
Comparative Evaluation of Plasma Metabolomic Data from Multiple Laboratories

 , ,
, ,  , , , , , , , , , ,
, , , , , , , , , ,  , and add
Show full author list
, and add
Show full author list
Abstract
:
1. Introduction
2. Results and Discussion
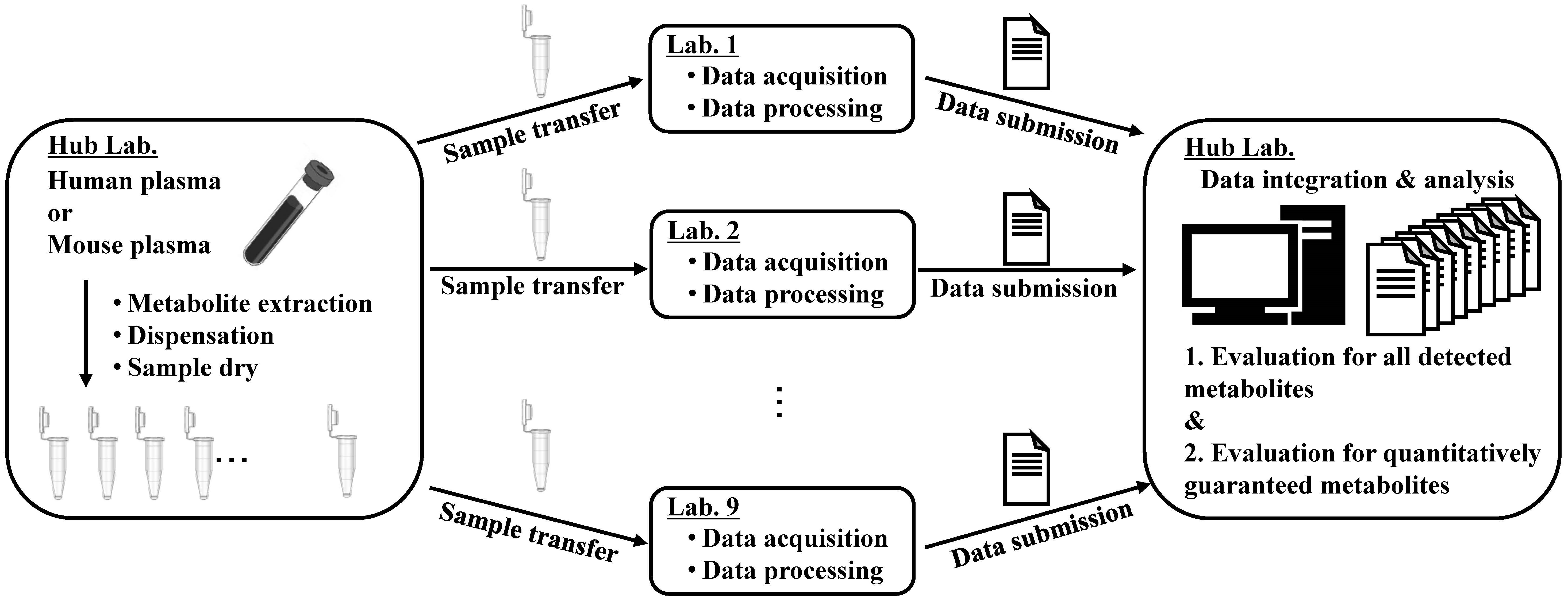
2.1. Analytical Procedure and Data Acquisition
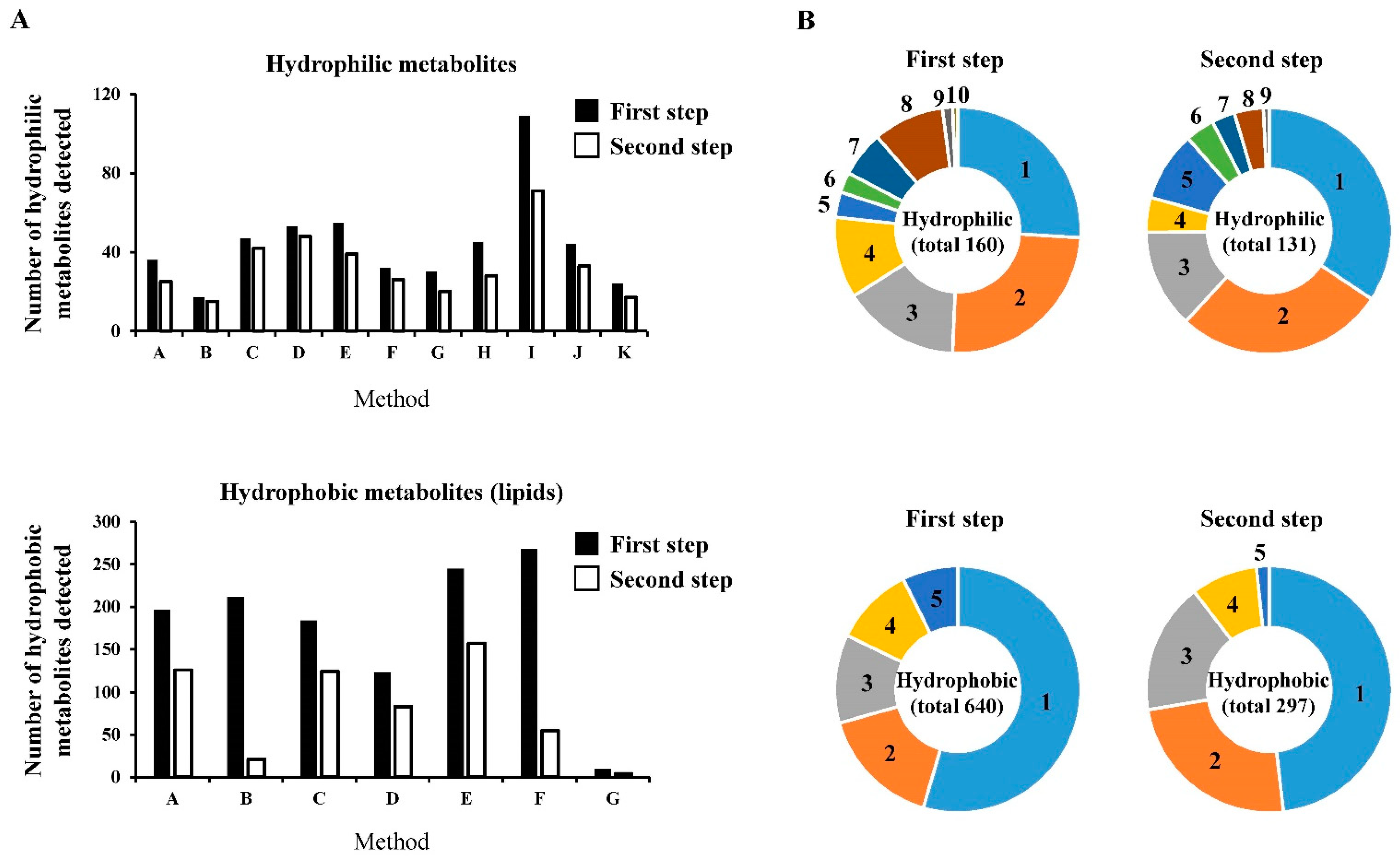
2.2. Data Summary and Comparisons of the Analytical Methods for the Relative Quantification for the Metabolites Detected (First Step)
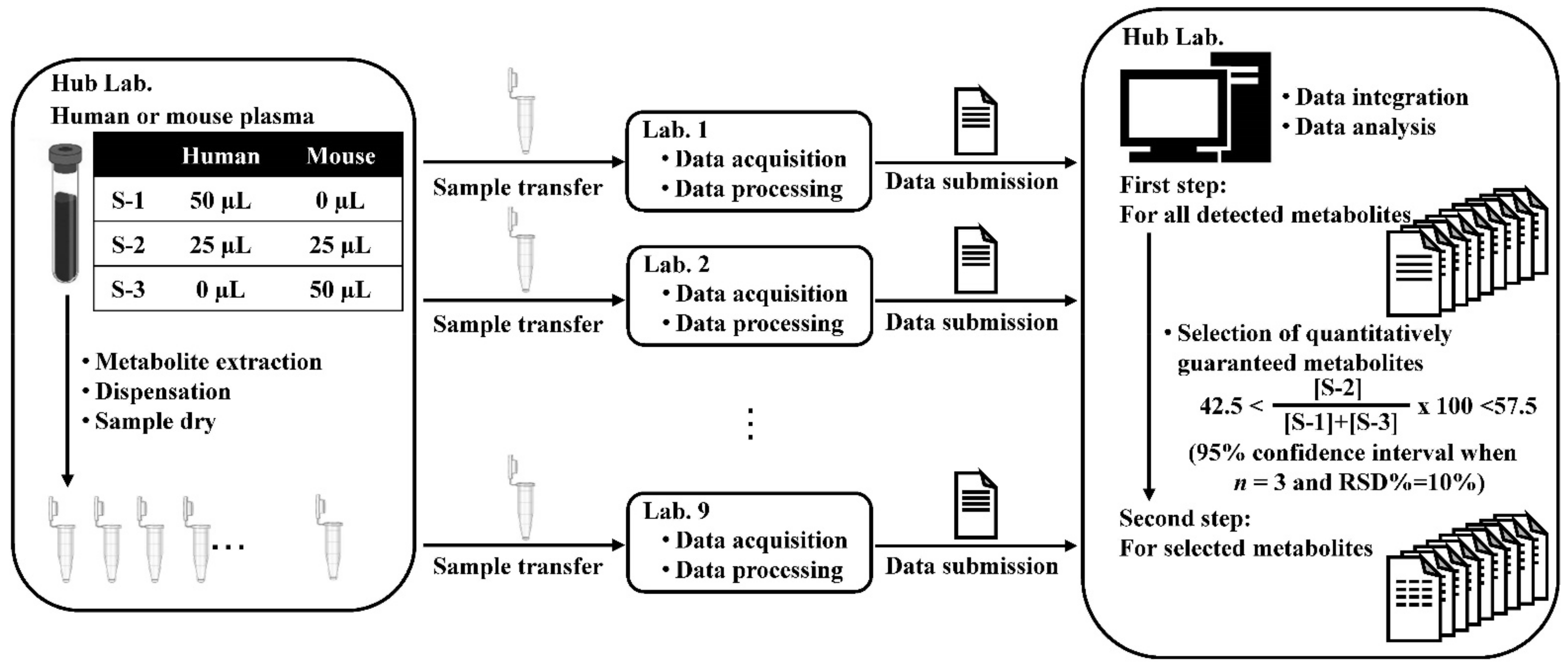
2.3. Data Summary and Comparisons of the Analytical Methods for the Relative Quantification of the Quantitatively Guaranteed Metabolites (Second Step)
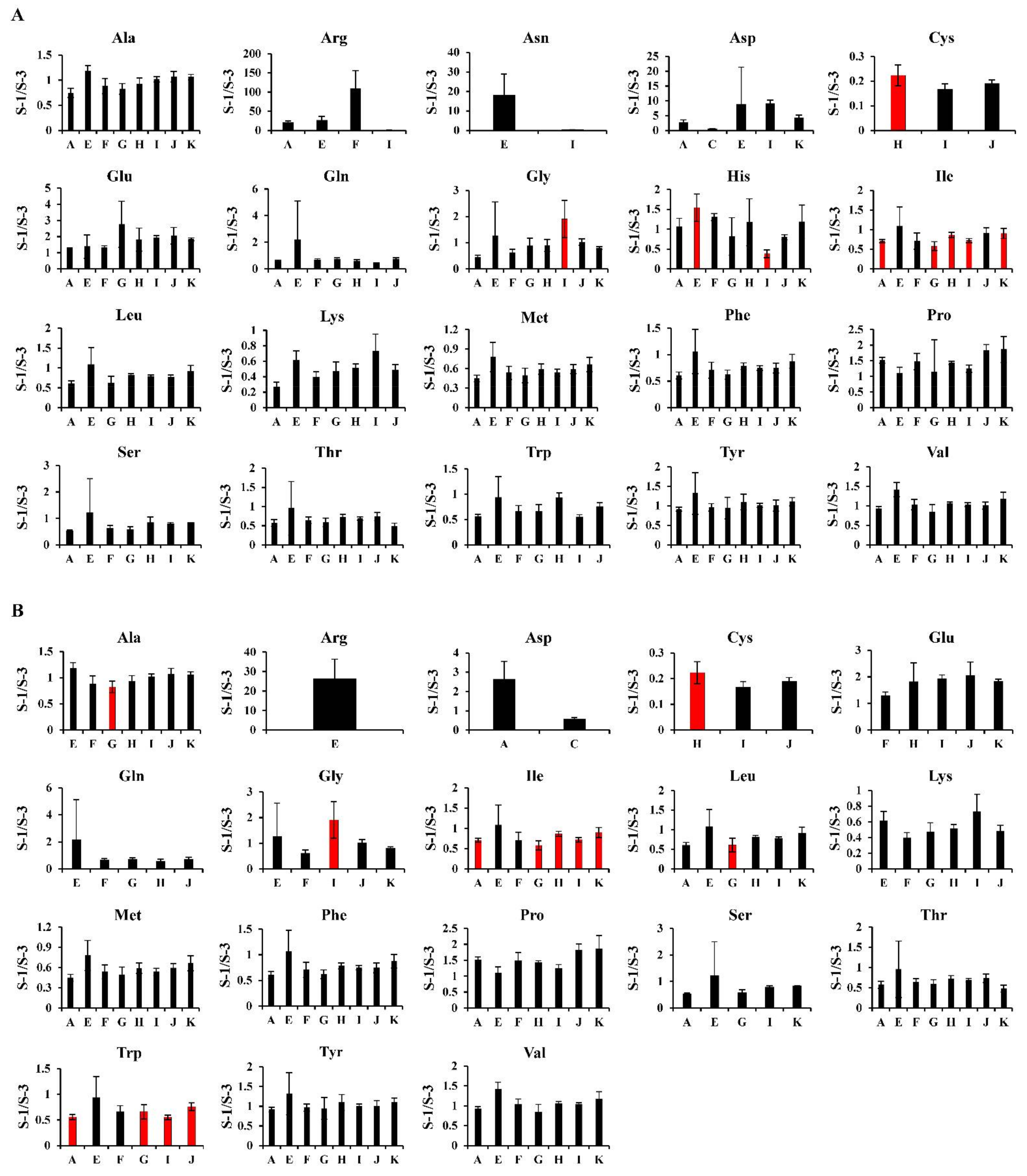
2.4. Possible Causes of the Differences in the Results from Different Laboratories/Machines in MS-Based Metabolomics
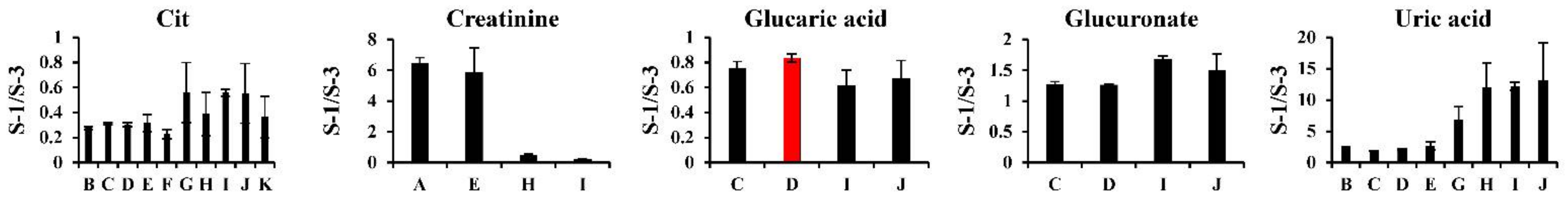
2.4.1. Hydrophilic Metabolites
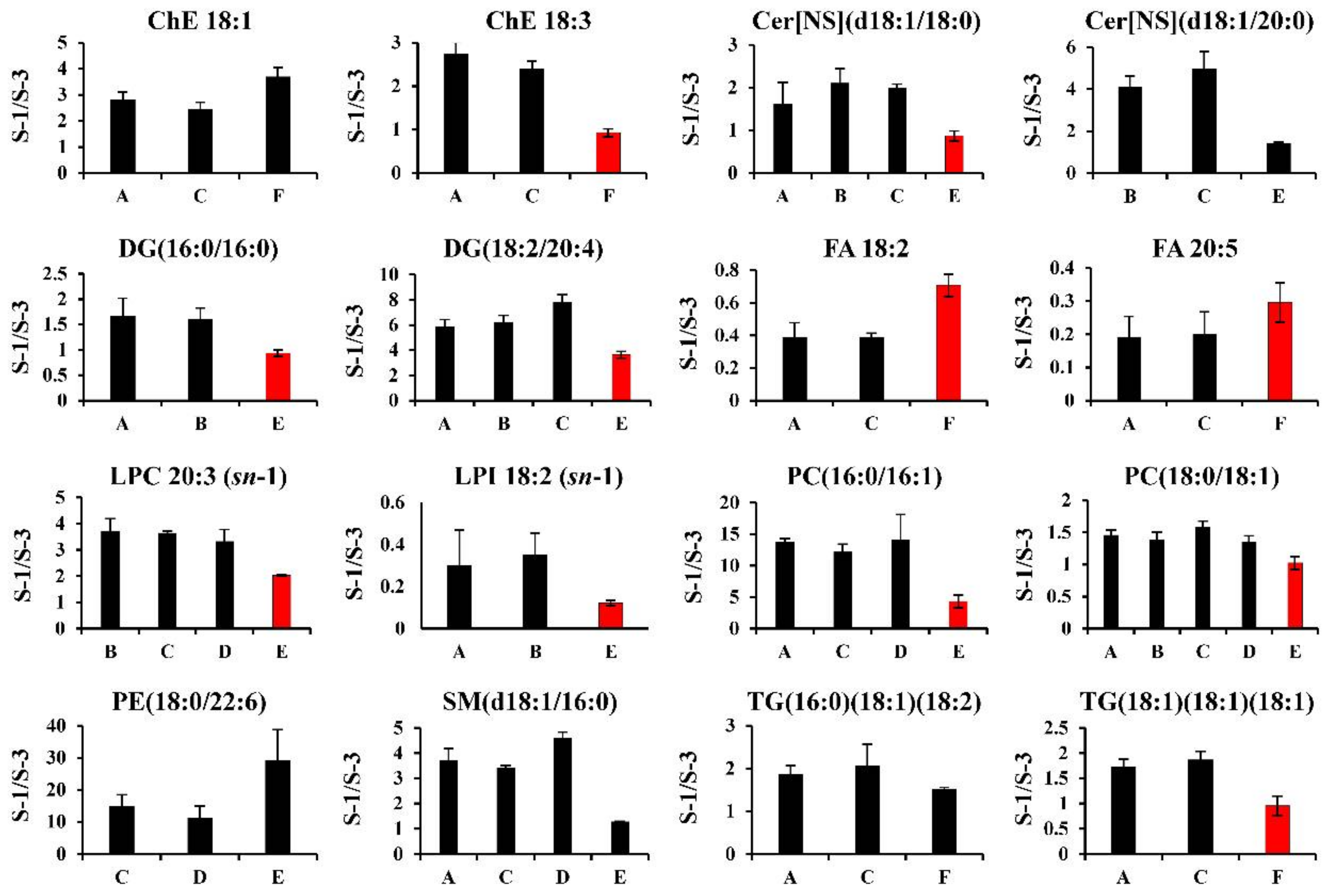
2.4.2. Hydrophobic Metabolites
2.4.3. Hydrophilic Metabolites versus Hydrophobic Metabolites
3. Materials and Methods
3.1. Materials
3.2. Sample Preparation
3.3. Analytical Procedure
3.4. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oyedeji, A.B.; Green, E.; Adebiyi, J.A.; Ogundele, O.M.; Gbashi, S.; Adefisoye, M.A.; Oyeyinka, S.A.; Adebo, O.A. Metabolomic approaches for the determination of metabolites from pathogenic microorganisms: A review. Food Res. Int. 2021, 140, 110042. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, D.B.; Brotman, Y.; Fernie, A.R. The utility of metabolomics as a tool to inform maize biology. Plant. Commun. 2021, 2, 100187. [Google Scholar] [CrossRef] [PubMed]
- Utpott, M.; Rodrigues, E.; Rios, A.O.; Mercali, G.D.; Flôres, S.H. Metabolomics: An analytical technique for food processing evaluation. Food Chem. 2021, 366, 130685. [Google Scholar] [CrossRef]
- Castelli, F.A.; Rosati, G.; Moguet, C.; Fuentes, C.; Marrugo-Ramírez, J.; Lefebvre, T.; Volland, H.; Merkoçi, A.; Simon, S.; Fenaille, F.; et al. Metabolomics for personalized medicine: The input of analytical chemistry from biomarker discovery to point-of-care tests. Anal. Bioanal. Chem. 2021, 414, 759–789. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.A.; Varma, V.R.; Huang, C.W.; An, Y.; Oommen, A.; Tanaka, T.; Ferrucci, L.; Elango, P.; Takebayashi, T.; Harada, S.; et al. Blood metabolite signature of metabolic syndrome implicates alterations in amino acid metabolism: Findings from the Baltimore longitudinal study of aging (BLSA) and the Tsuruoka metabolomics cohort study (TMCS). Int. J. Mol. Sci. 2020, 21, 1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmuganathan, M.; Kroezen, Z.; Gill, B.; Azab, S.; de Souza, R.J.; Teo, K.K.; Atkinson, S.; Subbarao, P.; Desai, D.; Anand, S.S.; et al. The maternal serum metabolome by multisegment injection-capillary electrophoresis-mass spectrometry: A high-throughput platform and standardized data workflow for large-scale epidemiological studies. Nat. Protoc. 2021, 16, 1966–1994. [Google Scholar] [CrossRef]
- Iida, M.; Harada, S.; Takebayashi, T. Application of metabolomics to epidemiological studies of atherosclerosis and cardiovascular disease. J. Atheroscler. Thromb. 2019, 26, 747–757. [Google Scholar] [CrossRef] [Green Version]
- García, C.J.; Yang, X.; Huang, D.; Tomás-Barberán, F.A. Can we trust biomarkers identified using different non-targeted metabolomics platforms? Multi-platform, inter-laboratory comparative metabolomics profiling of lettuce cultivars via UPLC-QTOF-MS. Metabolomics 2020, 16, 85. [Google Scholar] [CrossRef]
- Siskos, A.P.; Jain, P.; Römisch-Margl, W.; Bennett, M.; Achaintre, D.; Asad, Y.; Marney, L.; Richardson, L.; Koulman, A.; Griffin, J.L.; et al. Interlaboratory reproducibility of a targeted metabolomics platform for analysis of human serum and plasma. Anal. Chem. 2016, 89, 656–665. [Google Scholar] [CrossRef]
- Bowden, J.A.; Heckert, A.; Ulmer, C.Z.; Jones, C.M.; Koelmel, J.P.; Abdullah, L.; Ahonen, L.; Alnouti, Y.; Armando, A.M.; Asara, J.M.; et al. Harmonizing lipidomics: NIST interlaboratory comparison exercise for lipidomics using SRM 1950-Metabolites in frozen Human plasma. J. Lipid Res. 2017, 58, 2275–2288. [Google Scholar] [CrossRef] [Green Version]
- Liebisch, G.; Ekroos, K.; Hermansson, M.; Ejsing, C.S. Reporting of lipidomics data should be standardized. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Allwood, J.W.; Erban, A.; de Koning, S.; Dunn, W.B.; Luedemann, A.; Lommen, A.; Kay, L.; Löscher, R.; Kopka, J.; Goodacre, R. Inter-laboratory reproducibility of fast gas chromatography-electron impact-time of flight mass spectrometry (GC-EI-TOF/MS) based plant metabolomics. Metabolomics 2009, 5, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiehn, O. Metabolomics by gas chromatography-mass spectrometry: Combined targeted and untargeted profiling. Curr. Protoc. Mol. Biol. 2016, 114, 3041–30432. [Google Scholar] [CrossRef]
- Benton, H.P.; Want, E.; Keun, H.C.; Amberg, A.; Plumb, R.S.; Goldfain-Blanc, F.; Walther, B.; Reily, M.D.; Lindon, J.C.; Holmes, E.; et al. Intra- and interlaboratory reproducibility of ultra performance liquid chromatography–time-of-flight mass spectrometry for urinary metabolic profiling. Anal. Chem. 2012, 84, 2424–2432. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Matsuda, F.; Hirayama, A.; Ikeda, K.; Kita, Y.; Horie, K.; Saigusa, D.; Saito, K.; Sawada, Y.; Nakanishi, H.; et al. Inter-laboratory comparison of metabolite measurements for metabolomics data integration. Metabolites 2019, 9, 257. [Google Scholar] [CrossRef] [Green Version]
- Soga, T.; Ohashi, Y.; Ueno, Y.; Naraoka, H.; Tomita, M.; Nishioka, T. Quantitative metabolome analysis using capillary electrophoresis mass spectrometry. J. Proteome Res. 2003, 2, 488–494. [Google Scholar] [CrossRef]
- Soga, T.; Igarashi, K.; Ito, C.; Mizobuchi, K.; Zimmermann, H.P.; Tomita, M. Metabolomic profiling of anionic metabolites by capillary electrophoresis mass spectrometry. Anal. Chem. 2009, 81, 6165–6174. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, A.; Tabata, S.; Kudo, R.; Hasebe, M.; Suzuki, K.; Tomita, M.; Soga, T. The use of a double coaxial electrospray ionization sprayer improves the peak resolutions of anionic metabolites in capillary ion chromatography-mass spectrometry. J. Chromatogr. A 2020, 1619, 460914. [Google Scholar] [CrossRef]
- Fushimi, T.; Izumi, Y.; Takahashi, M.; Hata, K.; Murano, Y.; Bamba, T. Dynamic metabolome analysis reveals the metabolic fate of medium-chain fatty acid in AML12 cells. J. Agric. Food Chem. 2020, 68, 11997–12010. [Google Scholar] [CrossRef]
- Saigusa, D.; Hishinuma, E.; Matsukawa, N.; Takahashi, M.; Inoue, J.; Tadaka, S.; Motoike, I.N.; Hozawa, A.; Izumi, Y.; Bamba, T.; et al. Comparison of kit-based metabolomics with other methodologies in a large cohort, towards establishing reference values. Metabolites 2021, 11, 652. [Google Scholar] [CrossRef]
- Hata, K.; Soma, Y.; Yamashita, T.; Takahashi, M.; Sugitate, K.; Serino, T.; Miyagawa, H.; Suzuki, K.; Yamada, K.; Kawamukai, T.; et al. Calibration-curve-locking database for semi-quantitative metabolomics by gas chromatography/mass spectrometry. Metabolites 2021, 11, 207. [Google Scholar] [CrossRef] [PubMed]
- Nishiumi, S.; Shinohara, M.; Ikeda, A.; Yoshie, T.; Hatano, N.; Kakuyama, S.; Mizuno, S.; Sanuki, T.; Kutsumi, H.; Fukusaki, E.; et al. Serum metabolomics as a novel diagnostic approach for pancreatic cancer. Metabolomics 2010, 6, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Nishiumi, S.; Kobayashi, T.; Kawana, S.; Unno, Y.; Sakai, T.; Okamoto, K.; Yamada, Y.; Sudo, K.; Yamaji, T.; Saito, Y.; et al. Investigations in the possibility of early detection of colorectal cancer by gas chromatography/triple-quadrupole mass spectrometry. Oncotarget 2017, 8, 17115–17126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okahashi, N.; Kawana, S.; Iida, J.; Shimizu, H.; Matsuda, F. Fragmentation of dicarboxylic and tricarboxylic acids in the Krebs cycle using GC-EI-MS and GC-EI-MS/MS. Mass Spectrom. 2019, 8, A0073. [Google Scholar] [CrossRef]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef]
- Saito, K.; Ohno, Y.; Saito, Y. Enrichment of resolving power improves ion-peak quantification on a lipidomics platform. J. Chromatogr. B 2017, 1055–1056, 20–28. [Google Scholar] [CrossRef]
- Valentine, W.J.; Tokuoka, S.M.; Hishikawa, D.; Kita, Y.; Shindou, H.; Shimizu, T. LPAAT3 incorporates docosahexaenoic acid into skeletal muscle cell membranes and is upregulated by PPARδ activation. J. Lipid Res. 2018, 59, 184–194. [Google Scholar] [CrossRef] [Green Version]
- Takeda, H.; Izumi, Y.; Takahashi, M.; Paxton, T.; Tamura, S.; Koike, T.; Yu, Y.; Kato, N.; Nagase, K.; Shiomi, M.; et al. Widely-targeted quantitative lipidomics method by supercritical fluid chromatography triple quadrupole mass spectrometry. J. Lipid Res. 2018, 59, 1283–1293. [Google Scholar] [CrossRef] [Green Version]
- Chocholoušková, M.; Wolrab, D.; Jirásko, R.; Študentová, H.; Melichar, B.; Holčapek, M. Intra-laboratory comparison of four analytical platforms for lipidomic quantitation using hydrophilic interaction liquid chromatography or supercritical fluid chromatography coupled to quadrupole—Time-of-flight mass spectrometry. Talanta 2021, 231, 122367. [Google Scholar] [CrossRef]
- Pascale, R.; Acquavia, M.A.; Onzo, A.; Cataldi, T.R.I.; Calvano, C.D.; Bianco, G. Analysis of surfactants by mass spectrometry: Coming to grips with their diversity. Mass Spectrom. Rev. 2021. [Google Scholar] [CrossRef]
- Ishibashi, M.; Izumi, Y.; Sakai, M.; Ando, T.; Fukusaki, E.; Bamba, T. High-throughput simultaneous analysis of pesticides by supercritical fluid chromatography coupled with high-resolution mass spectrometry. J. Agric. Food Chem. 2015, 63, 4457–4463. [Google Scholar] [CrossRef] [PubMed]
- Burla, B.; Arita, M.; Arita, M.; Bendt, A.K.; Cazenave-Gassiot, A.; Dennis, E.A.; Ekroos, K.; Han, X.; Ikeda, K.; Liebisch, G.; et al. MS-based lipidomics of human blood plasma: A community-initiated position paper to develop accepted guidelines. J. Lipid Res. 2018, 59, 2001–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Caldwell, G.W.; Li, Y.; Lang, W.; Masucci, J. Inter-laboratory reproducibility of an untargeted metabolomics GC-MS assay for analysis of human plasma. Sci. Rep. 2020, 10, 10918. [Google Scholar]
- Collins, B.C.; Hunter, C.L.; Liu, Y.; Schilling, B.; Rosenberger, G.; Bader, S.L.; Chan, D.W.; Gibson, B.W.; Gingras, A.C.; Held, J.M.; et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat. Commun. 2017, 8, 291. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method ID | Lab ID | Analytical Method & Mode | Ref. |
|---|---|---|---|
| A | 1 | CE–TOFMS (cation mode, scan) | [16] |
| B | 1 | CE–TOFMS (anion mode, scan) | [17] |
| C | 1 | Capillary–IC/QExactive (scan) | [18] |
| D | 2 | IC/QExactive (scan) | [19] |
| E | 2 | PFPP–LC/QExactive (scan) | [19] |
| F | 3 | C18–LC/TQMS (MRM) | [20] |
| G | 2 | Derivatization and GC/QMS (scan) | [21] |
| H | 4 | Derivatization and GC/QMS (scan) | [22] |
| I | 3 | Derivatization and GC/TQMS (MRM) | [23] |
| J | 5 | Derivatization and GC/TQMS (MRM) | [23] |
| K | 6 | Derivatization and GC/QMS (SIM) | [24] |
| Method ID | Lab ID | Analytical Method | Ref. |
|---|---|---|---|
| A | 7 | C18–LC/QTOFMS (positive/negative, scan) | [25] |
| B | 8 | C18–LC/Q Exactive plus (positive/negative, scan) | – |
| C | 9 | C18–LC/Orbitrap Fusion (positive/negative, scan) | [26] |
| D | 4 | C8–LC/TQMS (positive/negative, MRM) | [27] |
| E | 2 | DEA–SFC/TQMS (positive/negative, MRM) | [28] |
| F | 2 | C18–SFC/TQMS (positive/negative, MRM) | [19] |
| G | 3 | FI/TQMS (positive, MRM) | [20] |
| Hydrophilic Metabolites | Hydrophobic Metabolites | Hydrophilic + Hydrophobic Metabolites | |
|---|---|---|---|
| 1. Number of identified metabolites from human plasma and/or mouse plasma samples using at least one analytical method | 160 | 660 | 820 |
| 2. Number of identified metabolites from both samples using ‘two or more’ methods | 111 | 291 | 402 |
| 3. Number of metabolites that were statistically significant between the human plasma and mouse plasma samples using multiple methods based on a two-sided Student’s t-test (α = 0.05) | 88 | 256 | 344 |
| 4. Number of metabolites with similar human plasma/mouse plasma levels among the methods, based on a two-sided Student’s t-test (α = 0.05) and a relative quantitative value of 1 | 82 (93.2%) | 243 (94.9%) | 325 (94.5%) |
| 5. Number of metabolites with statistically similar human plasma/mouse plasma levels among the multiple methods, using a one-way analysis of variance (ANOVA) (α = 0.05) | 40 (36.0%) | 62 (21.3%) | 102 (25.4%) |
| 6. Number of metabolites with statistically similar human plasma/mouse plasma levels among the multiple methods, ignoring one outlier method using a one-way ANOVA (α = 0.05) | 56 (50.5%) | 135 (46.4%) | 191 (47.5%) |
| Hydrophilic Metabolites | Hydrophobic Metabolites | Hydrophilic + Hydrophobic Metabolites | |
|---|---|---|---|
| 1. Number of identified metabolites from human plasma and/or mouse plasma samples by at least one analytical method | 131 | 297 | 428 |
| 2. Number of identified metabolites from both samples by ‘two or more’ multiple methods | 86 | 154 | 240 |
| 3. Number of metabolites that were statistically significant between the human plasma and mouse plasma samples from multiple methods based on a two-sided Student’s t-test (α = 0.05) | 66 | 123 | 189 |
| 4. Number of metabolites that showed similar human plasma/mouse plasma levels among the methods based on a two-sided Student’s t-test (α = 0.05) and a relative quantitative value of 1 | 60 (90.9%) | 117 (95.1%) | 177 (93.7%) |
| 5. Number of metabolites with statistically similar human plasma/mouse plasma levels among multiple methods using a one-way ANOVA (α = 0.05) | 30 (34.9%) | 49 (31.8%) | 79 (32.9%) |
| 6. Number of metabolites with statistically similar human plasma/mouse plasma levels among multiple methods, ignoring one outlier using a one-way ANOVA (α = 0.05) | 48 (55.8%) | 87 (56.5%) | 135 (56.3%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishiumi, S.; Izumi, Y.; Hirayama, A.; Takahashi, M.; Nakao, M.; Hata, K.; Saigusa, D.; Hishinuma, E.; Matsukawa, N.; Tokuoka, S.M.; et al. Comparative Evaluation of Plasma Metabolomic Data from Multiple Laboratories. Metabolites 2022, 12, 135. https://doi.org/10.3390/metabo12020135
Nishiumi S, Izumi Y, Hirayama A, Takahashi M, Nakao M, Hata K, Saigusa D, Hishinuma E, Matsukawa N, Tokuoka SM, et al. Comparative Evaluation of Plasma Metabolomic Data from Multiple Laboratories. Metabolites. 2022; 12(2):135. https://doi.org/10.3390/metabo12020135
Chicago/Turabian StyleNishiumi, Shin, Yoshihiro Izumi, Akiyoshi Hirayama, Masatomo Takahashi, Motonao Nakao, Kosuke Hata, Daisuke Saigusa, Eiji Hishinuma, Naomi Matsukawa, Suzumi M. Tokuoka, and et al. 2022. "Comparative Evaluation of Plasma Metabolomic Data from Multiple Laboratories" Metabolites 12, no. 2: 135. https://doi.org/10.3390/metabo12020135