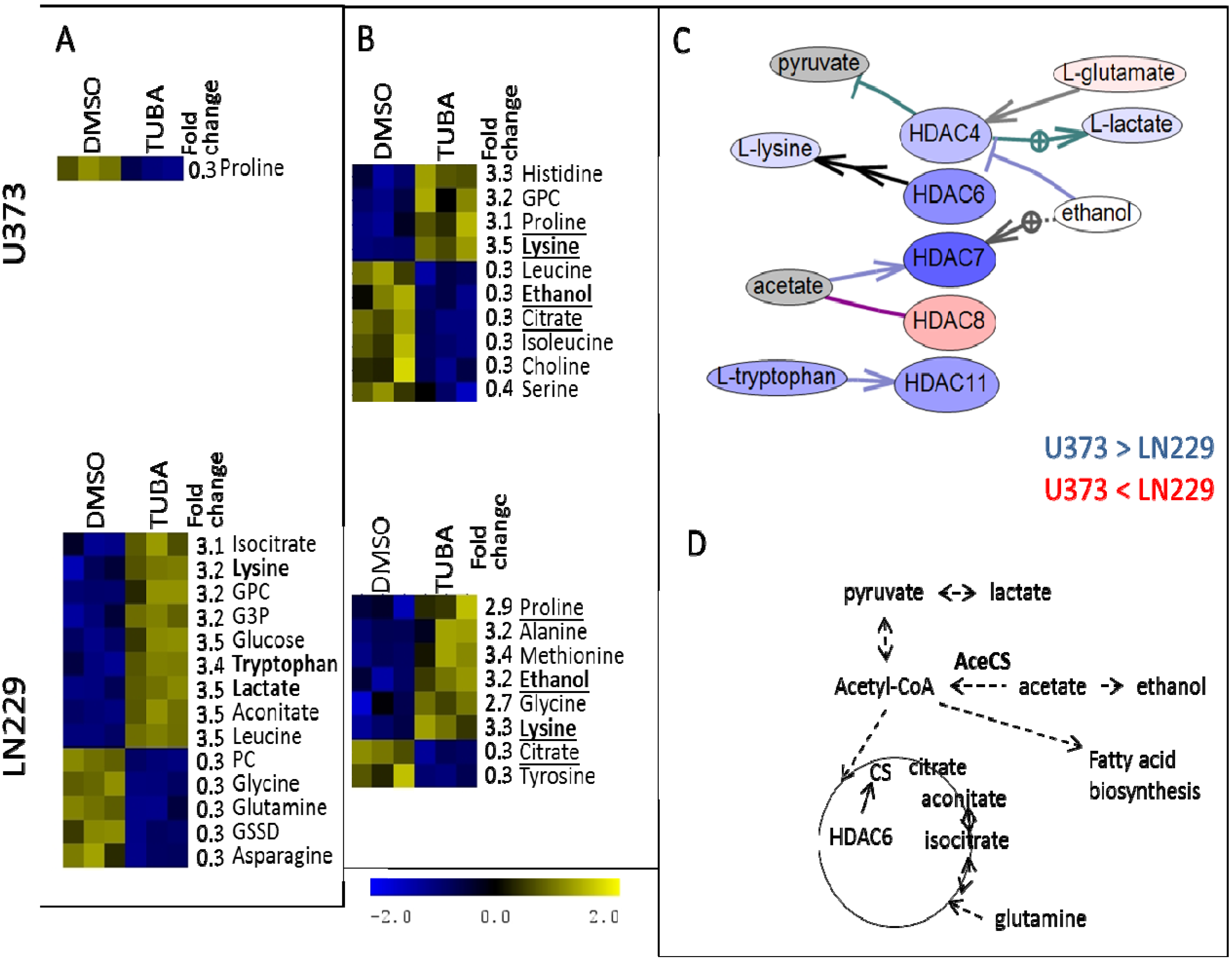
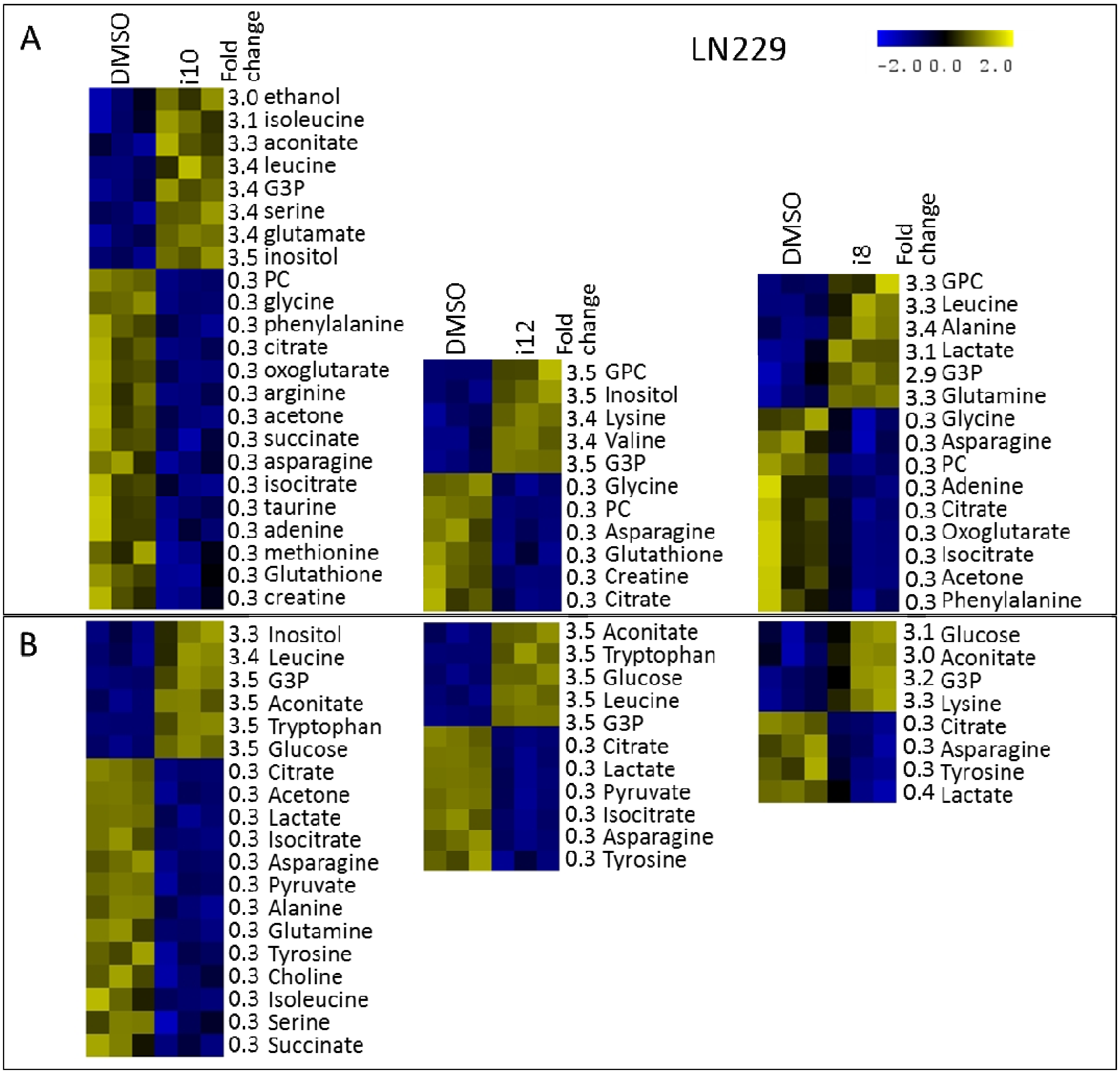
3. Experimental Section
3.1. Cell Culture and Treatment Procedures
3.2. Crystal Violet Assay Procedure
3.3. IC50 Calculations Methodology
3.4. Intracellular Hydrophilic Phase Metabolites Isolation Procedure
3.5. Extracellular Metabolites Isolation Procedure
3.6. NMR Experimentation and Preprocessing
3.7. Metabolite Quantification
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Dror, N.; Mandel, M.; Lavie, G. Unique anti-glioblastoma activities of hypericin are at the crossroad of biochemical and epigenetic events and culminate in tumor cell differentiation. PLoS One 2013, 8, e73625. [Google Scholar] [CrossRef]
- Rosato, R.R.; Almenara, J.A.; Dai, Y.; Grant, S. Simultaneous activation of the intrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitors and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) synergistically induces mitochondrial damage and apoptosis in human leukemia cells. Mol. Cancer Ther. 2003, 2, 1273–1284. [Google Scholar] [PubMed]
- Emanuele, S.; Lauricella, M.; Tesoriere, G. Histone deacetylase inhibitors: Apoptotic effects and clinical implications (Review). Int. J. Oncol. 2008, 33, 637–646. [Google Scholar] [PubMed]
- Zhang, J.; Kan, S.; Huang, B.; Hao, Z.; Mak, T.W.; Zhong, Q. Mule determines the apoptotic response to HDAC inhibitors by targeted ubiquitination and destruction of HDAC2. Genes Dev. 2011, 25, 2610–2618. [Google Scholar] [CrossRef]
- Robert, T.; Vanoli, F.; Chiolo, I.; Shubassi, G.; Bernstein, K.A.; Botrugno, O.A.; Parazzoli, D.; Oldani, A.; Minucci, S.; Foiani, M. HDACs link the DNA damage response, processing of doublestrand breaks and autophagy. Nature 2011, 471, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Gao, Z.; Marks, P.A.; Jiang, X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 18030–18035. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.; Jiang, X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.C.; Wei, K.C.; Tsai, C.N.; Huang, Y.C.; Chen, P.Y.; Chen, S.M.; Lu, Y.J.; Lee, S.T. Effect of valproic acid on the outcome of glioblastoma multiforme. Br. J. Neurosurg. 2012, 26, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Jaeckle, K.A.; Maurer, M.J.; Reid, J.M.; Ames, M.M.; Hardwick, J.S.; Reilly, J.F.; Loboda, A.; Nebozhyn, M.; Fantin, V.R.; et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: A north central cancer treatment group study. J. Clin. Oncol. 2009, 27, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Barker, C.A.; Bishop, A.J.; Chang, M.; Beal, K.; Chan, T.A. Valproic acid use during radiation therapy for glioblastoma associated with improved survival. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Shabason, J.E.; Tofilon, P.J.; Camphausen, K. Grand rounds at the National Institutes of Health: HDAC inhibitors as radiation modifiers, from bench to clinic. J. Cell Mol. Med. 2011, 15, 2735–2744. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.M.; Manton, C.A.; Bhat, K.P.; Tsai, W.W.; Aldape, K.; Barton, M.C.; Chandra, J. Inhibition of LSD1 sensitizes glioblastoma cells to histone deacetylase inhibitors. Neuro Oncol. 2011, 13, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Asklund, T.; Kvarnbrink, S.; Holmlund, C.; Wibom, C.; Bergenheim, T.; Henriksson, R.; Hedman, H. Synergistic killing of glioblastoma stem-like cells by bortezomib and HDAC inhibitors. Anticancer Res. 2012, 32, 2407–2413. [Google Scholar] [PubMed]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; von Deimling, A.; Fischer, M.; Witt, O. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 2009, 15, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Milde, T.; Oehme, I.; Korshunov, A.; Kopp-Schneider, A.; Remke, M.; Northcott, P.; Deubzer, H.E.; Lodrini, M.; Taylor, M.D.; von Deimling, A.; et al. HDAC5 and HDAC9 in medulloblastoma: novel markers for risk stratification and role in tumor cell growth. Clin. Cancer 2010, 16, 3240–3252. [Google Scholar]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Cuperlovic-Culf, M. NMR Metabolomics in Cancer Research; Woodheaven Publishing: Oxford, UK, 2013. [Google Scholar]
- Bensinger, S.J.; Christofk, H.R. New aspects of the Warburg effect in cancer cell biology. Semin. Cell Dev. Biol. 2012, 23, 352–361. [Google Scholar] [CrossRef]
- Ward, C.S.; Eriksson, P.; Izquierdo-Garcia, J.L.; Brandes, A.H.; Ronen, S.M. HDAC Inhibition induces increased choline uptake and elevated phosphocholine levels in MCF7 breast cancer cells. PLoS One 2013, 8, e62610. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.L.; Troy, H.; Kristeleit, R.; Aherne, W.; Jackson, L.E.; Atadja, P.; Griffiths, J.R.; Judson, I.R.; Workman, P.; Leach, M.O.; et al. Noninvasive magnetic resonance spectroscopic pharmacodynamic markers of a novel histone deacetylase inhibitor, LAQ824, in human colon carcinoma cells and xenografts. Neoplasia 2008, 10, 303–313. [Google Scholar]
- Beloueche-Babari, M.; Arunan, V.; Troy, H.; te Poele, R.H.; te Fong, A.C.W.; Jackson, L.E.; Payne, G.S.; Griffiths, J.R.; Judson, I.R.; Workman, P.; et al. Histone deacetylase inhibition increases levels of choline kinase α and phosphocholine facilitating noninvasive imaging in human cancers. Cancer Res. 2012, 72, 990–1000. [Google Scholar]
- Brandes, A.H.; Ward, C.S.; Ronen, S.M. 17-allyamino-17-demethoxygeldanamycin treatment results in a magnetic resonance spectroscopy-detectable elevation in choline-containing metabolites associated with increased expression of choline transporter SLC44A1 and phospholipase A2. Breast Cancer Res. 2010, 12, R84. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.J.; Moutinho, M.; Gama, M.J.; Rodrigues, C.M.P.; Rodrigues, E. Histone deacetylase inhibition decreases cholesterol levels in neuronal cells by modulating key genes in cholesterol synthesis, uptake and efflux. PLoS One 2013, 8, e53394. [Google Scholar] [CrossRef] [PubMed]
- Wardell, S.E.; Ilkayeva, O.R.; Wieman, H.L.; Frigo, D.E.; Rathmell, J.C.; Newgard, C.B.; McDonnell, D.P. Glucose metabolism as a target of histone deacetylase inhibitors. Mol. Endocrinol. 2009, 23, 388–401. [Google Scholar] [CrossRef] [PubMed]
- Amoêdo, N.D.; Rodrigues, M.F.; Pezzuto, P.; Galina, A.; da Costa, R.M.; de Almeida, F.C.; El-Bacha, T.; Rumjanek, F.D. Energy metabolism in H460 lung cancer cells: effects of histone deacetylase inhibitors. PLoS One 2011, 6, e22264. [Google Scholar]
- Kamemura, K.; Ogawa, M.; Ohkubo, S.; Ohtsuka, Y.; Shitara, Y.; Komiya, T.; Maeda, S.; Ito, A.; Yoshida, M. Depression of mitochondrial metabolism by downregulation of cytoplasmic deacetylase, HDAC6. FEBS Lett. 2012, 586, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Schwer, B.; Verdin, E. Conserved metabolic regulatory functions of sirtuins. Cell Metab. 2008, 7, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Hallows, W.C.; Lee, S.; Denu, J.M. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc. Natl. Acad. Sci. USA 2006, 103, 10230–10235. [Google Scholar] [CrossRef] [PubMed]
- Sebastián, C.; Zwaans, B.M.M.; Silberman, D.M.; Gymrek, M.; Goren, A.; Zhong, L.; Ram, O.; Truelove, J.; Guimaraes, A.R.; Toiber, D.; et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 2012, 151, 1185–1199. [Google Scholar]
- Lyssiotis, C.; Cantley, L.C. SIRT6 puts cancer metabolism in the driver’s seat. Cell 2012, 151, 1155–1156. [Google Scholar] [CrossRef] [PubMed]
- Borodovsky, A.; Seltzer, M.J.; Riggins, G.J. Altered cancer cell metabolism in gliomas with mutant IDH1 or IDH2. Curr. Opin. Oncol. 2012, 24, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Cuperlovic-Culf, M.; Ferguson, D.; Culf, A.; Morin, P.; Touaibia, M. 1H NMR metabolomics analysis of glioblastoma subtypes: Correlation between metabolomics and gene expression characteristics. J. Biol. Chem. 2012, 287, 20164–20175. [Google Scholar]
- Grant, S.; Dai, Y. Histone deacetylase inhibitors and rational combination therapies. Adv. Cancer Res. 2012, 116, 199–237. [Google Scholar] [PubMed]
- Ciampa-Crapoulet, S.; Léger, D.A.; Champigny, C.; Ghosh, A.; Ouellette, R.J.; Čuperlović-Culf, M.; Culf, A.S. Development and testing of substituted N1-Benzyl-glutaryl-N5-hydroxamic acids as HDAC6-selective inhibitors. 2014; unpublished work. [Google Scholar]
- Avalos, J.L.; Bever, K.M.; Wolberger, C. Mechanism of sirtuin inhibition by nicotinamide: Altering the NAD(+) cosubstrate specificity of a Sir2 enzyme. Mol. Cell 2005, 17, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Y.; Hirschey, M.D.; Shimazu, T.; Ho, L.; Verdin, E. Mitochondrial sirtuins. Biochim. Biophys. Acta 2010, 1804, 1645–1651. [Google Scholar]
- Marks, P.; Breslow, R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotech. 2007, 25, 84–90. [Google Scholar] [CrossRef]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nature reviews. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Bolden, J.E.; Shi, W.; Jankowski, K.; Kan, C.-Y.; Cluse, L.; Martin, B.P.; MacKenzie, K.L.; Smyth, G.K.; Johnstone, R.W. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis. 2013, 4, e519. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Chen, Y.P. Energy based pharmacophore mapping of HDAC inhibitors against class I HDAC enzymes. Biochim. Biophys. Acta 2013, 1834, 317–328. [Google Scholar] [CrossRef]
- Butler, K.; Kalin, J.; Brochier, C. Rational design and simple chemistry yield a superior neuroprotective hdac6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [PubMed]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.C.; Chu, P.C.; Wang, H.Y.; Huang, C.Y.; Chen, P.Y.; Tsai, H.C.; Lu, Y.J.; Lee, P.Y.; Tseng, I.C.; Feng, L.Y.; et al. Focused ultrasound-induced blood-brain barrier opening to enhance temozolomide delivery for glioblastoma treatment: a preclinical study. PLoS One 2013, 8, e58995. [Google Scholar] [CrossRef]
- Peter, Y.; Comellas, A.; Levantini, E.; Ingenito, E.P.; Shapiro, S.D. Epidermal growth factor receptor and claudin-2 participate in A549 permeability and remodeling: implications for non-small cell lung cancer tumor colonization. Mol. Carcinog. 2009, 48, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Cuperlovic-Culf, M.; Chute, I.C.; Culf, A.S.; Touaibia, M.; Ghosh, A.; Griffiths, S.; Tulpan, D.; Leger, S.; Belkaid, A.; Surette, M.E.; et al. 1H NMR metabolomics combined with gene expression analysis for the determination of major metabolic difference between subtypes of breast cell lines. Chem. Sci. 2011, 2, 2263–2270. [Google Scholar]
- Morin, P.; Ferguson, D.; LeBlanc, L.M.; Hébert, M.J.G.; Paré, A.F.; Jean-François, J.; Surette, M.E.; Touaibia, M.; Cuperlovic-Culf, M. NMR metabolomics analysis of the effects of 5-lipoxygenase inhibitors on metabolism in glioblastomas. J. Prot. Res. 2013, 12, 2165–2176. [Google Scholar] [CrossRef]
- Wiedemeyer, R.; Brennan, C.; Heffernan, T.P.; Xiao, Y.; Mahoney, J.; Protopopov, A.; Zheng, H.; Bignell, G.; Furnari, F.; Cavenee, W.K.; et al. Feedback circuit among INK4 tumor suppressors constrains human glioblastoma development. Cancer Cell 2008, 13, 355–364. [Google Scholar]
- Gallinari, P.; Di Marco, S.; Jones, P.; Pallaoro, M.; Steinkühler, C. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 2007, 17, 195–211. [Google Scholar] [PubMed]
- Hermisson, M.; Klumpp, A.; Wick, W.; Wischhusen, J.; Nagel, G.; Roos, W.; Kaina, B.; Weller, M. O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. J. Neurochem. 2006, 96, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Cahill, D.P.; Codd, P.J.; Batchelor, T.T.; Curry, W.T.; Louis, D.N. MSH6 inactivation and emergent temozolomide resistance in human glioblastomas. Clin. Neurosurg. 2008, 55, 165–171. [Google Scholar] [PubMed]
- Shinsato, Y.; Furukawa, T.; Yunoue, S.; Yonezawa, H.; Minami, K.; Nishizawa, Y.; Ikeda, R.; Kawahara, K.; Yamamoto, M.; Hirano, H.; et al. Reduction of MLH1 and PMS2 confers temozolomide resistance and is associated with recurrence of glioblastoma. Oncotarget 2013, 4, 2261–2270. [Google Scholar] [PubMed]
- Kitange, G.J.; Mladek, A.C.; Carlson, B.L.; Schroeder, M.A.; Pokorny, J.L.; Cen, L.; Decker, P.A.; Wu, W.; Lomberk, G.A.; Gupta, S.K.; et al. Inhibition of histone deacetylation potentiates the evolution of acquired temozolomide resistance linked to MGMT upregulation in glioblastoma xenografts. Clin. Cancer Res. 2012, 18, 4070–4079. [Google Scholar] [CrossRef] [PubMed]
- Cuperlovic-Culf, M.; Culf, A. Protein acetylation as an integral part of metabolism in cancer development and progression. Am. J. Cancer Rev. 2014, 2, 1–23. [Google Scholar]
- Lefort, N.; Brown, A.; Lloyd, V.; Ouellette, R.; Touaibia, M.; Culf, A.S.; Cuperlovic-Culf, M. 1H NMR metabolomics analysis of the effect of dichloroacetate and allopurinol on breast cancers. J. Pharma Biomed. Anal. 2014, 93, 77–85. [Google Scholar] [CrossRef]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116. [Google Scholar] [CrossRef] [PubMed]
- Sauve, A.; Wolberger, C.; Schramm, V.L.; Boeke, J.D. The biochemistry of sirtuins. Annu. Rev. Biochem. 2006, 75, 435–465. [Google Scholar] [CrossRef] [PubMed]
- Glunde, K.; Bhujwalla, Z.M.; Ronen, S.M. Choline metabolism in malignant transformation. Nat. Rev. Cancer 2011, 11, 835–848. [Google Scholar] [PubMed]
- Bowlby, S.C.; Thomas, M.J.; D’Agostino, R.B.; Kridel, S.J. Nicotinamide phosphoribosyl transferase (Nampt) is required for de novo lipogenesis in tumor cells. PLoS One 2012, 7, e40195. [Google Scholar] [CrossRef] [PubMed]
- Phang, J.M.; Liu, W.; Hancock, C. Bridging epigenetics and metabolism. Epigenetics 2013, 8, 231–236. [Google Scholar]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denny, B.J.; Wheelhouse, R.T.; Stevens, M.F.; Tsang, L.L.; Slack, J.A. NMR and molecular modeling investigation of the mechanism of activation of the antitumor drug temozolomide and its interaction with DNA. Biochemistry 1994, 33, 9045–9051. [Google Scholar]
- McBrian, M.A.; Behbahan, I.S.; Ferrari, R.; Su, T.; Huang, T.W.; Li, K.; Hong, C.S.; Christofk, H.R.; Vogelauer, M.; Seligson, D.B.; et al. Histone acetylation regulates intracellular pH. Mol. Cell. 2013, 49, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, R.M.; Gilon, P.; Henquin, J.C. Ketoisocaproic acid and leucine increase cytoplasmic pH in mouse pancreatic B cells: Role of cytoplasmic Ca2+ and pH-regulating exchangers. Endocrinology 1996, 137, 677–685. [Google Scholar]
- Ishii, N.; Maier, D.; Merlo, A.; Tada, M.; Sawamura, Y.; Diserens, A.C.; van Meir, E.G. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999, 9, 469–479. [Google Scholar] [CrossRef]
- MATLAB, vR2010b. Mathworks Inc.: Natick, MA, USA, 2010.
- Savorani, F.; Tomasi, G.; Engelsen, S.B. icoshift: A versatile tool for the rapid alignment of 1D NMR spectra. J. Magn. Reson. 2010, 202, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D.D.; Psychogios, N.; Dong, E.; Bouatra, S.; et al. HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 2009, 37, D603–D610. [Google Scholar] [CrossRef] [PubMed]
- Markley, J.L.; Anderson, M.E.; Cui, Q.; Eghbalnia, H.R.; Lewis, I.A.; Lewis, I.A.; Hegeman, A.D.; Li, J.; Schulte, C.F.; Sussman, M.R.; et al. New bioinformatics resources for metabolomics. Pac. Symp. Biocomput. 2007, 168, 157–168. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cuperlovic-Culf, M.; Touaibia, M.; St-Coeur, P.-D.; Poitras, J.; Morin, P., Jr; Culf, A.S. Metabolic Effects of Known and Novel HDAC and SIRT Inhibitors in Glioblastomas Independently or Combined with Temozolomide. Metabolites 2014, 4, 807-830. https://doi.org/10.3390/metabo4030807
Cuperlovic-Culf M, Touaibia M, St-Coeur P-D, Poitras J, Morin P Jr, Culf AS. Metabolic Effects of Known and Novel HDAC and SIRT Inhibitors in Glioblastomas Independently or Combined with Temozolomide. Metabolites. 2014; 4(3):807-830. https://doi.org/10.3390/metabo4030807
Chicago/Turabian StyleCuperlovic-Culf, Miroslava, Mohamed Touaibia, Patrick-Denis St-Coeur, Julie Poitras, Pier Morin, Jr, and Adrian S. Culf. 2014. "Metabolic Effects of Known and Novel HDAC and SIRT Inhibitors in Glioblastomas Independently or Combined with Temozolomide" Metabolites 4, no. 3: 807-830. https://doi.org/10.3390/metabo4030807
APA StyleCuperlovic-Culf, M., Touaibia, M., St-Coeur, P.-D., Poitras, J., Morin, P., Jr, & Culf, A. S. (2014). Metabolic Effects of Known and Novel HDAC and SIRT Inhibitors in Glioblastomas Independently or Combined with Temozolomide. Metabolites, 4(3), 807-830. https://doi.org/10.3390/metabo4030807