
Olaris Global Panel (OGP): A Highly Accurate and Reproducible Triple Quadrupole Mass Spectrometry-Based Metabolomics Method for Clinical Biomarker Discovery
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Material
2.2. U-13C Metabolite Yeast Extraction
2.3. Sample Preparation
2.4. Clinical Samples
2.5. Instrumentation and LC-MS Data Acquisition
2.6. Chromatographic Conditions
2.7. Data and Statistical Analysis
2.7.1. Analysis of the Impact of U-13C Metabolite Yeast Extract on Linearity of Response and Normalization
2.7.2. Assessment of Within- and Across-Batch Precision Using QC Samples
2.7.3. Comparison with NMR Data to Evaluate Data Accuracy
2.7.4. OGP Method Performance in a Clinical Sample Study
3. Results
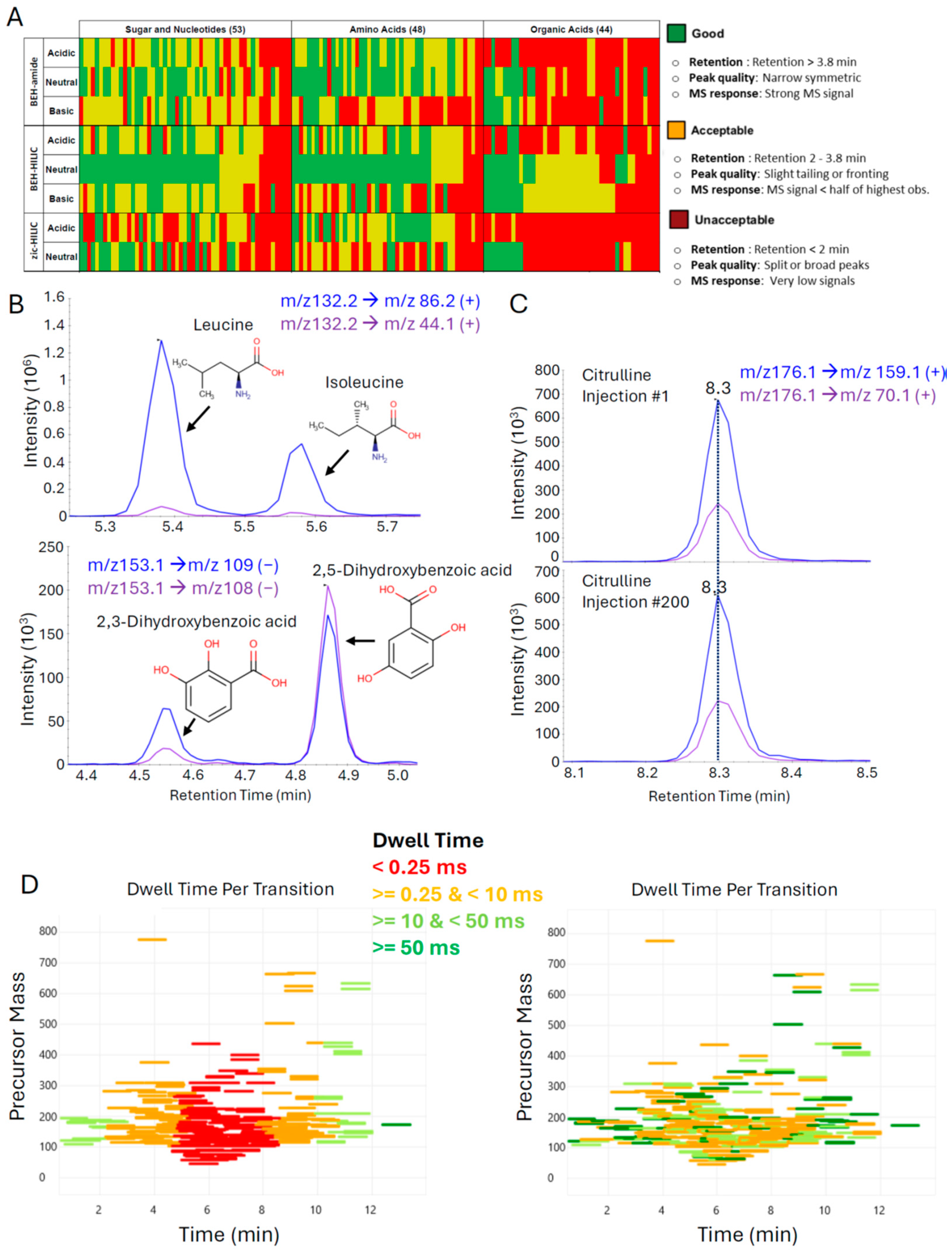
3.1. Optimization of Liquid Chromatography and Mass Spectrometry Parameters to Facilitate High-Quality Data Generation for Hundreds of Metabolites
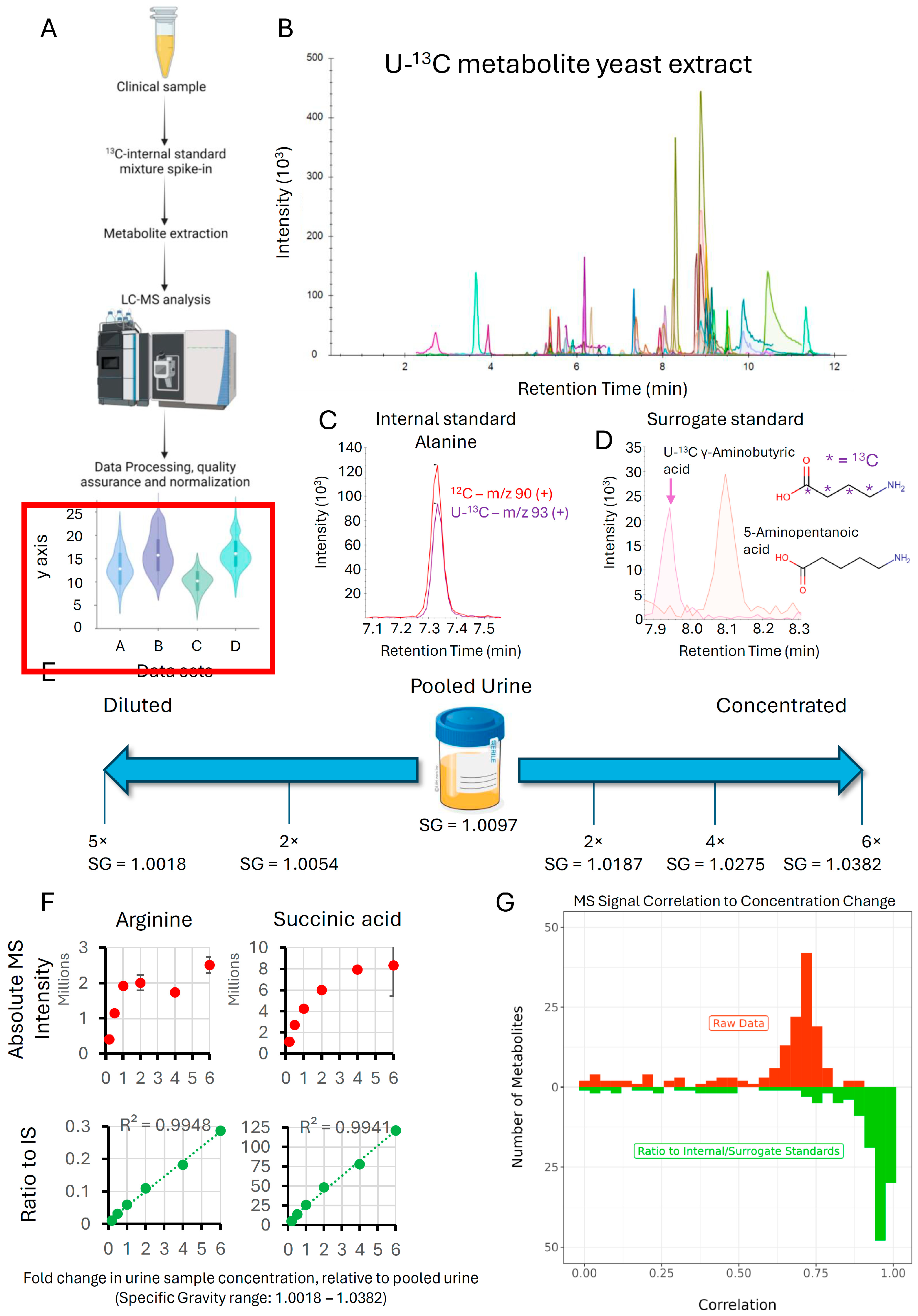
3.2. Impact of U-13C Metabolite Yeast Extract on Linearity of Response and Normalization
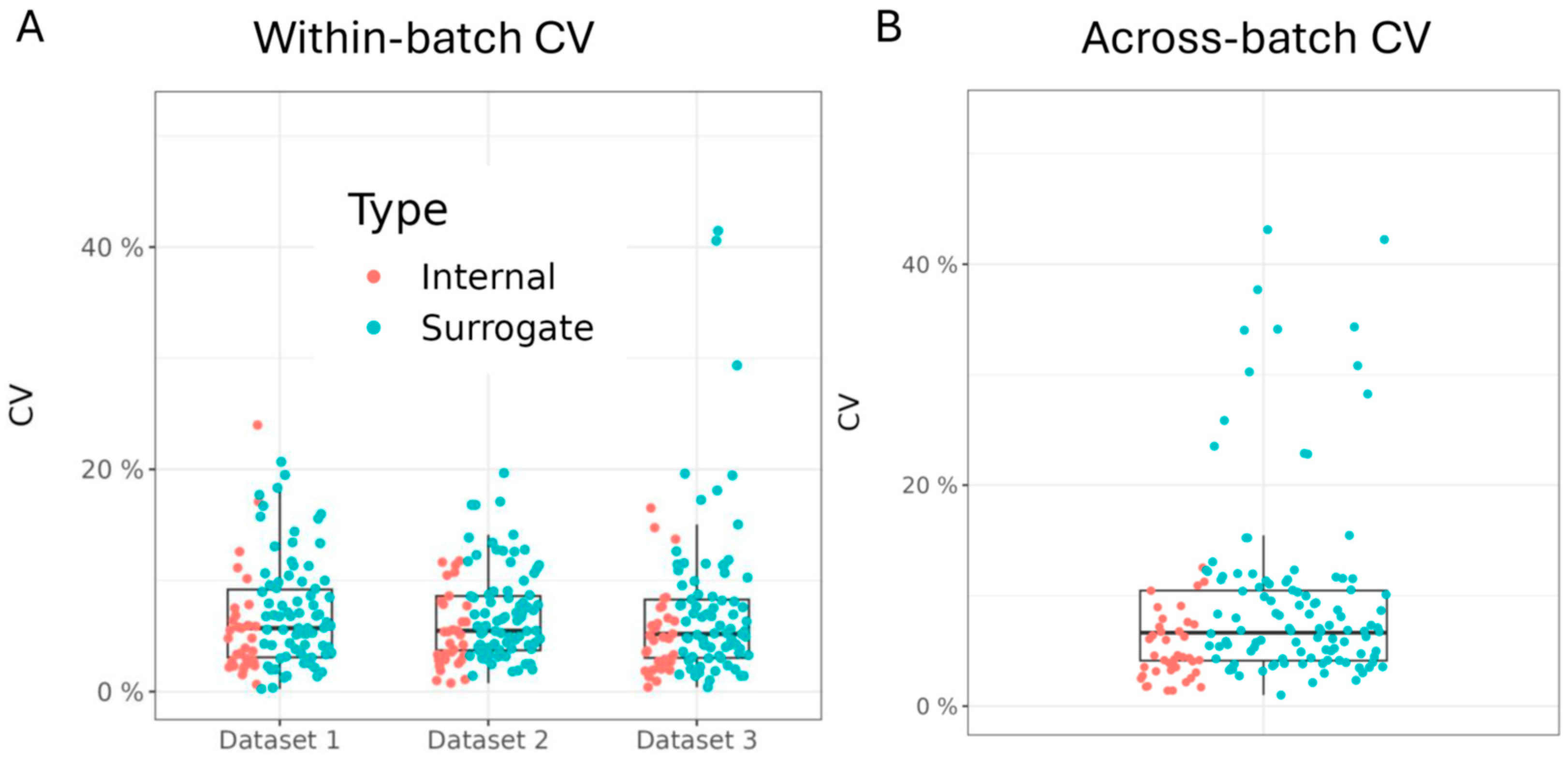
3.3. Assessment of Within- and Across-Batch Precision Using QC Samples
3.4. Comparison with NMR Data to Evaluate Data Accuracy
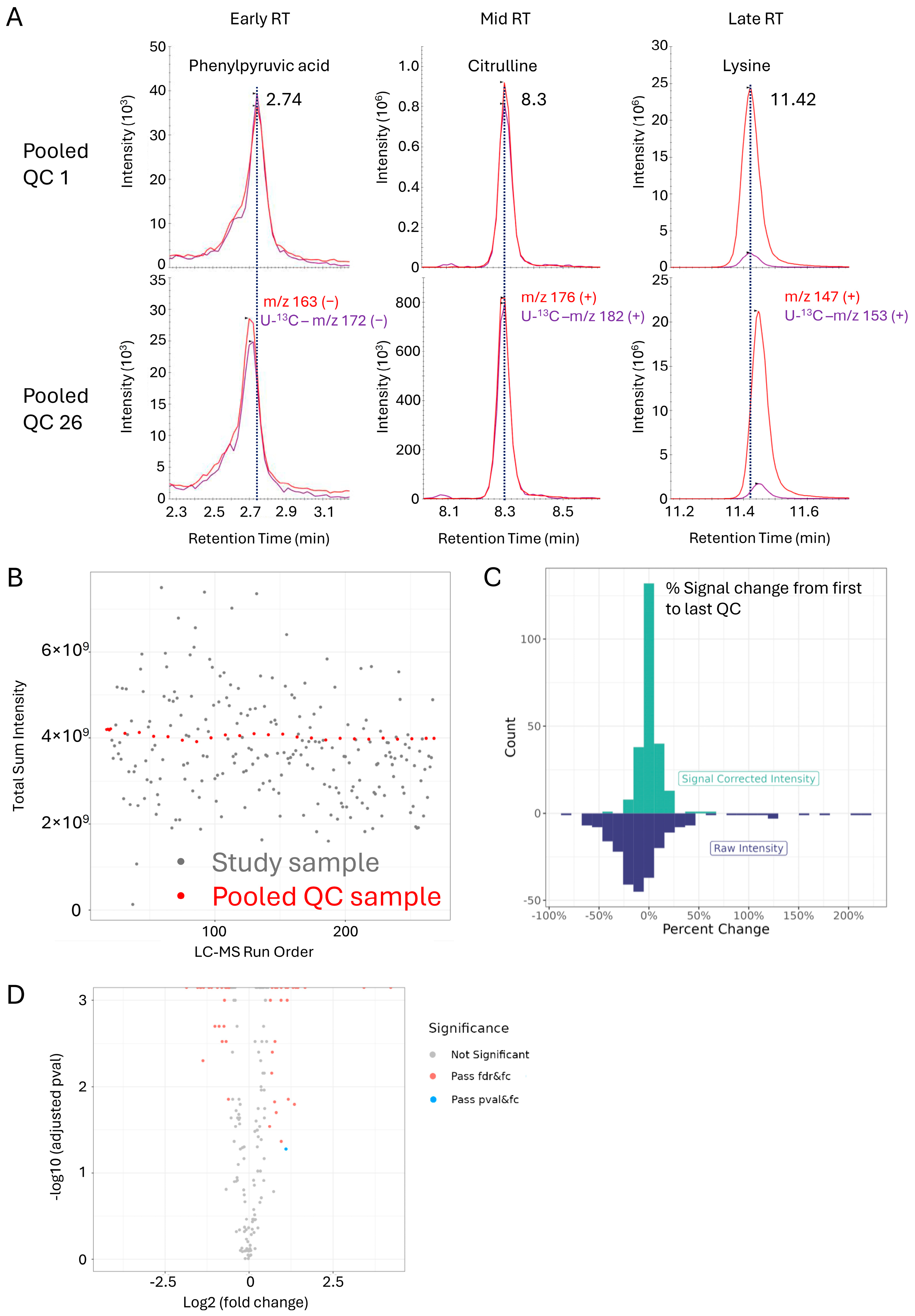
3.5. OGP Method Performance in a Clinical Sample Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ceglarek, U.; Leichtle, A.; Brügel, M.; Kortz, L.; Brauer, R.; Bresler, K.; Thiery, J.; Fiedler, G.M. Challenges and Developments in Tandem Mass Spectrometry Based Clinical Metabolomics. Mol. Cell Endocrinol. 2009, 301, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Chaby, L.E.; Lasseter, H.C.; Contrepois, K.; Salek, R.M.; Turck, C.W.; Thompson, A.; Vaughan, T.; Haas, M.; Jeromin, A. Cross-Platform Evaluation of Commercially Targeted and Untargeted Metabolomics Approaches to Optimize the Investigation of Psychiatric Disease. Metabolites 2021, 11, 609. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Cai, Y.; Yao, H.; Lin, C.; Xie, Y.; Tang, S.; Zhang, A. Small Molecule Metabolites: Discovery of Biomarkers and Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 132. [Google Scholar] [PubMed]
- Levin, A.; Stevens, P.E.; Bilous, R.W.; Coresh, J.; De Francisco, A.L.M.; De Jong, P.E.; Griffith, K.E.; Hemmelgarn, B.R.; Iseki, K.; Lamb, E.J.; et al. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. Suppl. 2013, 3, 1–150. [Google Scholar]
- Peng, H.; Liu, X.; Aoieong, C.; Tou, T.; Tsai, T.; Ngai, K.; Cheang, H.I.; Liu, Z.; Liu, P.; Zhu, H. Identification of Metabolite Markers Associated with Kidney Function. J. Immunol. Res. 2022, 2022, 6190333. [Google Scholar] [CrossRef] [PubMed]
- Ford, L.; Mitchell, M.; Wulff, J.; Evans, A.; Kennedy, A.; Elsea, S.; Wittmann, B.; Toal, D. Clinical Metabolomics for Inborn Errors of Metabolism. Adv. Clin. Chem. 2022, 107, 79–138. [Google Scholar] [PubMed]
- Long, J.; Yang, Z.; Wang, L.; Han, Y.; Peng, C.; Yan, C.; Yan, D. Metabolite Biomarkers of Type 2 Diabetes Mellitus and Pre-Diabetes: A Systematic Review and Meta-Analysis. BMC Endocr. Disord. 2020, 20, 174. [Google Scholar] [CrossRef] [PubMed]
- Cobb, J.; Gall, W.; Adam, K.-P.; Nakhle, P.; Button, E.; Hathorn, J.; Lawton, K.; Milburn, M.; Perichon, R.; Mitchell, M.; et al. A Novel Fasting Blood Test for Insulin Resistance and Prediabetes. J. Diabetes Sci. Technol. 2013, 7, 100–110. [Google Scholar] [CrossRef]
- Hilvo, M.; Meikle, P.J.; Pedersen, E.R.; Tell, G.S.; Dhar, I.; Brenner, H.; Schöttker, B.; Lääperi, M.; Kauhanen, D.; Koistinen, K.M.; et al. Development and Validation of a Ceramide- and Phospholipid-Based Cardiovascular Risk Estimation Score for Coronary Artery Disease Patients. Eur. Heart J. 2020, 41, 371–380. [Google Scholar] [CrossRef]
- Klatt, S.; Doecke, J.D.; Roberts, A.; Boughton, B.A.; Masters, C.L.; Horne, M.; Roberts, B.R. A Six-Metabolite Panel as Potential Blood-Based Biomarkers for Parkinson’s Disease. NPJ Parkinsons Dis. 2021, 7, 94. [Google Scholar] [CrossRef]
- Quintero, M.E.; de Moraes Pontes, J.G.; Tasic, L. Metabolomics in Degenerative Brain Diseases. Brain Res. 2021, 1773, 147704. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Honrao, C.; Rodrigues, L.O.; Wolf, J.; Sheehan, K.B.; Surface, M.; Alcalay, R.N.; O’day, E.M. Plasma Metabolite Signature Classifies Male LRRK2 Parkinson’s Disease Patients. Metabolites 2022, 12, 149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhang, Y.; Zhao, W.; Deng, K.; Wang, Z.; Yang, C.; Ma, L.; Openkova, M.S.; Hou, Y.; Li, K. Metabolomics for Biomarker Discovery in the Diagnosis, Prognosis, Survival and Recurrence of Colorectal Cancer: A Systematic Review. Oncotarget 2017, 8, 35460–35472. [Google Scholar] [CrossRef] [PubMed]
- Long, N.P.; Yoon, S.J.; Anh, N.H.; Nghi, T.D.; Lim, D.K.; Hong, Y.J.; Hong, S.S.; Kwon, S.W. A Systematic Review on Metabolomics-Based Diagnostic Biomarker Discovery and Validation in Pancreatic Cancer. Metabolomics 2018, 14, 109. [Google Scholar] [CrossRef] [PubMed]
- Kdadra, M.; Höckner, S.; Leung, H.; Kremer, W.; Schiffer, E. Metabolomics Biomarkers of Prostate Cancer: A Systematic Review. Diagnostics 2019, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Nagana Gowda, G.A.; Djukovic, D. Overview of Mass Spectrometry-Based Metabolomics: Opportunities and Challenges. Methods Mol. Biol. 2014, 1198, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Nagana Gowda, G.A.; Raftery, D. Overview of NMR Spectroscopy-Based Metabolomics: Opportunities and Challenges. Methods Mol. Biol. 2019, 2037, 3. [Google Scholar] [CrossRef] [PubMed]
- Nikolskiy, I.; Siuzdak, G.; Patti, G.J. Discriminating Precursors of Common Fragments for Large-Scale Metabolite Profiling by Triple Quadrupole Mass Spectrometry. Bioinformatics 2015, 31, 2017–2023. [Google Scholar] [CrossRef]
- Schwaiger-Haber, M.; Stancliffe, E.; Arends, V.; Thyagarajan, B.; Sindelar, M.; Patti, G.J. A Workflow to Perform Targeted Metabolomics at the Untargeted Scale on a Triple Quadrupole Mass Spectrometer. ACS Meas. Sci. Au 2021, 1, 35–45. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, T.T.; Li, Y.; Li, J.; Fan, Y.; Huang, F.Q.; Cai, Y.Y.; Ma, G.; Liu, J.F.; Chen, Q.Q.; et al. Functional Metabolomics Characterizes a Key Role for N-Acetylneuraminic Acid in Coronary Artery Diseases. Circulation 2018, 137, 1374–1390. [Google Scholar] [CrossRef]
- Ding, J.; Feng, Y.Q. Mass Spectrometry-Based Metabolomics for Clinical Study: Recent Progresses and Applications. TrAC Trends Anal. Chem. 2023, 158, 116896. [Google Scholar] [CrossRef]
- Sindelar, M.; Patti, G.J. Chemical Discovery in the Era of Metabolomics. J. Am. Chem. Soc. 2020, 142, 9097–9105. [Google Scholar] [CrossRef] [PubMed]
- Dührkop, K.; Nothias, L.F.; Fleischauer, M.; Reher, R.; Ludwig, M.; Hoffmann, M.A.; Petras, D.; Gerwick, W.H.; Rousu, J.; Dorrestein, P.C.; et al. Systematic Classification of Unknown Metabolites Using High-Resolution Fragmentation Mass Spectra. Nat. Biotechnol. 2021, 39, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Mahieu, N.G.; Patti, G.J. Systems-Level Annotation of a Metabolomics Data Set Reduces 25,000 Features to Fewer than 1000 Unique Metabolites. Anal. Chem 2017, 89, 10397–10406. [Google Scholar] [CrossRef] [PubMed]
- Peterman, S.M.; Percy, A.J.; Bird, S.S.; Zhao, J.; Dorrani, M.; Bekhti, N.; Kamphorst, J.J. A Guide to Implementing Targeted and Standardized Clinical Metabolomics Using Stable Isotope-Labeled Standards and Triple Quadrupole Mass Spectrometry Mass Spectrometry. Available online: https://assets.thermofisher.com/TFS-Assets/CMD/Reference-Materials/wp-002706-ms-tsq-altis-targeted-clinical-metabolomics-lcms-wp00270-en.pdf (accessed on 9 May 2024).
- López-Hernández, Y.; Lima-Rogel, V.; Mandal, R.; Zheng, J.; Zhang, L.; Oler, E.; García-López, D.A.; Torres-Calzada, C.; Mejía-Elizondo, A.R.; Poelsner, J.; et al. The Urinary Metabolome of Newborns with Perinatal Complications. Metabolites 2024, 14, 41. [Google Scholar] [CrossRef] [PubMed]
- Cavus, E.; Karakas, M.; Ojeda, F.M.; Kontto, J.; Veronesi, G.; Ferrario, M.M.; Linneberg, A.; Jørgensen, T.; Meisinger, C.; Thorand, B.; et al. Association of Circulating Metabolites with Risk of Coronary Heart Disease in a European Population: Results from the Biomarkers for Cardiovascular Risk Assessment in Europe (BiomarCaRE) Consortium. JAMA Cardiol. 2019, 4, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Southam, A.D.; Haglington, L.D.; Najdekr, L.; Jankevics, A.; Weber, R.J.M.; Dunn, W.B. Assessment of Human Plasma and Urine Sample Preparation for Reproducible and High-Throughput UHPLC-MS Clinical Metabolic Phenotyping. Analyst 2020, 145, 6511–6523. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.J.; Pratt, B.; Bose, N.; Dubois, L.G.; St. John-Williams, L.; Perrott, K.M.; Ky, K.; Kapahi, P.; Sharma, V.; Maccoss, M.J.; et al. Skyline for Small Molecules: A Unifying Software Package for Quantitative Metabolomics. J. Proteome Res. 2020, 19, 1447–1458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Powers, R.; O’Day, E.M. Evaluation of Non-Uniform Sampling 2D1H–13C HSQC Spectra for Semi-Quantitative Metabolomics. Metabolites 2020, 10, 203. [Google Scholar] [CrossRef]
- Wishart, D.S.; Guo, A.C.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef]
- Contrepois, K.; Jiang, L.; Snyder, M. Optimized Analytical Procedures for the Untargeted Metabolomic Profiling of Human Urine and Plasma by Combining Hydrophilic Interaction (HILIC) and Reverse-Phase Liquid Chromatography (RPLC)-Mass Spectrometry. Mol. Cell. Proteom. 2015, 14, 1684–1695. [Google Scholar] [CrossRef]
- Hosseinkhani, F.; Huang, L.; Dubbelman, A.C.; Guled, F.; Harms, A.C.; Hankemeier, T. Systematic Evaluation of HILIC Stationary Phases for Global Metabolomics of Human Plasma. Metabolites 2022, 12, 165. [Google Scholar] [CrossRef]
- Hermann, G.; Schwaiger, M.; Volejnik, P.; Koellensperger, G. 13C-Labelled Yeast as Internal Standard for LC–MS/MS and LC High Resolution MS Based Amino Acid Quantification in Human Plasma. J. Pharm. Biomed. Anal. 2018, 155, 329–334. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed Minimum Reporting Standards for Chemical Analysis: Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Salek, R.M.; Neumann, S.; Schober, D.; Hummel, J.; Billiau, K.; Kopka, J.; Correa, E.; Reijmers, T.; Rosato, A.; Tenori, L.; et al. COordination of Standards in MetabOlomicS (COSMOS): Facilitating Integrated Metabolomics Data Access. Metabolomics 2015, 11, 1587–1597. [Google Scholar] [CrossRef]
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K.S.; et al. Metabolomics Workbench: An International Repository for Metabolomics Data and Metadata, Metabolite Standards, Protocols, Tutorials and Training, and Analysis Tools. Nucleic Acids Res. 2016, 44, D463–D470. [Google Scholar] [CrossRef]
- Haug, K.; Salek, R.M.; Conesa, P.; Hastings, J.; De Matos, P.; Rijnbeek, M.; Mahendraker, T.; Williams, M.; Neumann, S.; Rocca-Serra, P.; et al. MetaboLights—An Open-Access General-Purpose Repository for Metabolomics Studies and Associated Meta-Data. Nucleic Acids Res. 2013, 41, D781–D786. [Google Scholar] [CrossRef]
- Zheng, J.; Zhang, L.; Johnson, M.; Mandal, R.; Wishart, D.S. Comprehensive Targeted Metabolomic Assay for Urine Analysis. Anal. Chem. 2020, 92, 10627–10634. [Google Scholar] [CrossRef]
- Jia, Z.; Qiu, Q.; He, R.; Zhou, T.; Chen, L. Identification of Metabolite Interference Is Necessary for Accurate LC-MS Targeted Metabolomics Analysis. Anal. Chem. 2023, 95, 7985–7992. [Google Scholar] [CrossRef] [PubMed]
- Li, X. Amino Acid Metabolism in the Kidneys: Nutritional and Physiological Significance; Springer: Cham, Switzerland, 2020; Volume 1265. [Google Scholar]
- Baylis, C. Arginine, Arginine Analogs and Nitric Oxide Production in Chronic Kidney Disease. Nat. Clin. Pract. Nephrol. 2006, 2, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Boirie, Y.; Albright, R.; Bigelow, M.; Nair, K.S. Impairment of Phenylalanine Conversion to Tyrosine in End-Stage Renal Disease Causing Tyrosine Deficiency. Kidney Int. 2004, 66, 591–596. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on Uremic Toxins: Classification, Concentration, and Interindividual Variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dorrani, M.; Zhao, J.; Bekhti, N.; Trimigno, A.; Min, S.; Ha, J.; Han, A.; O’Day, E.; Kamphorst, J.J. Olaris Global Panel (OGP): A Highly Accurate and Reproducible Triple Quadrupole Mass Spectrometry-Based Metabolomics Method for Clinical Biomarker Discovery. Metabolites 2024, 14, 280. https://doi.org/10.3390/metabo14050280
Dorrani M, Zhao J, Bekhti N, Trimigno A, Min S, Ha J, Han A, O’Day E, Kamphorst JJ. Olaris Global Panel (OGP): A Highly Accurate and Reproducible Triple Quadrupole Mass Spectrometry-Based Metabolomics Method for Clinical Biomarker Discovery. Metabolites. 2024; 14(5):280. https://doi.org/10.3390/metabo14050280
Chicago/Turabian StyleDorrani, Masoumeh, Jifang Zhao, Nihel Bekhti, Alessia Trimigno, Sangil Min, Jongwon Ha, Ahram Han, Elizabeth O’Day, and Jurre J. Kamphorst. 2024. "Olaris Global Panel (OGP): A Highly Accurate and Reproducible Triple Quadrupole Mass Spectrometry-Based Metabolomics Method for Clinical Biomarker Discovery" Metabolites 14, no. 5: 280. https://doi.org/10.3390/metabo14050280