Recent Applications of Metabolomics Toward Cyanobacteria


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
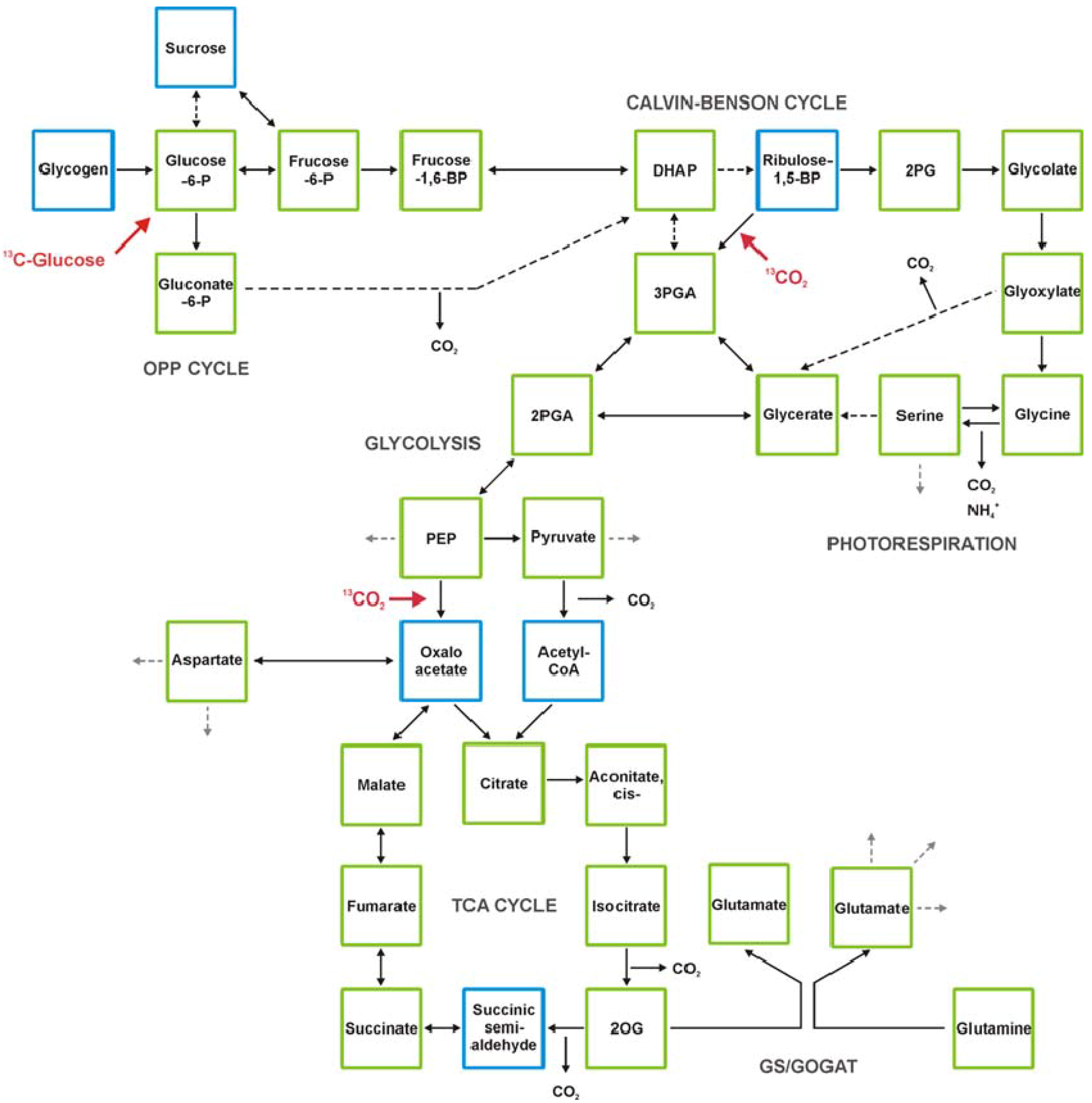
2. Technical Development towards Comprehensive Analysis of the Cyanobacterial Metabolome
2.1. Sampling and Metabolic Inactivation
2.2. Technologies for the Profiling of the Cyanobacterial Metabolome
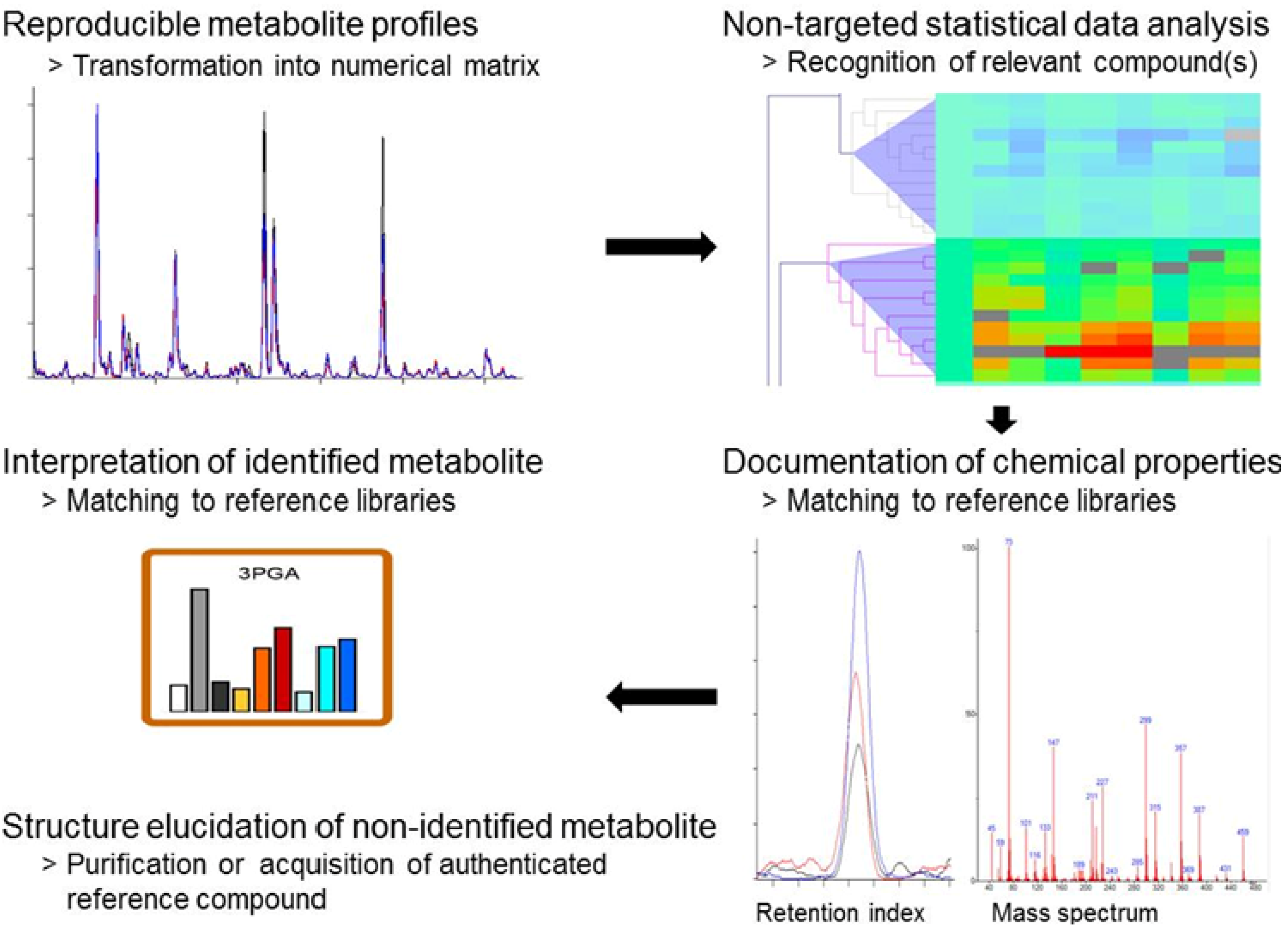
2.3. Standardized Data Processing and Specific Data Visualization
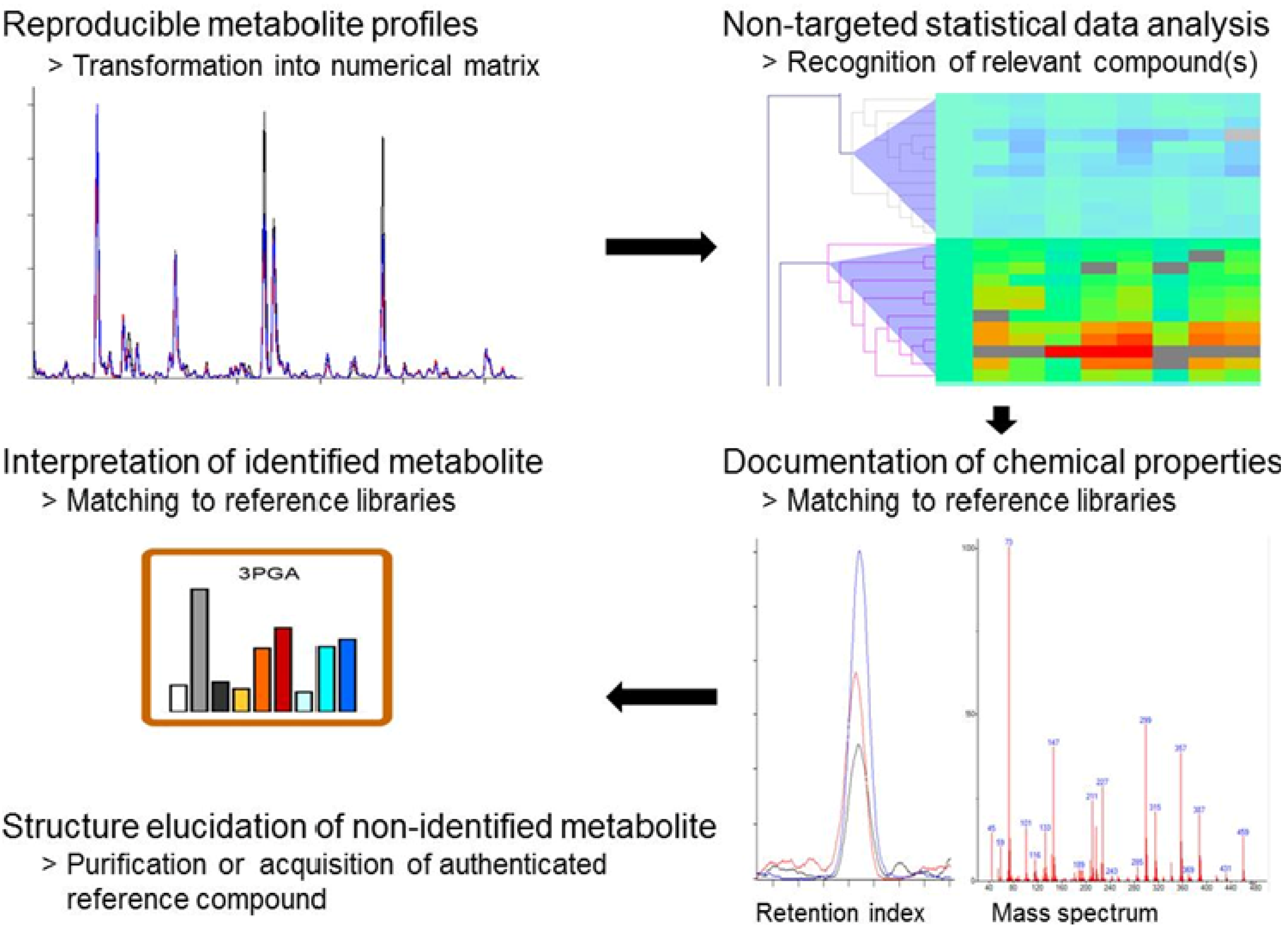
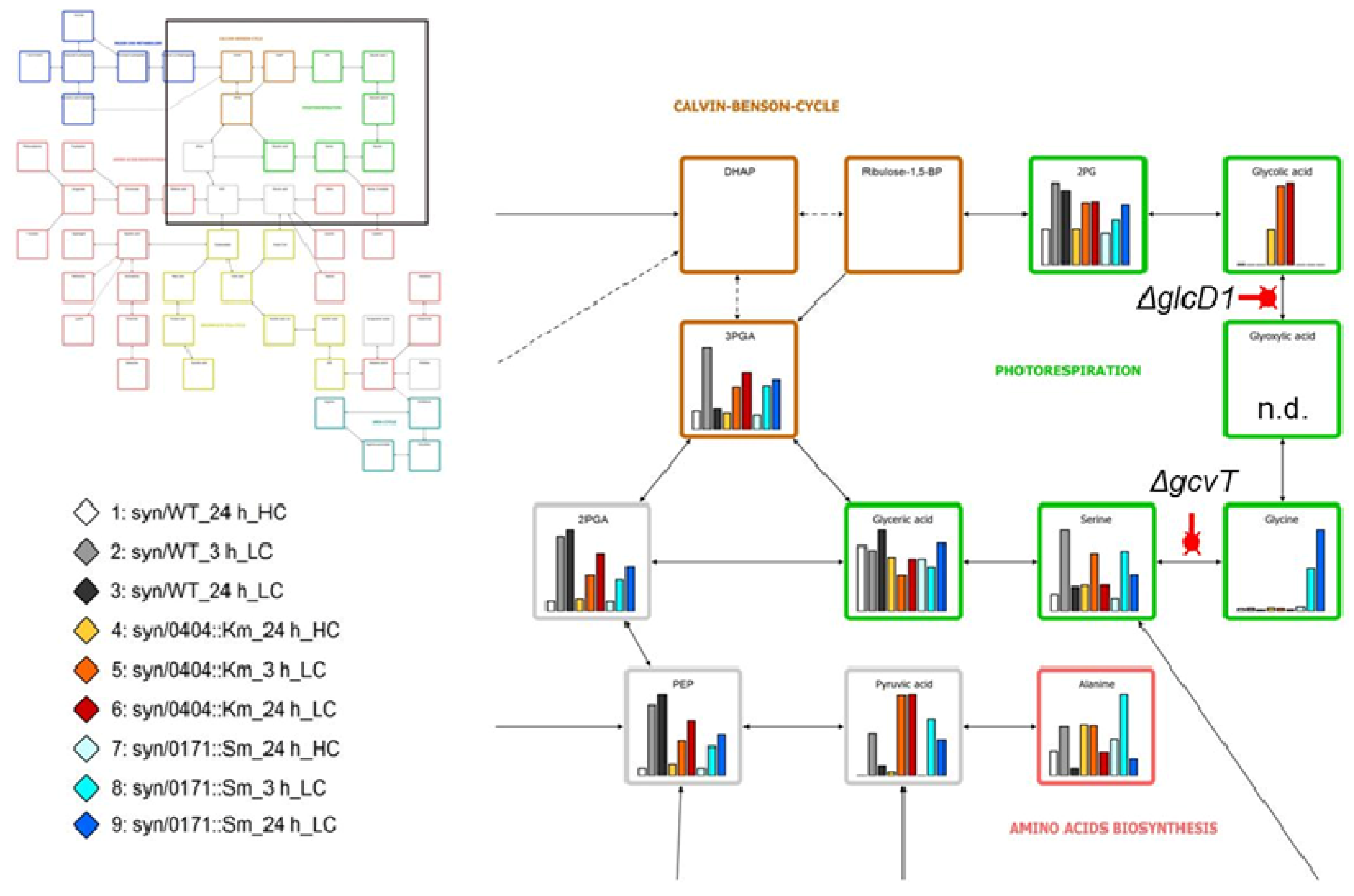
2.4. Discovery of Still Unknown Metabolites
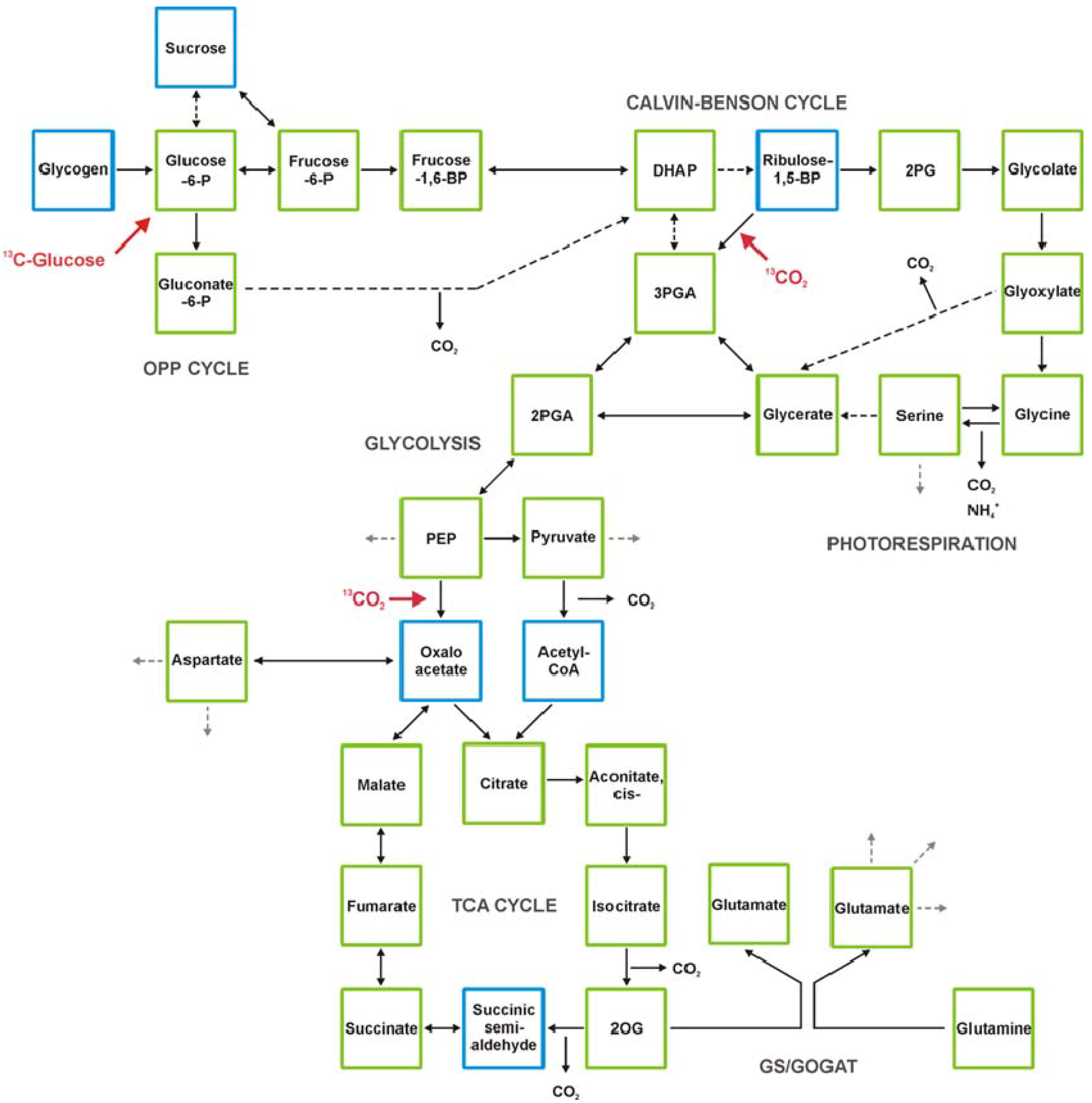
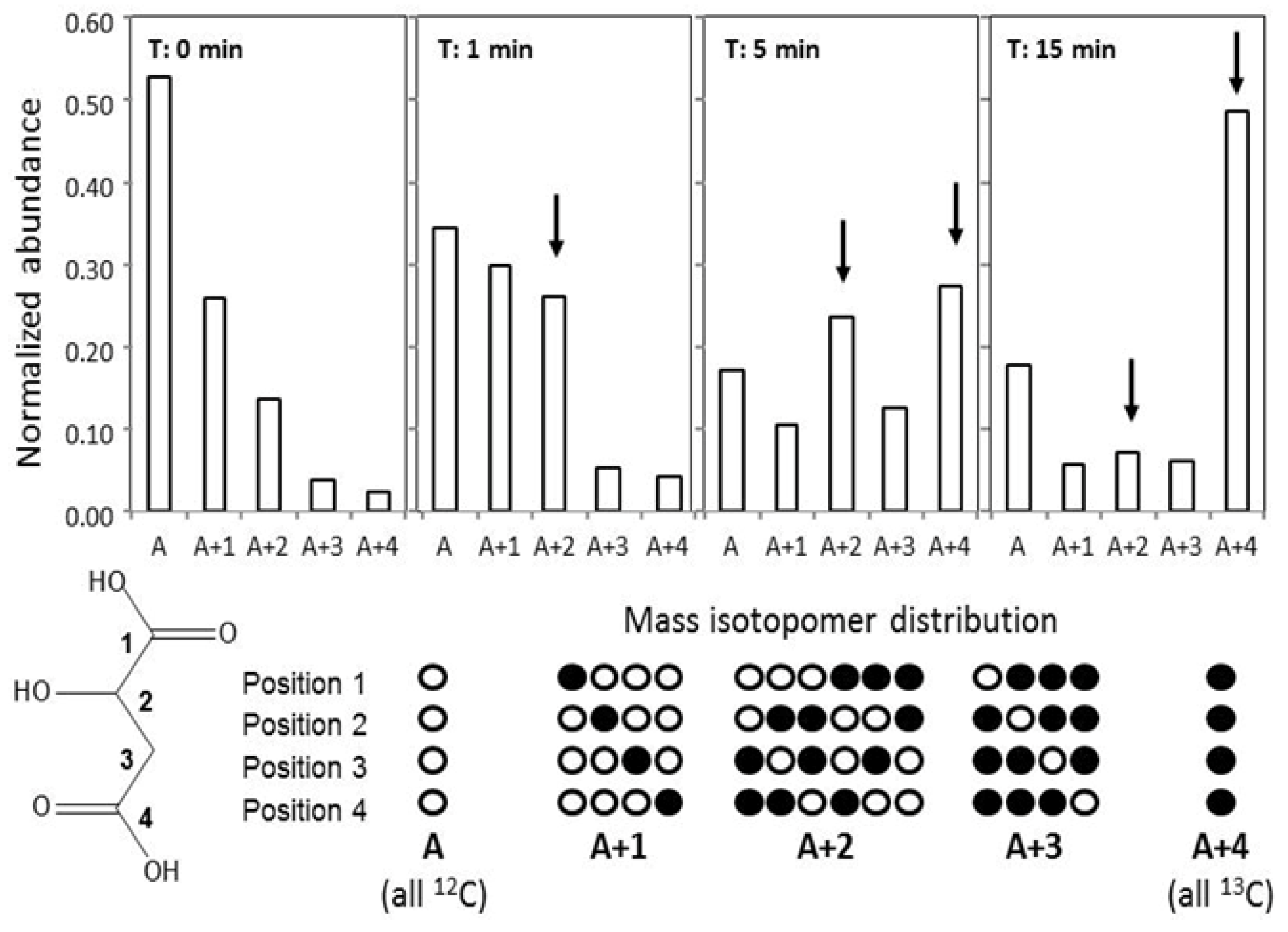
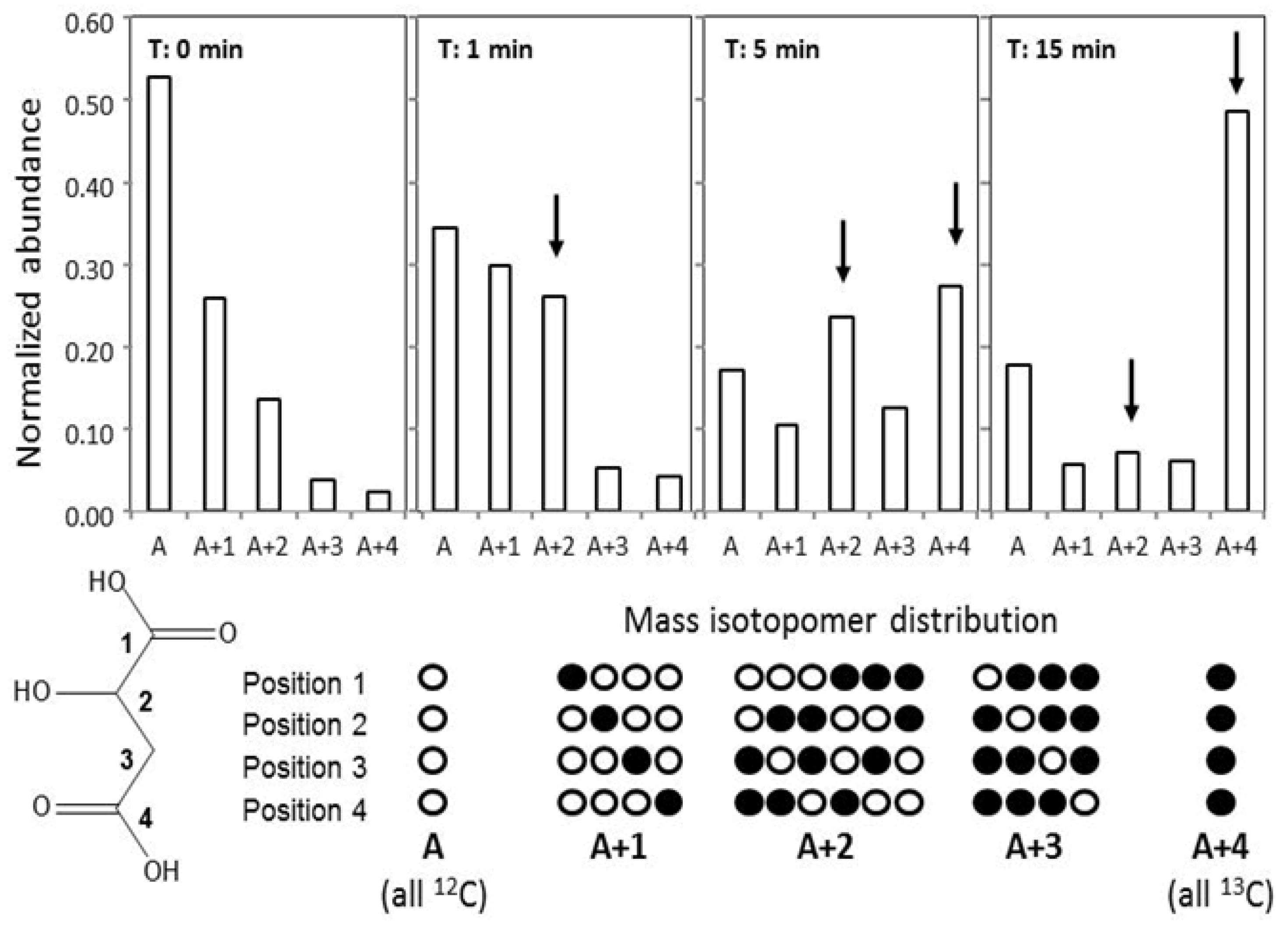
2.5. GC-MS-Based Metabolic Profiling Combined with Isotopic Tracing and Flux Analysis
3. Metabolomics with Cyanobacteria
3.1. Early Approaches to Analyze Cyanobacterial Metabolites
3.2. Metabolic Target Analysis—Bioactive Compounds
3.3. Metabolic Profiling—Fatty Acids
3.4. Metabolic Fingerprinting
3.5. Metabolic Footprinting
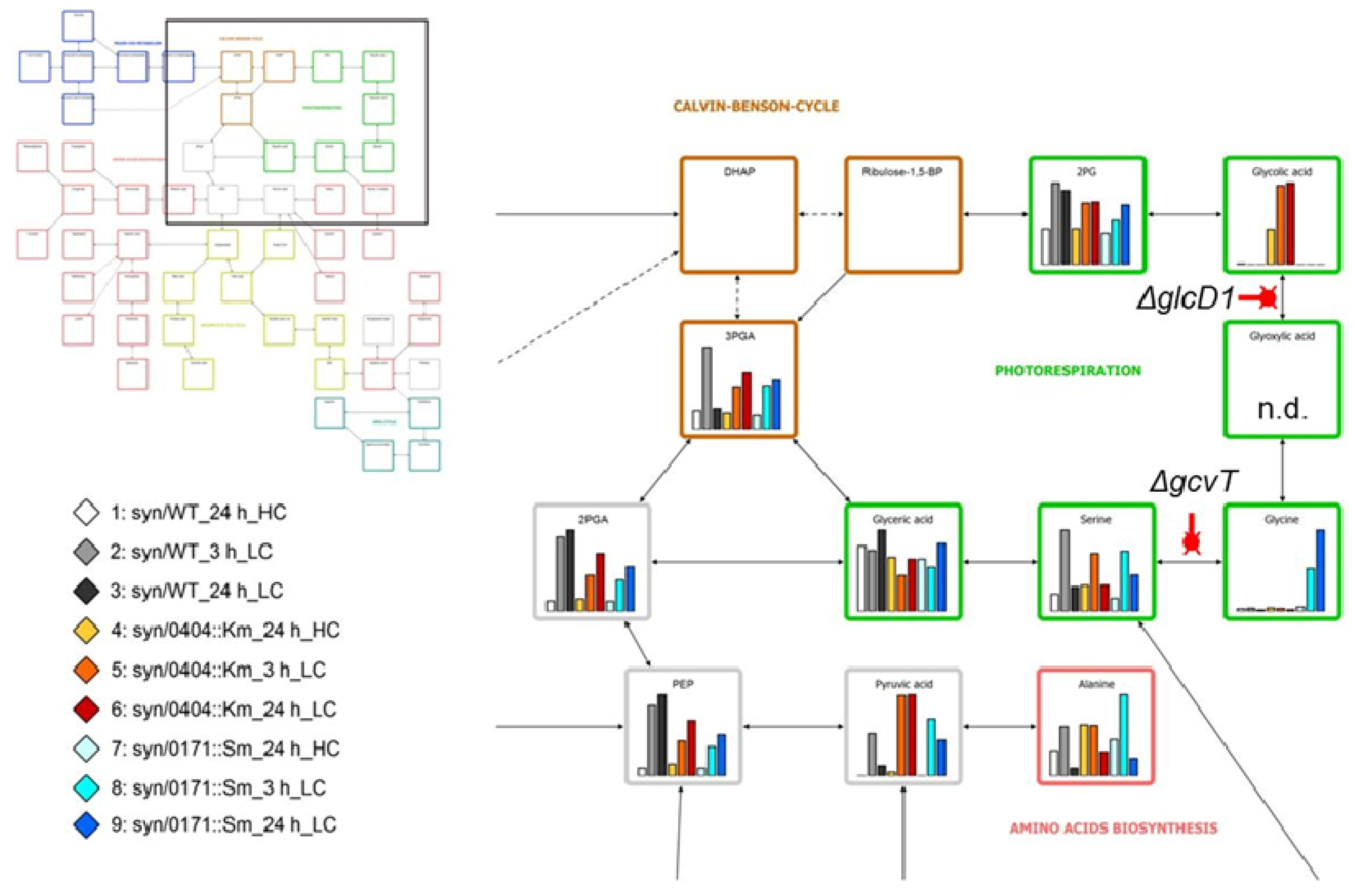
3.6. Flux Analysis
4. Future Perspectives
Acknowledgements
Conflict of Interest
References
- Oliver, S.G.; Winson, M.K.; Kell, D.B.; Baganz, F. Systemic functional analysis of the yeast genome. Trends Biotechnol. 1998, 16, 373–378. [Google Scholar]
- Fiehn, O.; Kopka, J.; Dörmann, P.; Altmann, T.; Trethewey, R.N.; Willmitzer, L. Metabolite profiling for plant functional genomics. Nat. Biotechnol. 2000, 18, 1157–1161. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics – the link between genotypes and phenotypes. Plant. Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Brunengraber, H.; Kelleher, J.K.; des Rosiers, C. Applications of mass isotopomer analysis to nutrition research. Annu. Rev. Nutr. 1997, 17, 559–596. [Google Scholar] [CrossRef]
- Roessner, U.; Wagner, C.; Kopka, J.; Trethewey, R.N.; Willmitzer, L. Simultaneous analysis of metabolites in potato by gas chromatography-mass spectrometry. Plant. J. 2000, 23, 131–142. [Google Scholar] [CrossRef]
- Lei, Z.; Huhman, D.V.; Summer, L.W. Mass spectrometry strategies in metabolomics. J. Biol. Chem. 2011, 286, 25435–25442. [Google Scholar]
- Desbrosses, G.G.; Kopka, J.; Udvardi, M.K. Lotus japonicus metabolic profiling. Development of gas chromatography-mass spectrometry resources for the study of plant-microbe interactions. Plant Physiol. 2005, 137, 1302–1318. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar]
- Fernie, A.R.; Aharoni, A.; Willmitzer, L.; Stitt, M.; Tohge, T.; Kopka, J.; Carroll, A.J.; Saito, K.; Fraser, P.D.; DeLuca, V. Recommendations for reporting metabolite data. Plant. Cell. 2011, 23, 2477–2482. [Google Scholar] [CrossRef]
- Hohmann-Marriott, M.F.; Blankenship, R.E. Evolution of photosynthesis. Annu. Rev. Plant. Biol. 2011, 62, 515–548. [Google Scholar] [CrossRef]
- Martin, W.; Rujan, T.; Richly, E.; Hansen, A.; Cornelsen, S.; Lins, T.; Leister, D.; Stoebe, B.; Hasegawa, M.; Penny, D. Evolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phyogeny and thousands of cyanobacterial genes in the nucleus. Proc. Natl. Acad. Soi. USA 2002, 99, 12246–12251. [Google Scholar]
- Kaneko, T.; Sato, S.; Kotani, H.; Tanaka, A.; Asamizu, E.; Nakamura, Y.; Miyajima, N.; Hirosawa, M.; Sugiura, M.; Sasamoto, S.; et al. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis. sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 1996, 3, 109–136. [Google Scholar] [CrossRef]
- Stanley, D.N.; Raines, C.A.; Kerfeld, C.A. Comparative analysis of 126 cyanobacterial genomes reveals evidence of functional diversity among homologs of the redox-regulated CP12 protein. Plant. Physiol. 2012. [Google Scholar] [CrossRef]
- Marin, K.; Suzuki, I.; Yamaguchi, K.; Ribbeck, K.; Yamamoto, H.; Kanesaki, Y.; Hagemann, M.; Murata, N. Identification of histidine kinases that act as sensors in the perception of salt stress in Synechocystis. sp. PCC 6803. Proc. Natl. Acad. Soi. USA 2003, 100, 9061–9066. [Google Scholar] [CrossRef]
- Los, D.A.; Suzuki, I.; Zinchenko, V.V.; Murata, N. Stress responses in Synechocytis. Regulated genes and regulatory systems. In The Cyanobacteria: Molecular Biology, Genomics and Evolution; Herrero, A., Flores, E., Eds.; Caister Academic Press: Norfolk, UK, 2008; pp. 117–157. [Google Scholar]
- Mitschke, J.; Georg, J.; Scholz, I.; Sharma, C.M.; Dienst, D.; Bantscheff, J.; Voss, B.; Steglich, C.; Wilde, A.; Vogel, J.; et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis. sp. PCC6803. Proc. Natl. Acad. Soi. USA 2011, 108, 2124–2129. [Google Scholar] [CrossRef]
- Mitschke, J.; Vioque, A.; Haas, F.; Hess, W.R.; Muro-Pastor, A.M. Dynamics of transcriptional start site selection during nitrogen stress-induced cell differentiation in Anabaena sp. PCC7120. Proc. Natl. Acad. Soi. USA 2011, 108, 20130–20135. [Google Scholar] [CrossRef]
- Ludwig, M.; Bryant, D.A. Acclimation of the global transcripome of the cyanobacterium Synechoccocus. sp. PCC 7002 to nutrient limitation and different nitrogen sources. Front. Microbiol. 2012, 3, 145. [Google Scholar]
- Fulda, S.; Mikkat, S.; Huang, F.; Huckauf, J.; Marin, K.; Norling, B.; Hagemann, M. Proteome analysis of salt stress response in the cyanobacterium Synechocystis. sp. Strain PCC 6803. Proteomics 2006, 6, 2733–2745. [Google Scholar] [CrossRef]
- Ishino, Y.; Okada, H.; Ikeuchi, M.; Taniguchi, H. Mass spectrometry-based prokaryote gene annotation. Proteomics 2007, 7, 4053–4065. [Google Scholar] [CrossRef]
- Yang, C.; Hua, Q.; Shimizu, K. Metabolic flux analysis in Synechocystis. using isotope distribution from 13C-labeled glucose. Metab. Eng. 2002, 4, 202–216. [Google Scholar] [CrossRef]
- Eisenhut, M.; Huege, J.; Schwarz, D.; Bauwe, H.; Kopka, J.; Hagemann, M. Metabolome phenotyping of inorganic carbon limitation in cells of wild type and photorespiratory mutants of the cyanobacterium Synechocystis. sp. strain PCC 6803. Plant. Physiol. 2008, 148, 2109–2120. [Google Scholar]
- Takahashi, H.; Uchimiya, H.; Hihara, Y. Difference in the metabolite levels between photoautotrophic and photomixotrophic cultures of Synechocystis. sp. PCC 6803 examined by capillary electrophoresis electrospray ionization mass spectrometry. J. Exp. Bot. 2008, 59, 3009–3018. [Google Scholar] [CrossRef]
- Dunn, W.B.; Ellis, D.I. Metabolomics: Current analytical platforms and methodologies. Trend Anal. Chem. 2005, 24, 285–294. [Google Scholar] [CrossRef]
- Fiehn, O. Combining genomics, metabolome analysis, and biochemical modelling to understand metabolic networks. Compar. Funct. Genom. 2001, 2, 155–168. [Google Scholar] [CrossRef]
- Allen, J.; Davey, H.M.; Broadhurst, D.; Heald, J.K.; Rowland, J.J.; Oliver, S.G.; Kell, D.B. High-throughput classification of yeast mutants for functional genomics using metabolic footprinting. Nat. Biotechnol. 2003, 21, 692–696. [Google Scholar]
- Wiechert, W.; Schweissgut, O.; Takanaga, H.; Frommer, W.B. Fluxomics: mass spectrometry versus quantitative imaging. Curr. Opin. Plant. Biol. 2007, 10, 323–330. [Google Scholar] [CrossRef]
- Ducat, D.C.; Way, J.C.; Silver, P.A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 2011, 29, 95–103. [Google Scholar]
- McNeely, K.; Xu, Y.; Bennette, N.; Bryant, D.A.; Dismukes, G.C. Redirecting reductant flux into hydrogen production via metabolic engineering of fermentative carbon metabolism in a cyanobacterium. Appl. Environ. Microbiol. 2010, 76, 5032–5038. [Google Scholar]
- Quintana, N.; van der Kooy, F.; van de Rhee, M.D.; Voshol, G.P.; Verpoorte, R. Renewable energy from cyanobacteria. Energy production optimization by metabolic pathway engineering. Appl. Microbiol. Biotechnol. 2011, 91, 471–490. [Google Scholar] [CrossRef]
- Sumner, L.W.; Mendes, P.; Dixon, R.A. Plant metabolomics: large-scale phytochemistry in the functional genomics era. Phytochemistry 2003, 62, 817–836. [Google Scholar] [CrossRef]
- Bino, R.J.; Hall, R.D.; Fiehn, O.; Kopka, J.; Saito, K.; Draper, J.; Nikolau, B.J.; Mendes, P.; Roessner-Tunali, U.; Beale, M.H.; et al. Potential of metabolomics as a functional genomics tool. Trends Plant. Sci. 2004, 9, 418–425. [Google Scholar] [CrossRef]
- Birkemeyer, C.; Luedemann, A.; Wagner, C.; Erban, A.; Kopka, J. Metabolome analysis: the potential of in vivo labeling with stable isotopes for metabolite profiling. Trends Biotechnol. 2005, 23, 28–33. [Google Scholar]
- Kopka, J. Current challenges and developments in GC-MS based metabolite profiling technology. J. Biotechnol. 2006, 124, 312–322. [Google Scholar] [CrossRef]
- Schauer, N.; Fernie, A.R. Plant metabolomics: towards biological function and mechanism. Trends Plant. Sci. 2006, 11, 508–516. [Google Scholar] [CrossRef]
- Steinhauser, D.; Kopka, J. Methods, applications and concepts of metabolite profiling: Primary metabolism. In Plant Systems Biology; Baginsky, S., Fernie, A.R., Eds.; Experientia Supplementum: Basel, Switzerland, 2007; Volume 97, pp. 171–194. [Google Scholar]
- Allwood, J.W.; de Vos, R.C.; Moing, A.; Deborde, C.; Erban, A.; Kopka, J.; Goodacre, R.; Hall, R.D. Plant metabolomics and its potential for systems biology research: Background concepts, technology, and methodology. In Methods in Enzymology; Abelson, J.N., Simon, M.I., Eds.; Elsevier-Academic Press: Amsterdam, The Netherlands, 2011; Volume 500, pp. 299–336. [Google Scholar]
- Wagner, C.; Sefkow, M.; Kopka, J. Construction and application of a mass spectral and retention time index database generated from plant GC/EI-TOF-MS metabolite profiles. Phytochemistry 2003, 62, 887–900. [Google Scholar]
- Steinhauser, D.; Usadel, B.; Luedemann, A.; Thimm, O.; Kopka, J. CSB.DB: A comprehensive systems-biology database. Bioinformatics 2004, 20, 3647–3651. [Google Scholar]
- Schauer, N.; Steinhauser, D.; Strelkov, S.; Schomburg, D.; Allison, G.; Moritz, T.; Lundgren, K.; Roessner-Tunali, U.; Forbes, M.G.; Willmitzer, L.; Fernie, A.R.; Kopka, J. GC-MS libraries for the rapid indentification of metabolites in complex biological samples. FEBS Lett. 2005, 59, 1332–1337. [Google Scholar]
- Hummel, J.; Strehmel, N.; Selbig, J.; Walther, D.; Kopka, J. Decision tree supported substructure prediction of metabolites from GC-MS profiles. Metabolomics 2010, 6, 322–333. [Google Scholar] [CrossRef]
- Haug, K.; Salek, R.M.; Conesa, P.; Hastings, J.; de Matos, P.; Rijnbeek, M.; Mahendraker, T.; Williams, M.; Neumann, S.; Rocca-Serra, P.; et al. MetaboLights—An open-access general purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 2013, 41, 781–786. [Google Scholar]
- Baran, R.; Bowen, B.P.; Northen, T.R. Untargeted metabolic footprinting reveals a surprising breadth of metabolite uptake and release by Synechococcus. sp. PCC 7002. Mol. Biosyst. 2011, 7, 3200–3206. [Google Scholar] [CrossRef]
- Calvin, M. The path of carbon in photosynthesis. Science 1962, 135, 879–889. [Google Scholar]
- Young, J.D.; Shastri, A.A.; Stephanopoulos, G.; Morgan, J.A. Mapping photoautotrophic metabolism with isotopicallynonstationary13C flux analysis. Metab. Eng. 2011, 13, 656–665. [Google Scholar]
- Krall, L.; Huege, J.; Catchpole, G.; Steinhauser, D.; Willmitzer, L. Assessment of sampling strategies for gas chromatography-mass spectrometry (GC-MS) based metabolomics of cyanobacteria. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 2952–2960. [Google Scholar]
- Huege, J.; Goetze, J.; Schwarz, D.; Bauwe, H.; Hagemann, M.; Kopka, J. Modulation of the major paths of carbon in photorespiratory mutants of Synechocystis. PLoS One 2011, 6, e16278. [Google Scholar]
- Narainsamy, K.; Cassier-Chauvat, C.; Junot, C.; Chauvat, F. High performance analysis of the cyanobacterial metabolism via liquid chromatography coupled to a LTQ-Orbitrap mass spectrometer: evidence that glucose reprograms the whole carbon metabolism and triggers oxidative stress. Metabolomics 2013, 9, 21–32. [Google Scholar] [CrossRef]
- Bennette, N.B.; Eng, J.F.; Dismukes, G.C. An LC-MS-based chemical and analytical method for targeted metabolite quantification in the model cyanobacterium Synechococcus. sp. PCC 7002. Anal. Chem. 2011, 83, 3808–3816. [Google Scholar] [CrossRef]
- Lin, Y.; Schiavo, S.; Orjala, J.; Vouros, P.; Kautz, R. Microscale LC-MS-NMR platform applied to the identification of active cyanobacterial metabolites. Anal. Chem. 2008, 80, 8045–8054. [Google Scholar] [CrossRef]
- Schwarz, D.; Nodop, A.; Hüge, J.; Purfürst, S.; Forchhammer, K.; Michel, K.P.; Bauwe, H.; Kopka, J.; Hagemann, M. Metabolic and transcriptomic phenotyping of inorganic carbon acclimation in the cyanobacterium Synechococcus. elongates PCC 7942. Plant. Physiol. 2011, 155, 1640–1655. [Google Scholar] [CrossRef]
- Lisec, J.; Schauer, N.; Kopka, J.; Willmitzer, L.; Fernie, A.R. Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat. Protoc. 2006, 1, 387–396. [Google Scholar]
- Erban, A.; Schauer, N.; Fernie, A.R.; Kopka, J. Non-supervised construction and application of mass spectral and retention time index libraries from time-of-flight GC-MS metabolite profiles. In Metabolomics: Methods and protocols. Methods in Molecular Biology; Weckwerth, W., Ed.; Humana Press: Totowa, New Jersey, USA, 2007; Volume 358, pp. 19–38. [Google Scholar]
- Luedemann, A.; Strassbrug, K.; Erban, A.; Kopka, J. TagFinder for the quantitative analysis of gas chromatography-mass spectrometry (GC-MS)-based metabolite profiling experiments. Bioinformatics 2008, 24, 732–737. [Google Scholar]
- Luedemann, A.; von Malotky, L.; Erban, A.; Kopka, J. TagFinder: Preprocessing software for the fingerprinting and the profiling of gas chromatography-mass spectrometry based metabolome Analyses. In Plant Metabolomics: Methods and Protocols. Methods in Molecular Biology; Hardy, N.W., Hall, R.D., Eds.; Humana Press: Totowa, New Jersey, USA, 2012; Volume 860, pp. 255–286. [Google Scholar]
- Knoop, H.; Zilliges, Y.; Lockau, W.; Steuer, R. The metabolic network of Synechocystis. sp. PCC 6803: Systemic properties of autotrophic growth. Plant Physiol. 2010, 154, 410–422. [Google Scholar] [CrossRef]
- Luedemann, A.; Weicht, D.; Selbig, J.; Kopka, J. PaVESy: Pathway visualization and editing systems. Bioinformatics 2004, 20, 2841–2844. [Google Scholar] [CrossRef]
- Junker, B.H.; Klukas, C.; Schreiber, F. VANTED: a system for advanced data analysis and visualization in the context of biological networks. BMC Bioinformatics 2006, 7, 109. [Google Scholar]
- Klukas, C.; Schreiber, F. Integration of -omics data and networks for biochemical research with VANTED. J. Integr. Bioinform. 2010, 7, 112. [Google Scholar]
- Kopka, J. Gas chromatography mass spectrometry. In Plant Metabolomics. Biotechnology in Agriculture and Forestry; Nagata, T., Loerz, H., Widholm, J.M., Saito, K., Dixon, R.A., Willmitzer, L., Eds.; Springer-Verlag: Berlin, Heidelberg, Germany, 2007; Volume 57, pp. 3–20. [Google Scholar]
- Huege, J.; Sulpice, R.; Gibon, Y.; Lisec, J.; Koehl, K.; Kopka, J. GC-EI-TOF-MS analysis of in vivo carbon-partitioning into soluble metabolite pools of higher plants by monitoring isotope dilution after 13CO2 labelling. Phytochemistry. 2007, 68, 2258–2272. [Google Scholar] [CrossRef]
- Pelroy, R.A.; Bassham, J.A. Photosynthetic and dark carbon metabolism in unicellular blue-green algae. Arch. Microbiol. 1972, 86, 25–38. [Google Scholar]
- Tabita, F.R. The biochemistry and molecular regulation of carbon dioxide metabolism in cyanobacteria. In The Molecular Biology of Cyanobacteria; Bryant, D.A., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1994; pp. 437–467. [Google Scholar]
- Pelroy, R.A.; Rippka, R.; Stanier, R.Y. The mechanism of glucose by unicellular blue-green algae. Arch. Microbiol. 1972, 87, 303–322. [Google Scholar]
- Smith, A.J.; London, J.; Stanier, R.Y. Biochemical basis of obligate autotrophy in blue-green algae and thiobacilli. J. Bacteriol. 1967, 94, 972–983. [Google Scholar]
- Pearce, J.; Carr, N.G. The metabolism of acetate by the blue-green algae, Anabaena variabilis and Anacystis. nidulans. J. Gen. Microbiol. 1967, 49, 301–313. [Google Scholar]
- Pearce, J.; Leach, C.K.; Carr, N.G. The incomplete tricarboxylic acid cycle in the blue-green alga Anabaena variabilis. J. Gen. Microbiol. 1969, 55, 371–378. [Google Scholar]
- Flores, E.; Herrero, A. Nitrogen assimilation and nitrogen control in cyanobacteria. Biochem. Soc. Trans. 2005, 33, 164–167. [Google Scholar] [CrossRef]
- Forchhammer, K. PII signal transducers: novel functional and structural insights. Trends Microbiol. 2008, 16, 65–72. [Google Scholar] [CrossRef]
- Zhang, S.; Bryant, D.A. The tricarboxylic acid cycle in cyanobacteria. Science 2011, 334, 1551–1553. [Google Scholar]
- Cooley, J.W.; Howitt, C.A.; Vermaas, W.F. Succinate:quinol oxidoreductases in the cyanobacterium Synechocystis. sp. PCC 6803: Presence and function in metabolism and electron transport. J. Bacteriol. 2000, 182, 714–722. [Google Scholar] [CrossRef]
- Steinhauser, D.; Fernie, A.R.; Araújo, W.L. Unusual cyanobacterial TCA cycles: not broken just different. Trends Plant. Sci. 2012, 17, 503–509. [Google Scholar] [CrossRef]
- Carmichael, W.W. Cyanobacteria secondary metabolites—the cyanotoxins. J. ApplBacteriol 1992, 72, 445–459. [Google Scholar] [CrossRef]
- Dittmann, E.; Fewer, D.P.; Neilan, B.A. Cyanobacterial toxins: biosynthetic routes and evolutionary roots. FEMS Microbiol. Rev. 2013, 37, 23–43. [Google Scholar] [CrossRef]
- Humpage, A.R.; Magalhaes, V.F.; Froscio, S.M. Comparison of analytical tools and biological assays for detection of paralytic shellfish poisoning toxins. Anal. Bioanal. Chem. 2010, 397, 1655–1671. [Google Scholar] [CrossRef]
- Hiller, S.; Krock, B.; Cembella, A.; Luckas, B. Rapid detection of cyanobacterial toxins in precursor ion mode by liquid chromatography tandem mass spectrometry. J. Mass Spectrom. 2007, 42, 1238–1250. [Google Scholar] [CrossRef]
- Welker, M.; Fastner, J.; Erhard, M.; von Döhren, H. Applications of MALDI-TOF MS analysis in cyanotoxin research. Environ. Toxicol. 2002, 17, 367–374. [Google Scholar] [CrossRef]
- Esquenazi, E.; Coates, C.; Simmons, L.; Gonzalez, D.; Gerwick, W.H.; Dorrestein, P.C. Visualizing the spatial distribution of secondary metabolites produced by marine cyanobacteria and sponges via MALDI-TOF imaging. Mol. Bio Syst. 2008, 4, 562–570. [Google Scholar]
- Esquenazi, E.; Jones, A.C.; Byrum, T.; Dorrestein, P.C.; Gerwick, W.H. Temporal dynamics of natural product biosynthesis in marine cyanobacteria. Proc. Natl. Acad. Soi. USA 2011, 108, 5226–5231. [Google Scholar]
- Kaasalainen, U.; Fewer, D.P.; Jokela, J.; Wahlsten, M.; Sivonen, K.; Rikkinen, J. Cyanobacteria produce a high variety of hepatotoxic peptides in lichen symbiosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5886–5891. [Google Scholar]
- Oren, A. Cyanobacterial systematics and nomenclature as featured in the International Bulletin of Bacteriological Nomenclature and Taxonomy/International Journal of Systematic Bacteriology/International Journal of Systematic and Evolutionary Microbiology. Int. J. Syst. Evol. Microbiol. 2011, 61, 10–15. [Google Scholar] [CrossRef]
- Castenholz, R.W. General characteristics of the cyanobacteria. In Bergey's Manual of Systematic Bacteriology, 2nd; Boone, D.R., Castenholz, R.W., Eds.; Springer: New York, NY, USA, 2001; Volume 1, pp. 474–487. [Google Scholar]
- Rippka, R.; Deruelles, J.; Waterbury, J.B. Generic assignments, strains histories and properties of pure cultures of cyanobacteria. J. Gen. Microbiol. 1979, 111, 1–61. [Google Scholar]
- Stanier, R.Y.; Sistrom, W.R.; Hansen, T.A.; Whitton, B.A.; Castenholz, R.W; Pfennig, N.; Gorlenko, V.N.; Kondratieva, E.N.; Eimhjellen, K.E.; Whittenbury, R.; et al. Proposal to place the nomenclature of the cyanobacteria (blue-green algae) under the rules of the international code of nomenclature of bacteria. Int. J. Syst. Bacteriol. 1978, 28, 335–336. [Google Scholar]
- Cleenwerck, I.; de Vos, P. Polyphasic taxonomy of acetic acid bacteria: an overview of the currently applied methodology. Int. J. Food Microbiol. 2008, 125, 2–14. [Google Scholar] [CrossRef]
- Gugger, M.; Lyra, C.; Suominen, I.; Tsitko, I.; Humbert, J.F.; Salkinoja-Salonen, M.S.; Sivonen, K. Cellular fatty acids as chemotaxonomic markers of the genera Anabaena, Aphanizomenon., Microcystis., Nostoc. and Planktothrix. (cyanobacteria). Int. J. Syst. Evol. Microbiol. 2002, 52, 1007–1015. [Google Scholar] [CrossRef]
- Krüger, G.H.J.; deWet, H.; Kock, H.L.F.; Pieterse, A.J.H. Fatty acid composition as a taxonomic characteristic for Microcystis. and other coccoid cyanobacteria (blue-green alga) isolates. Hydrobiologia 1995, 308, 145–151. [Google Scholar] [CrossRef]
- Li, R.; Yokota, A.; Sugiyama, J.; Watanabe, M.; Hiroki, M.; Watanabe, M.M. Chemotaxonomy of planktonic cyanobacteria based on non-polar and 3-hydroxy fatty acid composition. Phycol. Res. 1998, 46, 21–28. [Google Scholar] [CrossRef]
- Liu, X.; Sheng, J.; Curtiss, R., 3rd. Fatty acid production in genetically modified cyanobacteria. Proc. Natl. Acad. Sci. USA 2011, 108, 6899–6904. [Google Scholar]
- Guan, W.; Zhao, H.; Lu, X.; Wang, C.; Yang, M.; Bai, F. Quantitative analysis of fatty-acid-based biofuels produced by wild-type and genetically engineered cyanobacteria by gas chromatography-mass spectrometry. J. Chromatogr. A 2011, 1218, 8289–8293. [Google Scholar] [CrossRef]
- Tandeau de Marsac, N.; Houmard, J. Adaptation of cyanobacteria to environmental stimuli: New steps towards molecular mechanisms. FEMS Microbiol. Rev. 1993, 104, 119–189. [Google Scholar] [CrossRef]
- Hagemann, M. Molecular biology of cyanobacterial salt acclimation. FEMS Microbiol. Rev. 2011, 35, 87–123. [Google Scholar] [CrossRef]
- Wang, H.L.; Postier, B.L.; Burnap, R.L. Alterations in global patterns of gene expression in Synechocystis. sp. PCC 6803 in response to inorganic carbon limitation and the inactivation of ndhR, a LysR family regulator. J. Biol. Chem. 2004, 279, 5739–5751. [Google Scholar]
- Eisenhut, M.; von Wobeser, E.A.; Jonas, L.; Schubert, H.; Ibelings, B.W.; Bauwe, H.; Matthijs, H.C.; Hagemann, M. Long-term response toward carbon limitation in wild type and glycolate turnover mutants of the cyanobacterium Synechocystis. sp. strain PCC 6803. Plant. Physiol. 2007, 144, 1946–1959. [Google Scholar] [CrossRef]
- Battchikova, N.; Vainonen, J.P.; Vorontsova, N.; Keranen, M.; Carmel, D.; Aro, E.M. Dynamic changes in the proteome of Synechocystis. 6803 in response to CO2 limitation revealed by quantitative proteomics. J. Proteome Res. 2010, 9, 5896–5912. [Google Scholar] [CrossRef]
- Kaplan, A.; Hagemann, M.; Bauwe, H.; Kahlon, S.; Ogawa, T. Carbon acquisition by cyanobacteria: mechanism, comparative genomics and evolution. In The Cyanobacteria: Molecular Biology, Genomics and Evolution; Herrero, A., Flores, E., Eds.; Caister Academic Press: Norfolk, UK, 2008; pp. 305–334. [Google Scholar]
- Muro-Pastor, M.I.; Reyes, J.C.; Florencio, F.J. Cyanobacteria perceive nitrogen status by sensing intracellular 2-oxoglutarate levels. J. Biol. Chem. 2001, 276, 38320–38328. [Google Scholar]
- Yang, C.; Hua, Q.; Shimizu, K. Integration of the information from gene expression and metabolic fluxes for the analysis of the regulatory mechanisms in Synechocystis. Appl. Microbiol. Biotechnol. 2002, 58, 813–822. [Google Scholar] [CrossRef]
- Kahlon, S.; Beeri, K.; Ohkawa, H.; Hihara, Y.; Murik, O.; Suzuki, I.; Ogawa, T.; Kaplan, A. A putative sensor kinase, Hik31, is involved in the response of Synechocysti. sp. PCC 6803 to the presence of glucose. Microbiology 2006, 152, 647–655. [Google Scholar]
- Herranen, M.; Battchikova, N.; Zhang, P.; Graf, A.; Sirpiö, S.; Paakkarinen, V.; Aro, E.M. Towards functional proteomics of membrane protein complexes in Synechocystis. sp. PCC 6803. Plant. Physiol. 2004, 134, 470–481. [Google Scholar] [CrossRef]
- Miranda, H.; Cheregi, O.; Netotea, S.; Hvidsten, T.R.; Moritz, T.; Funk, C. Co-expression analysis, proteomic and metabolomic study on the impact of a Deg/HtrA protease triple mutant in Synechocystis. sp. PCC 6803 exposed to temperature and high light stress. J. Proetomics 2012. [Google Scholar] [CrossRef]
- Osanai, T.; Oikawa, A.; Azuma, M.; Tanaka, K.; Saito, K.; Hirai, M.Y.; Ikeuchi, M. Genetic aengineering of group 2 sigma factor SigE widely activates expression of sugar catabolic genes in Synechocystis. species PCC 6803. J. Biol. Chem. 2011, 286, 30962–30971. [Google Scholar]
- Gründel, M.; Scheunemann, R.; Lockau, W.; Zilliges, Y. Impaired glycogen synthesis causes metabolic overflow reactions and affects stress responses in the cyanobacterium Synechocystis. sp. PCC 6803. Microbiology 2012, 158, 3032–3043. [Google Scholar] [CrossRef]
- Hackenberg, C.; Huege, J.; Engelhardt, A.; Wittink, F.; Laue, M.; Matthijs, H.C.; Kopka, J.; Bauwe, H.; Hagemann, M. Low-carbon acclimation in carboxysome-less and photorespiratory mutants of the cyanobacterium Synechocystis. sp. strain PCC 6803. Microbiology 2012, 158, 398–413. [Google Scholar] [CrossRef]
- McNeeley, K.; Xu, Y.; Bennette, N.; Bryant, D.A.; Dismukes, G.C. Redirecting reductant flux into hydrogen production via metabolic engineering of fermentative carbon metabolism in a cyanobacterium. Appl. Environ. Mircobiol. 2010, 76, 5032–5038. [Google Scholar] [CrossRef]
- Schmetterer, G.R. Sequence conservation among the glucose transporter from the cyanobacterium Synechocystis. sp. PCC 6803 and mammalian glucose transporters. Plant. Mol. Biol. 1990, 14, 697–706. [Google Scholar] [CrossRef]
- Zubkov, M.V.; Fuchs, B.M.; Tarran, G.A.; Burkill, P.H.; Amann, R. High rate of uptake of organic nitrogen compounds by Prochlorococcus. cyanobacteria as a key to their dominance in oligotrophic oceanic waters. Appl. Environ. Microbiol. 2003, 69, 1299–1304. [Google Scholar] [CrossRef]
- Norman, E.G.; Colman, B. Evidence for an incomplete glycolate pathway in cyanobacteria. J. Plant. Physiol. 1988, 132, 766–768. [Google Scholar] [CrossRef]
- Glibert, P.M.; Bronk, D.A. Release of dissolved organic nitrogen by marine diazotrophic cyanobacteria, Trichodesmium. spp. Appl. Environ. Microbiol. 1994, 60, 3996–4000. [Google Scholar]
- Yang, C.; Hua, Q.; Shimizu, K. Quantitative analysis of intracellular metabolic fluxes using GC-MS and two-dimensional NMR spectroscopy. J. Biosci. Bioeng. 2002, 93, 78–87. [Google Scholar]
- Saha, R.; Verseput, A.T.; Berla, B.M.; Mueller, T.J.; Pakrasi, H.B.; Maranas, C.D. Reconstruction and comparison of the metabolic potential of cyanobacteria Cyanothece. sp. ATCC 51142 and Synechocystis. sp. PCC 6803. PLoS One 2012, 7, e48285. [Google Scholar]
- Feng, X.; Bandyopadhyay, A.; Berla, B.; Page, L.; Wu, B.; Pakrasi, H.B.; Tang, Y.J. Mixotrophic and photoheterotrophic metabolism in Cyanothece. sp. ATCC 51142 under continuous light. Microbiology 2010, 156, 2566–2574. [Google Scholar] [CrossRef]
- Wu, B.; Zhang, B.; Feng, X.; Rubens, J.R.; Huang, R.; Hicks, L.M.; Pakrasi, H.B.; Tang, Y.J. Alternative isoleucine synthesis pathway in cyanobacterial species. Microbiology 2010, 156, 596–602. [Google Scholar] [CrossRef]
- Nogales, J.; Gudmundsson, S.; Thiele, I. Toward systems metabolic engineering in cyanobacteria: Opportunities and bottlenecks. Bioengineered. 2012, 4. Available online: http://www.landesbioscience.com/journals/bioe/article/22792/.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schwarz, D.; Orf, I.; Kopka, J.; Hagemann, M. Recent Applications of Metabolomics Toward Cyanobacteria. Metabolites 2013, 3, 72-100. https://doi.org/10.3390/metabo3010072
Schwarz D, Orf I, Kopka J, Hagemann M. Recent Applications of Metabolomics Toward Cyanobacteria. Metabolites. 2013; 3(1):72-100. https://doi.org/10.3390/metabo3010072
Chicago/Turabian StyleSchwarz, Doreen, Isabel Orf, Joachim Kopka, and Martin Hagemann. 2013. "Recent Applications of Metabolomics Toward Cyanobacteria" Metabolites 3, no. 1: 72-100. https://doi.org/10.3390/metabo3010072
APA StyleSchwarz, D., Orf, I., Kopka, J., & Hagemann, M. (2013). Recent Applications of Metabolomics Toward Cyanobacteria. Metabolites, 3(1), 72-100. https://doi.org/10.3390/metabo3010072