Characterization of the Interaction Between the Small Regulatory Peptide SgrT and the EIICBGlc of the Glucose-Phosphotransferase System of E. coli K-12

Abstract
:1. Introduction
2. Results and Discussion
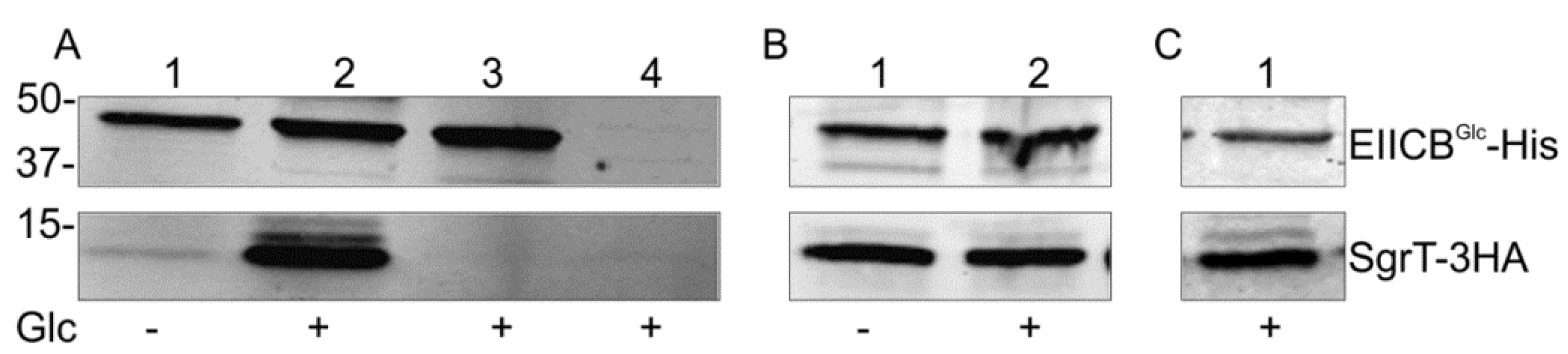
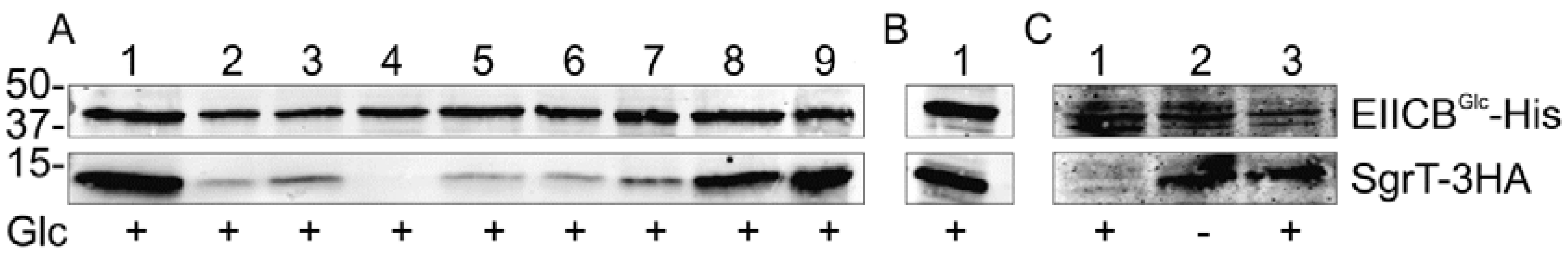
2.1. SgrT Binds to Dephosphorylated EIICBGlc in in vivo Crosslinking assays

- (a) Lanes 1 and 2 show crosslinking experiments with strain JKA12 (LJ110ΔptsG::cat ΔsgrRST::neo) transformed with two plasmids expressing EIICBGlc-His (pRR48GH) and SgrT-3HA (pACYC184sgrT3HA). Cells were grown in the absence or presence of glucose as indicated; molecular weight markers are given on the left side (in kDa). The results show an interaction of SgrT and EIICBGlc in the presence of glucose. Control experiments are illustrated in lane 3 (JKA12 transformed with pRR48GH and pACYC184) and lane 4 (JKA12 transformed with pRR48 and pACYC184sgrT3HA). In both cases, no signals for SgrT-3HA could be observed.
- (b) Lanes 1 and 2 show crosslinking experiments with the ptsHIcrr deletion strain LJ140 transformed with pRR48GH and pACYC184sgrT3HA. Cells were grown in the absence or presence of glucose as indicated.
- (c) Lane 1 shows a crosslinking experiment with the dgsA deletion strain LJB17 transformed with pRR48GH and pACYC184sgrT3HA. Cells were grown in the presence of glucose. This result indicates an Mlc-independent interaction between EIICBGlc and SgrT.
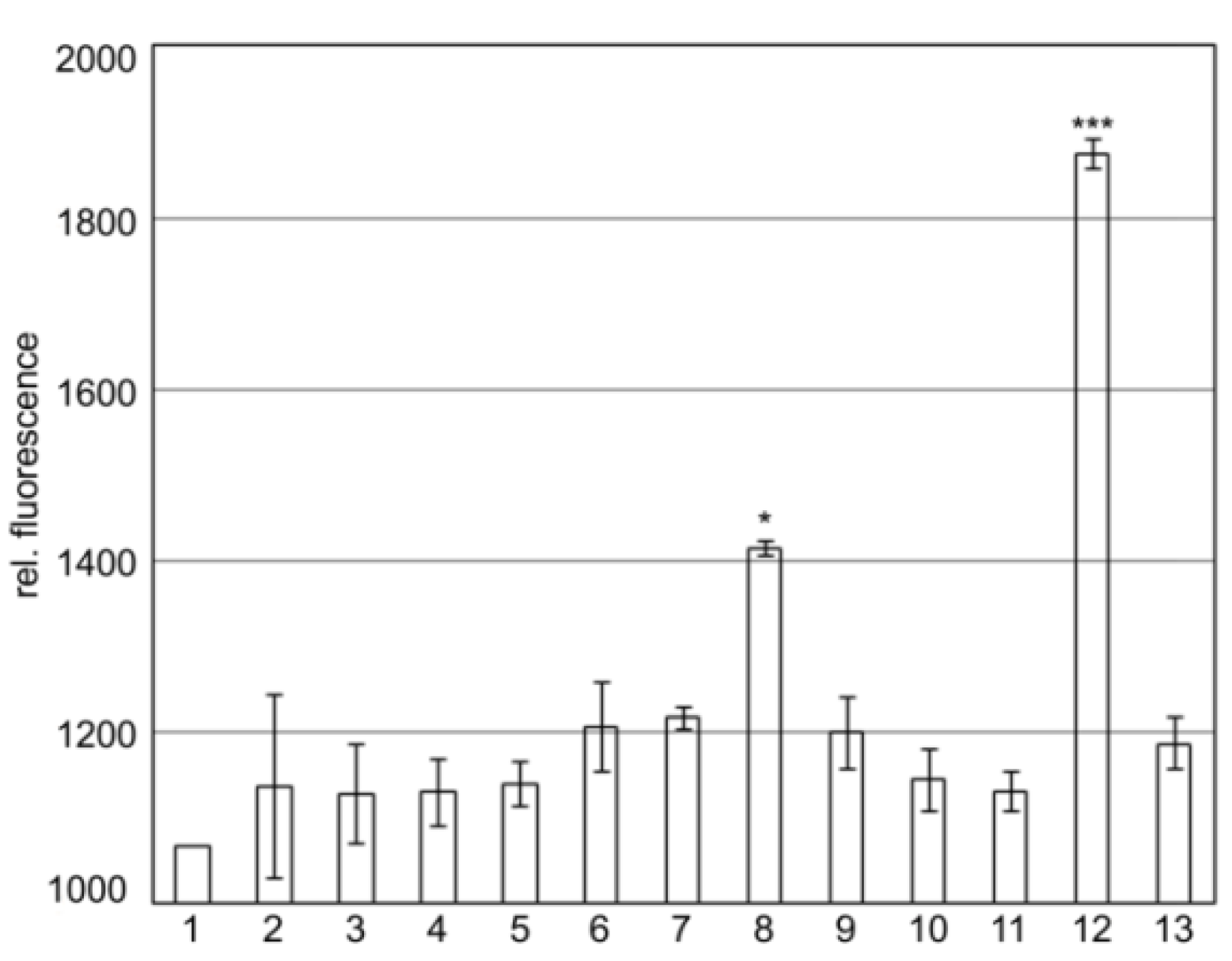
2.2. SgrT Binds to Full Length EIICBGlc and to Its Truncated EIIC-Linker Derivative in Bimolecular Fluorescence Complementation Assays
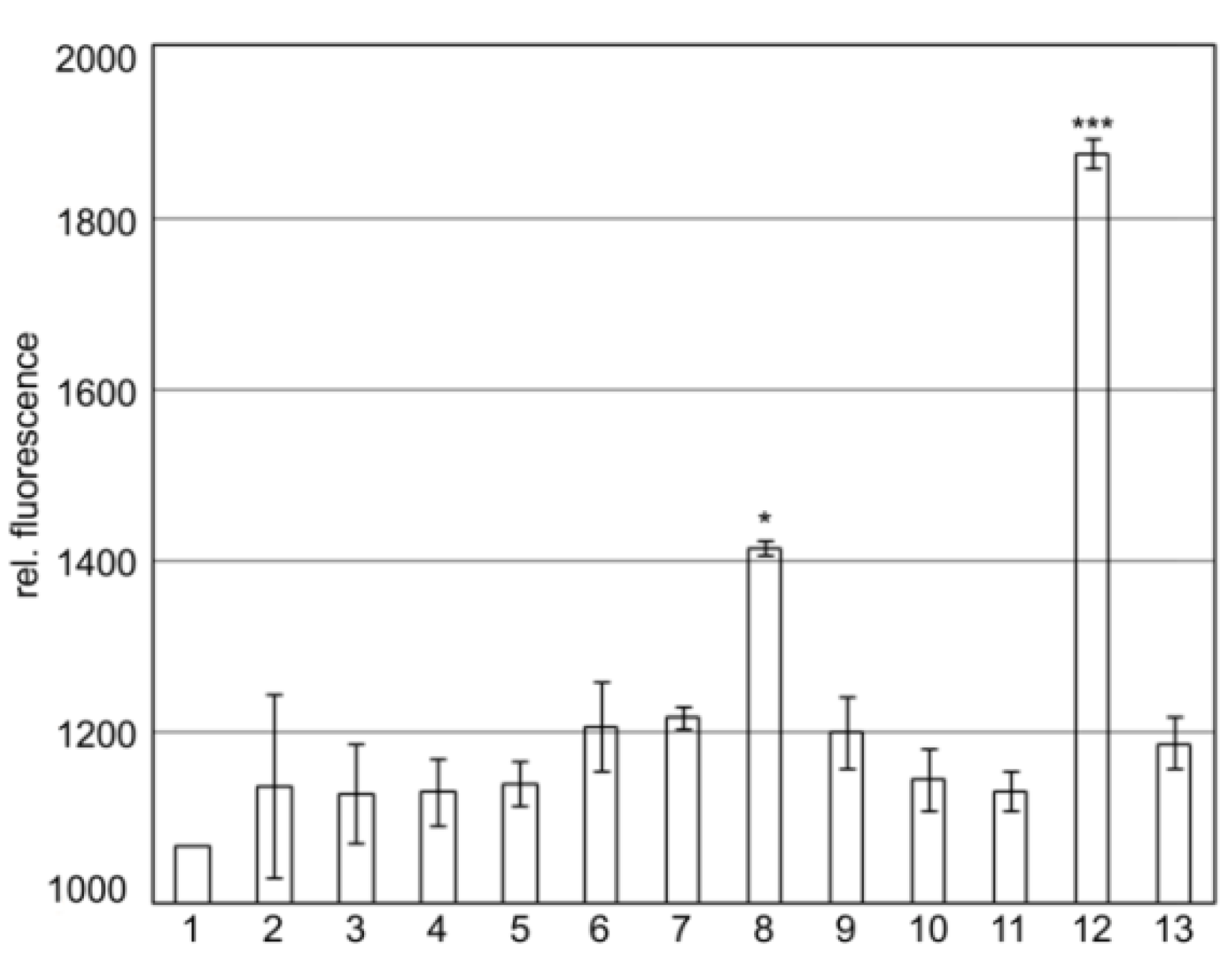
2.3. The KTPGRED Motif in the Linker Region of EIICBGlc is the Main SgrT Target Sequence
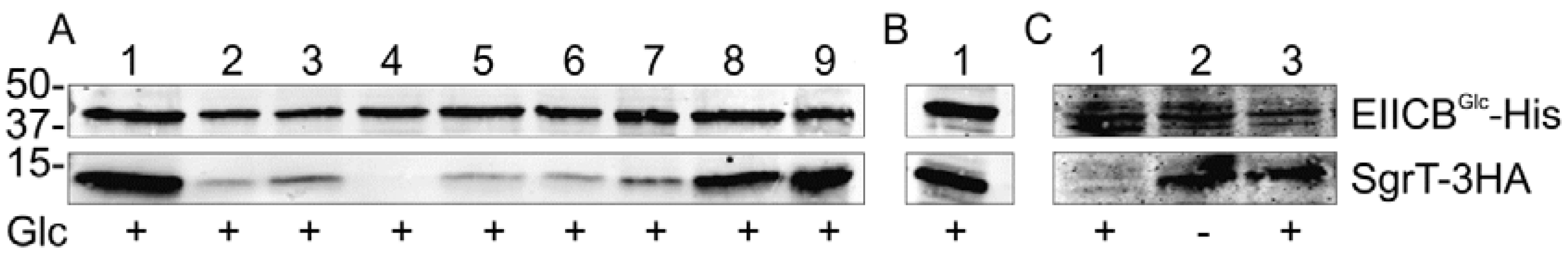
- (a) This part of the figure shows crosslinking experiments with strain JKA12 (LJ110ΔptsG::cat ΔsgrRST::neo) transformed with two plasmids expressing SgrT-3HA (pACYC184sgrT3HA) and wild type EIICBGlc-His (lane 9) or different EIICBGlc-His-derivatives (lanes 1-8). Cells were grown in the presence of glucose as indicated; molecular weight markers are given on the left side (in kDa). The following EIICBGlc derivatives were used in combination with SgrT-3HA:EIICBGlc-K382A-His; 2. EIICBGlc-T383A-His; 3. EIICBGlc-P384A-His; 4. EIICBGlc-P384R-His; 5. EIICBGlc-G385A-His; 6. EIICBGlc-R386A-His; 7. EIICBGlc-E387A-His; 8. EIICBGlc-D388A-His; 9. EIICBGlc-His (wild type). These results indicate that the crucial residues for the interaction between the two proteins are in the center of the KTPGRED motif.
- (b) Lane 1 shows a crosslinking experiment with strain JKA12 expressing SgrT-3HA (pACYC184sgrT3HA) and the so called “relaxed” mutant EIICBGlc-V12F-His (pRR48GH-V12F). Cells were grown in the presence of glucose. These results indicate an interaction between SgrT and the “relaxed” derivative of EIICBGlc.
- (c) This part of the figure shows crosslinking experiments between SgrT-3HA and the “locked in” mutant EIICBGlc-K150E-His (pRR48GH-K150E) in different genetic backgrounds. Lane 1 shows a sample of strain JKA12 expressing SgrT-3HA and EIICBGlc-K150E-His, lanes 2 and 3 exhibit samples of LJ140 expressing the same proteins. Cells were grown in the absence or presence of glucose as indicated. These results indicate no interaction between SgrT and EIICBGlcK150E in a PTS-positive strain, but a strong interaction in a ptsHIcrr deletion background.
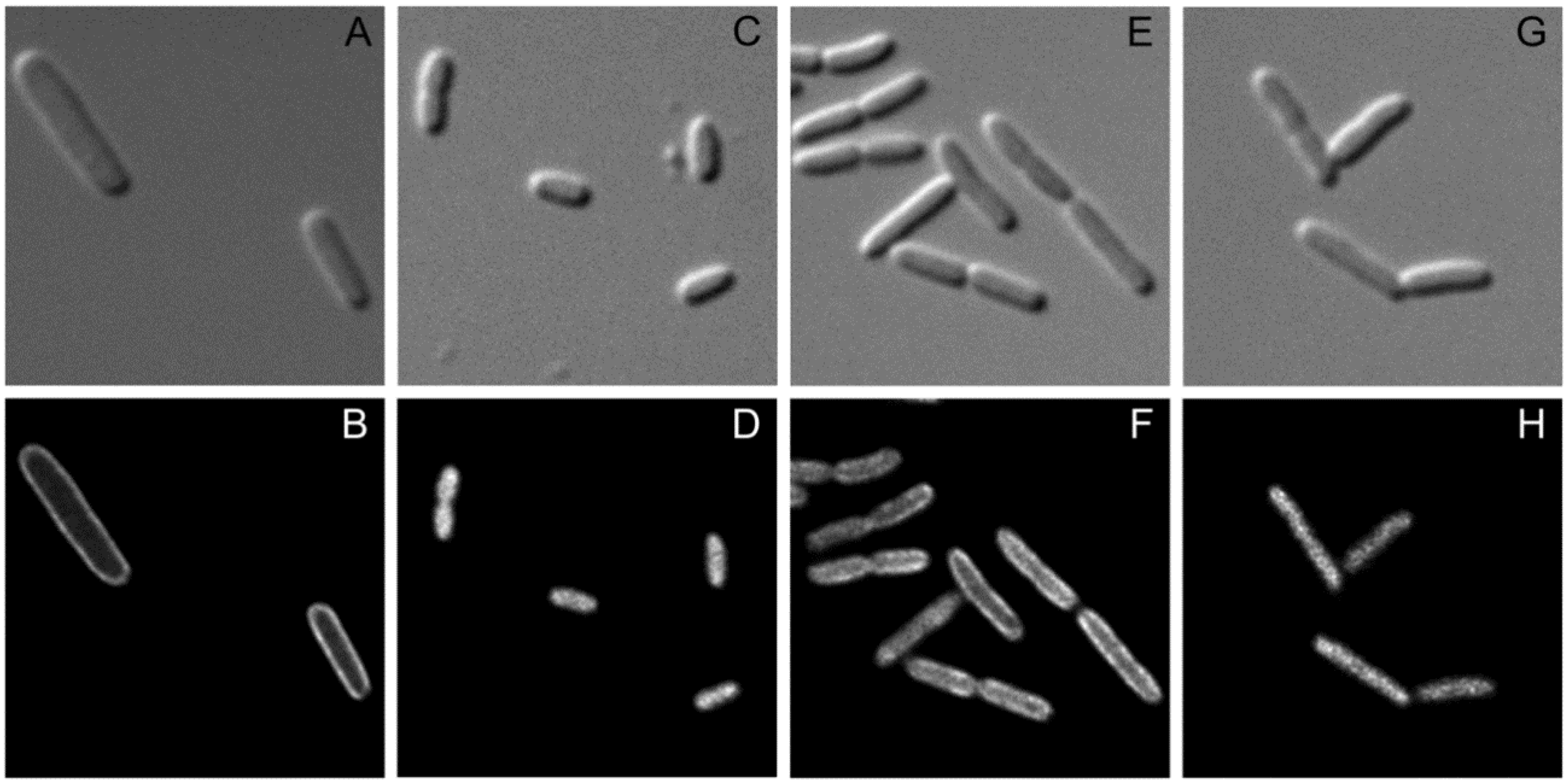
2.4. Recruitment of SgrT to the Membrane by EIICBGlc Can Be Visualized By in vivo Fluorescence Microscopy
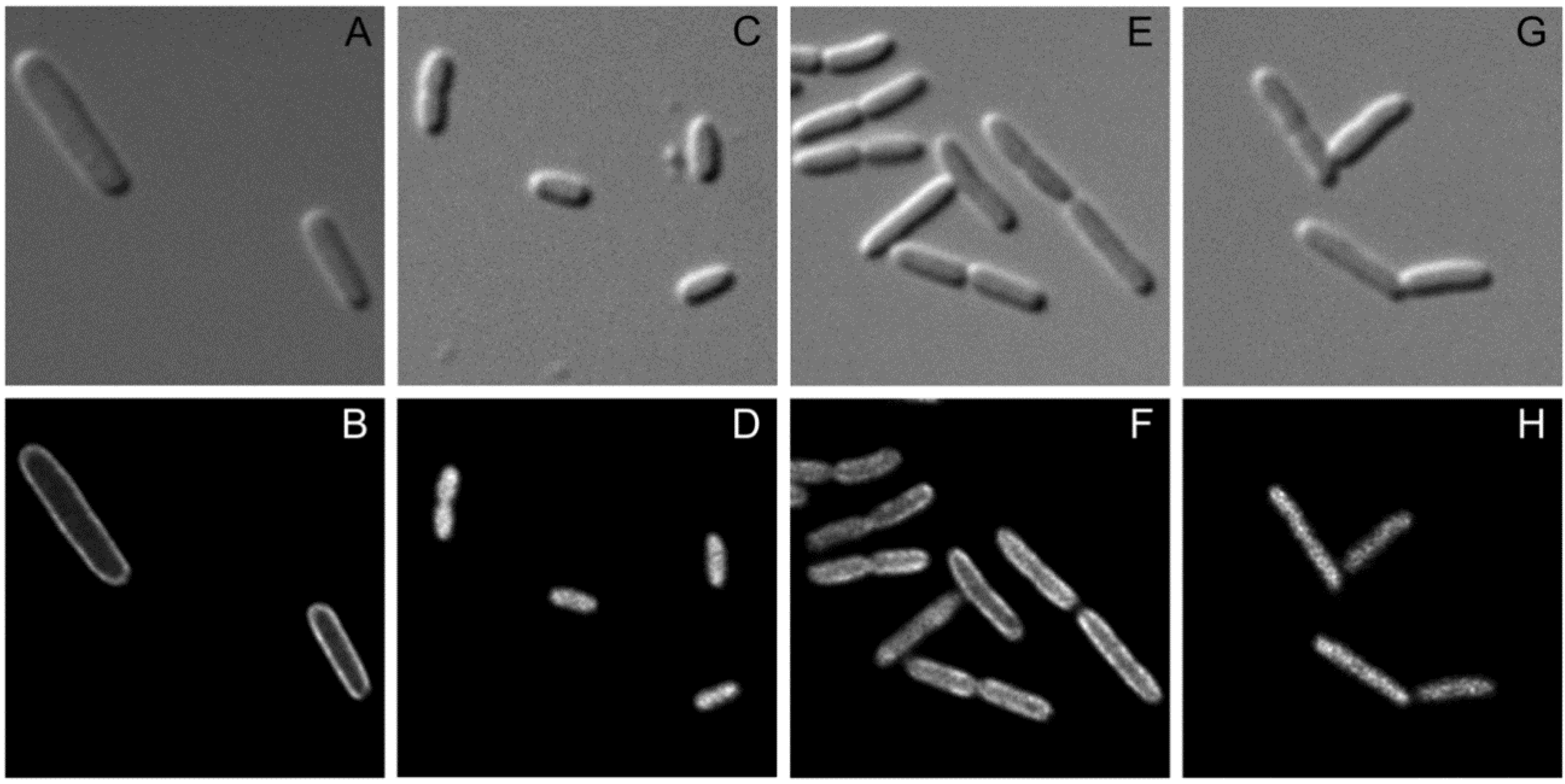
2.5. Discussion
3. Experimental Section

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Relevant Genotype or Phenotype | Source or Reference |
|---|---|---|
| Escherichia coli | ||
| BL21 (λDE3) | fhuA2 [lon] ompT gal (λ DE3) [dcm] ΔhsdS λ DE3 = λ sBamHIo ΔEcoRI-B int::(lacI::PlacUV5::T7 gene1) i21 Δnin5 | [43] |
| BW25113 | lacIq rrnBT14 ΔlacZWJ16 hsdR514 ΔaraBADAH33 ΔrhaBADLD78 | [44] |
| JKA1 | LJ110 ΔsgrRST::cat+ (CamR) | this study |
| JKA12 | LJ120 ΔsgrRST:: neo+ (KanR) | this study |
| JKA17 | BL21 (λDE3) ΔptsG::cat+ (CamR) | this study |
| JKA18 | LJB5 ΔsgrRST:: neo+ (KanR) | this study |
| JM109 | thi-1Δ(lac-proA,B)U169 gyrA1,96 recA1 endA1 relA1 hsdR17 supE44/F´traD36 proA+B+lacIqlacZΔM15 | [45] |
| K-12 | Wildtyp | K. Jahreis, lab stock |
| LJ110 | W3110 Fnr+ | [16] |
| LJ120 | LJ110 ΔptsG::cat+ (CamR) | [16] |
| LJ140 | LJ110 ΔptsHIcrr::neo+ (KanR) | [16] |
| LJ231 | LJ110 ΔmanXYZ::cat csc+ | K. Jahreis, lab stock |
| LJB5 | LJ231 ptsGP384R Tn10tet+(TetR) | [46] |
| LJB17 | LJ110 dgsA::cat | [47] |
| MG1655 | F-, λ-, rph-1 | Yale E.coli Stock Center |
| W3110 | F-λ- IN(rrnD-rrnE)1 rph-1 | Yale E.coli Stock Center |
| Bacteriophage | ||
| P1kc | lysogen | [48] |
| Name | Resistance | Properties | Source or Reference |
|---|---|---|---|
| Escherichia coli vectors | |||
| pACYC184 | TcR | [49] | |
| pACYC184sgrT3HA | TcR | SgrT-3HA (tacPO) | this study |
| pBAD24 | ApR | [50] | |
| pBLP2 | ApR | EIICBGlc-GFP | [51] |
| pET11a-link-NGFP | ApR | [52] | |
| pET11a-Z-NGFP | ApR | NGFP-Leucinzipper | [52] |
| pETS | ApR | NGFP-SgrT | this study |
| pKD3 | ApR, CmR | [44] | |
| pKD46 | ApR | [44] | |
| pMRB | KnR | EIIBGlc-CGFP | this study |
| pMRBAD-link-CGFP | KnR | [52] | |
| pMRBAD-Z-CGFP | KnR | Leucinzipper-CGFP | [52] |
| pMRC | KnR | EIICGlc-CGFP | this study |
| pMRCL | KnR | EIICGlc-linker-CGFP | this study |
| pMRCL-P384R | KnR | EIICGlc-linker-CGFP-P384R | this study |
| pMRG | KnR | EIICBGlc-CGFP | this study |
| pMRLB | KnR | Linker-EIIBGlc-CGFP | this study |
| pMRLB-P384R | KnR | Linker-EIIBGlc-CGFP-P384R | this study |
| pRR48 | ApR | Parkinson, LKS | |
| pRR48G | ApR | EIICBGlc | [53] |
| pRR48GH | ApR | EIICBGlc-5His | [53] |
| pRR48GH-D388A | ApR | EIICBGlc-D388A-5His | this study |
| pRR48GH-E387A | ApR | EIICBGlc-E387A-5His | this study |
| pRR48GH-G385A | ApR | EIICBGlc-G385A-5His | this study |
| pRR48GH-I296N | ApR | EIICBGlc-I296N-5His | this study |
| pRR48GH-K150E | ApR | EIICBGlc-K150E-5His | this study |
| pRR48GH-K382A | ApR | EIICBGlc-K382A-5His | this study |
| pRR48GH-P384A | ApR | EIICBGlc-384A-5His | this study |
| pRR48GH-P384R | ApR | EIICBGlc-384R-5His | this study |
| pRR48GH-R386A | ApR | EIICBGlc-R386A-5His | this study |
| pRR48GH-T383A | ApR | EIICBGlc-T383A-5His | this study |
| pRR48GH-V12F | ApR | EIICBGlc-V12F-5His | this study |
| pTM30 | ApR | [54] | |
| pTM30sgrT | ApR | SgrT | this study |
| pTM30sgrT3HA | ApR | SgrT-3HA | this study |
| pTM30sgrTgfp | ApR | SgrT-GFP | this study |
4. Conclusions
Acknowledgments
References
- Ladisch, M.R.; Kohlmann, K.L. Recombinant human insulin. Biotechnol. Prog. 1992, 8, 469–478. [Google Scholar] [CrossRef]
- Contiero, J.; Beatty, C.; Kumari, S.; DeSanti, C.L.; Strohl, W.R.; Wolfe, A. Effects of mutations in acetate metabolism on high-cell-density growth of Escherichia coli. J. Ind. Microbiol. Biot. 2000, 24, 421–430. [Google Scholar] [CrossRef]
- Lee, S.Y. High cell-density culture of Escherichia coli. Trends Biotechnol. 1996, 14, 98–105. [Google Scholar] [CrossRef]
- Phue, J.N.; Lee, S.J.; Kaufman, J.B.; Negrete, A.; Shiloach, J. Acetate accumulation through alternative metabolic pathways in ackA - pta - poxB - triple mutant in Escherichia coli B (BL21). Biotechnol. Lett. 2010, 32, 1897–1903. [Google Scholar] [CrossRef]
- De Anda, R.; Lara, A.R.; Hernandez, V.; Hernandez-Montalvo, V.; Gosset, G.; Bolivar, F.; Ramirez, O.T. Replacement of the glucose phosphotransferase transport system by galactose permease reduces acetate accumulation and improves process performance of Escherichia coli for recombinant protein production without impairment of growth rate. Metab. Eng. 2006, 8, 281–290. [Google Scholar] [CrossRef]
- Flores, N.; Leal, L.; Sigala, J.C.; de Anda, R.; Escalante, A.; Martinez, A.; Ramirez, O.T.; Gosset, G.; Bolivar, F. Growth recovery on glucose under aerobic conditions of an Escherichia coli strain carrying a phosphoenolpyruvate:carbohydrate phosphotransferase system deletion by inactivating arcA and overexpressing the genes coding for glucokinase and galactose permease. J. Mol. Microbiol. Biotechnol. 2007, 13, 105–116. [Google Scholar] [CrossRef]
- Jahreis, K.; Pimentel-Schmitt, E.F.; Bruckner, R.; Titgemeyer, F. Ins and outs of glucose transport systems in eubacteria. FEMS Microbiol. Rev. 2008, 32, 891–907. [Google Scholar] [CrossRef]
- Lengeler, J.W.; Jahreis, K. Bacterial PEP-dependent carbohydrate: phosphotransferase systems couple sensing and global control mechanisms. Contrib. Microbiol. 2009, 16, 65–87. [Google Scholar] [CrossRef]
- Lengeler, J.W.; Jahreis, K. Phosphotransferase systems or PTSs as carbohydrate transport and as signal transduction systems. In Handbook of Biological Physics; Konings, W.N., Kaback, H.S., Lolkema, J.S., Eds.; Elsevier Science: Amsterdam, the Netherlands, 1996; Vol. 2, pp. 573–598. [Google Scholar]
- Erni, B. Glucose Transport by the Bacterial Phosphotransferase System (PTS): An Interface between Energy- and Signal Transduction. In Transport Systems; Winkelmann, G., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2003. [Google Scholar] [CrossRef]
- Buhr, A.; Flukiger, K.; Erni, B. The glucose transporter of Escherichia coli. Overexpression, purification, and characterization of functional domains. J. Biol. Chem. 1994, 269, 23437–23443. [Google Scholar]
- Siebold, C.; Flukiger, K.; Beutler, R.; Erni, B. Carbohydrate transporters of the bacterial phosphoenolpyruvate: sugar phosphotransferase system (PTS). FEBS Lett. 2001, 504, 104–111. [Google Scholar]
- Lee, S.J.; Boos, W.; Bouche, J.P.; Plumbridge, J. Signal transduction between a membrane-bound transporter, PtsG, and a soluble transcription factor, Mlc, of Escherichia coli. Embo. J. 2000, 19, 5353–5361. [Google Scholar] [CrossRef]
- Nam, T.W.; Cho, S.H.; Shin, D.; Kim, J.H.; Jeong, J.Y.; Lee, J.H.; Roe, J.H.; Peterkofsky, A.; Kang, S.O.; Ryu, S.; et al. The Escherichia coli glucose transporter enzyme IICB(Glc) recruits the global repressor Mlc. Embo. J. 2001, 20, 491–498. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kimata, K.; Aiba, H. A novel regulatory role of glucose transporter of Escherichia coli: membrane sequestration of a global repressor Mlc. Embo. J. 2000, 19, 5344–5352. [Google Scholar] [CrossRef]
- Zeppenfeld, T.; Larisch, C.; Lengeler, J.W.; Jahreis, K. Glucose transporter mutants of Escherichia coli K-12 with changes in substrate recognition of IICB(Glc) and induction behavior of the ptsG gene. J. Bacteriol. 2000, 182, 4443–4452. [Google Scholar] [CrossRef]
- Jahreis, K. cAMP Signaling in Prokaryotes. In Bacterial Signaling; Krämer, R., Jung, K., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 357–375. [Google Scholar]
- Jeong, J.Y.; Kim, Y.J.; Cho, N.; Shin, D.; Nam, T.W.; Ryu, S.; Seok, Y.J. Expression of ptsG encoding the major glucose transporter is regulated by ArcA in Escherichia coli. J. Biol. Chem. 2004, 279, 38513–38518. [Google Scholar]
- Rungrassamee, W.; Liu, X.; Pomposiello, P.J. Activation of glucose transport under oxidative stress in Escherichia coli. Arch. Microbiol. 2008, 190, 41–49. [Google Scholar] [CrossRef]
- Shin, D.; Cho, N.; Heu, S.; Ryu, S. Selective regulation of ptsG expression by Fis. Formation of either activating or repressing nucleoprotein complex in response to glucose. J. Biol. Chem. 2003, 278, 14776–14781. [Google Scholar]
- Shin, D.; Lim, S.; Seok, Y.J.; Ryu, S. Heat shock RNA polymerase (E sigma(32)) is involved in the transcription of mlc and crucial for induction of the Mlc regulon by glucose in Escherichia coli. J. Biol. Chem. 2001, 276, 25871–25875. [Google Scholar]
- Seeto, S.; Notley-McRobb, L.; Ferenci, T. The multifactorial influences of RpoS, Mlc and cAMP on ptsG expression under glucose-limited and anaerobic conditions. Res. Microbiol. 2004, 155, 211–215. [Google Scholar] [CrossRef]
- Morita, T.; El-Kazzaz, W.; Tanaka, Y.; Inada, T.; Aiba, H. Accumulation of glucose 6-phosphate or fructose 6-phosphate is responsible for destabilization of glucose transporter mRNA in Escherichia coli. J. Biol. Chem. 2003, 278, 15608–15614. [Google Scholar]
- Vanderpool, C.K.; Gottesman, S. Involvement of a novel transcriptional activator and small RNA in post-transcriptional regulation of the glucose phosphoenolpyruvate phosphotransferase system. Mol. Microbiol. 2004, 54, 1076–1089. [Google Scholar] [CrossRef]
- Wadler, C.S.; Vanderpool, C.K. A dual function for a bacterial small RNA: SgrS performs base pairing-dependent regulation and encodes a functional polypeptide. Proc. Natl. Acad. Sci. USA 2007, 104, 20454–20459. [Google Scholar] [CrossRef]
- Negrete, A.; Majdalani, N.; Phue, J.N.; Shiloach, J. Reducing acetate excretion from E. coli K-12 by over-expressing the small RNA SgrS. N. Biotechnol. 2011. [Google Scholar] [CrossRef]
- Gabor, E.; Göhler, A.K.; Kosfeld, A.; Staab, A.; Kremling, A.; Jahreis, K. The phosphoenolpyruvate-dependent glucose-phosphotransferase system from Escherichia coli K-12 as the center of a network regulating carbohydrate flux in the cell. Eur. J. Cell Biol. 2011, 90, 711–720. [Google Scholar] [CrossRef]
- Schmid, K.; Ebner, R.; Altenbuchner, J.; Schmitt, R.; Lengeler, J.W. Plasmid-mediated sucrose metabolism in Escherichia coli K12: mapping of the scr genes of pUR400. Mol. Microbiol. 1988, 2, 1–8. [Google Scholar] [CrossRef]
- Lengeler, J.W.; Jahreis, K.; Wehmeier, U.F. Enzymes II of the phosphoenol pyruvate-dependent phosphotransferase systems: their structure and function in carbohydrate transport. Biochim. Biophys. Acta 1994, 1188, 1–28. [Google Scholar] [CrossRef]
- Herzberg, C.; Weidinger, L.A.; Dorrbecker, B.; Hubner, S.; Stulke, J.; Commichau, F.M. SPINE: a method for the rapid detection and analysis of protein-protein interactions in vivo. Proteomics 2007, 7, 4032–4035. [Google Scholar] [CrossRef]
- Magliery, T.J.; Wilson, C.G.; Pan, W.; Mishler, D.; Ghosh, I.; Hamilton, A.D.; Regan, L. Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap: scope and mechanism. J. Am. Chem. Soc. 2005, 127, 146–157. [Google Scholar]
- Xu, B.; Jahic, M.; Enfors, S.O. Modeling of overflow metabolism in batch and fed-batch cultures of Escherichia coli. Biotechnol. Prog. 1999, 15, 81–90. [Google Scholar] [CrossRef]
- De Mey, M.; De Maeseneire, S.; Soetaert, W.; Vandamme, E. Minimizing acetate formation in E. coli fermentations. J. Ind. Microbiol. Biotechnol. 2007, 34, 689–700. [Google Scholar] [CrossRef] [Green Version]
- Morita, T.; Aiba, H. Small RNAs making a small protein. Proc. Natl. Acad. Sci. USA 2007, 104, 20149–20150. [Google Scholar] [CrossRef]
- Vanderpool, C.K.; Gottesman, S. The Novel Transcription Factor SgrR Coordinates the Response to Glucose-Phosphate Stress. J. Bacteriol. 2007, 189, 2238–2248. [Google Scholar] [CrossRef]
- Lolkema, J.S.; Dijkstra, D.S.; Robillard, G.T. Mechanics of solute translocation catalyzed by enzyme IImtl of the phosphoenolpyruvate-dependent phosphotransferase system of Escherichia coli. Biochemistry 1992, 31, 5514–5521. [Google Scholar] [CrossRef]
- Notley-McRobb, L.; Ferenci, T. Substrate specificity and signal transduction pathways in the glucose-specific enzyme II (EII(Glc)) component of the Escherichia coli phosphotransferase system. J. Bacteriol. 2000, 182, 4437–4442. [Google Scholar] [CrossRef]
- Kornberg, H.L.; Lambourne, L.T.; Sproul, A.A. Facilitated diffusion of fructose via the phosphoenolpyruvate/glucose phosphotransferase system of Escherichia coli. Proc. Natl. Acad. Sci. USA 2000, 97, 1808–1812. [Google Scholar]
- Oh, H.; Park, Y.; Park, C. A mutated PtsG, the glucose transporter, allows uptake of D-ribose. J. Biol. Chem. 1999, 274, 14006–14011. [Google Scholar]
- Begley, G.S.; Warner, K.A.; Arents, J.C.; Postma, P.W.; Jacobson, G.R. Isolation and characterization of a mutation that alters the substrate specificity of the Escherichia coli glucose permease. J. Bacteriol. 1996, 178, 940–942. [Google Scholar]
- Ruijter, G.J.; van Meurs, G.; Verwey, M.A.; Postma, P.W.; van Dam, K. Analysis of mutations that uncouple transport from phosphorylation in enzyme IIGlc of the Escherichia coli phosphoenolpyruvate-dependent phosphotransferase system. J. Bacteriol. 1992, 174, 2843–2850. [Google Scholar]
- Buhr, A.; Daniels, G.A.; Erni, B. The glucose transporter of Escherichia coli. Mutants with impaired translocation activity that retain phosphorylation activity. J. Biol. Chem. 1992, 267, 3847–3851. [Google Scholar]
- Studier, F.W.; Rosenberg, A.H.; Dunn, J.J.; Dubendorff, J.W. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990, 185, 60–89. [Google Scholar]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef]
- Yanisch-Perron, C.; Vieira, J.; Messing, J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 1985, 33, 103–119. [Google Scholar] [CrossRef]
- Becker, A.K. Das EIICBGlc als Glukose-Sensor in E.coli K-12: Eine molekulargenetische Analyse der Funktion des Enzyms und der Regulation des zugehörigen Gens ptsG. Master Thesis, Universität Osnabrück, Osnabrück, Saxony, Germany, 2000. [Google Scholar]
- Becker, A.K.; Zeppenfeld, T.; Staab, A.; Seitz, S.; Boos, W.; Morita, T.; Aiba, H.; Mahr, K.; Titgemeyer, F.; Jahreis, K. YeeI, a novel protein involved in modulation of the activity of the glucose-phosphotransferase system in Escherichia coli K-12. J. Bacteriol. 2006, 188, 5439–5449. [Google Scholar] [CrossRef]
- Lennox, E.S. Transduction of linked genetic characters of the host by bacteriophage P1. Virology 1955, 1, 190–206. [Google Scholar] [CrossRef]
- Chang, A.C.; Cohen, S.N. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J. Bacteriol. 1978, 134, 1141–1156. [Google Scholar]
- Guzman, L.M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar]
- Siepelmeyer, J. Entwicklung von Systemen zur quantitativen Messung der intrazellulären cAMP-Konzentration und der in vivo Aktivität der Adenylatzyklase CyaA in einer isogenen Stammreihe von Escherichia coli K-12. Ph.D. Thesis, Universität Osnabrück, Osnabrück, Saxony, Germany, 2000. [Google Scholar]
- Wilson, C.G.; Magliery, T.J.; Regan, L. Detecting protein-protein interactions with GFP-fragment reassembly. Nat. Methods 2004, 1, 255–262. [Google Scholar] [CrossRef]
- Gabor, E. Molekularbiologische Untersuchungen verschiedener Komponenten des Glukose-Phosphotransferasesystems in Escherichia coli K-12 mit Schwerpunkt auf der Strukturanalyse des Transportproteins EIICBGlc. Ph.D. Thesis, Universität Osnabrück, Thesis, Universität Osnabrück, Osnabrück, Saxony, Germany, 2011. [Google Scholar]
- Morrison, T.B.; Parkinson, J.S. Liberation of an interaction domain from the phosphotransfer region of CheA, a signaling kinase of Escherichia coli. Proc. Natl. Acad. Sci. USA 1994, 91, 5485–5489. [Google Scholar] [CrossRef]
- Ausubel, F.M.; Brent, R.; Kingston, R.E.; Moore, D.D.; Seidmann, J.G.; Smith, J.A.; Struhl, K. Current Protocols in Molecular Biology; Greene Publishing and Wiley-Interscience: New York, NY, USA, 1990. [Google Scholar]
- Tanaka, S.; Lerner, S.A.; Lin, E.C. Replacement of a phosphoenolpyruvate-dependent phosphotransferase by a nicotinamide adenine dinucleotide-linked dehydrogenase for the utilization of mannitol. J. Bacteriol. 1967, 93, 642–648. [Google Scholar]
- Saiki, R.K.; Gelfand, D.H.; Stoffel, S.; Scharf, S.J.; Higuchi, R.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 1988, 239, 487–491. [Google Scholar]
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kosfeld, A.; Jahreis, K. Characterization of the Interaction Between the Small Regulatory Peptide SgrT and the EIICBGlc of the Glucose-Phosphotransferase System of E. coli K-12. Metabolites 2012, 2, 756-774. https://doi.org/10.3390/metabo2040756
Kosfeld A, Jahreis K. Characterization of the Interaction Between the Small Regulatory Peptide SgrT and the EIICBGlc of the Glucose-Phosphotransferase System of E. coli K-12. Metabolites. 2012; 2(4):756-774. https://doi.org/10.3390/metabo2040756
Chicago/Turabian StyleKosfeld, Anne, and Knut Jahreis. 2012. "Characterization of the Interaction Between the Small Regulatory Peptide SgrT and the EIICBGlc of the Glucose-Phosphotransferase System of E. coli K-12" Metabolites 2, no. 4: 756-774. https://doi.org/10.3390/metabo2040756