Two Metabolomics Phenotypes of Human Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease According to Fibrosis Severity

 and
and
Abstract
:1. Introduction
2. Results
2.1. Patients Characteristics
2.2. Identification of Discriminant Metabolites
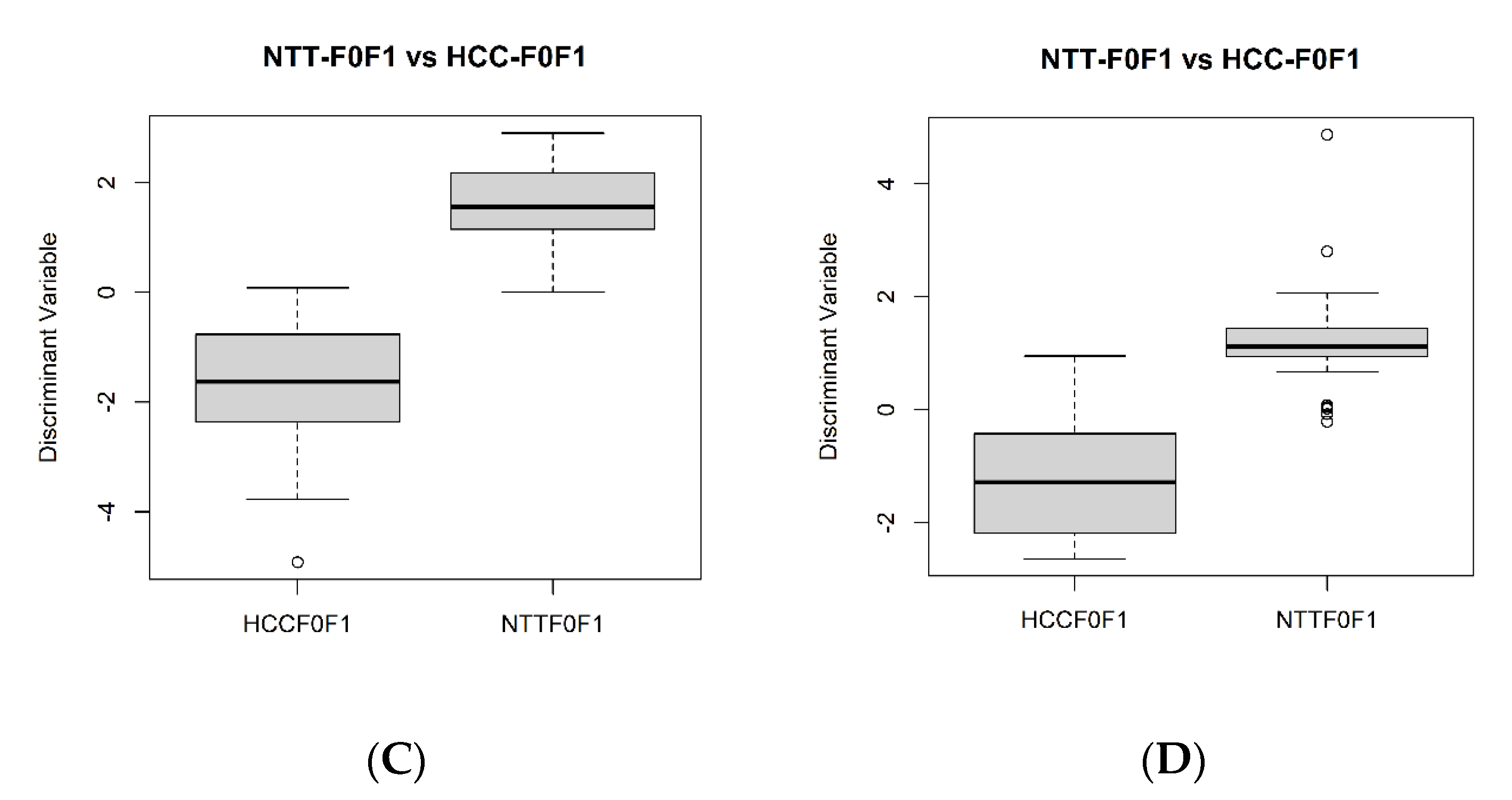
2.3. Differential Metabolites between NAFLD-HCC-F0F1 vs. NTT-F0F1
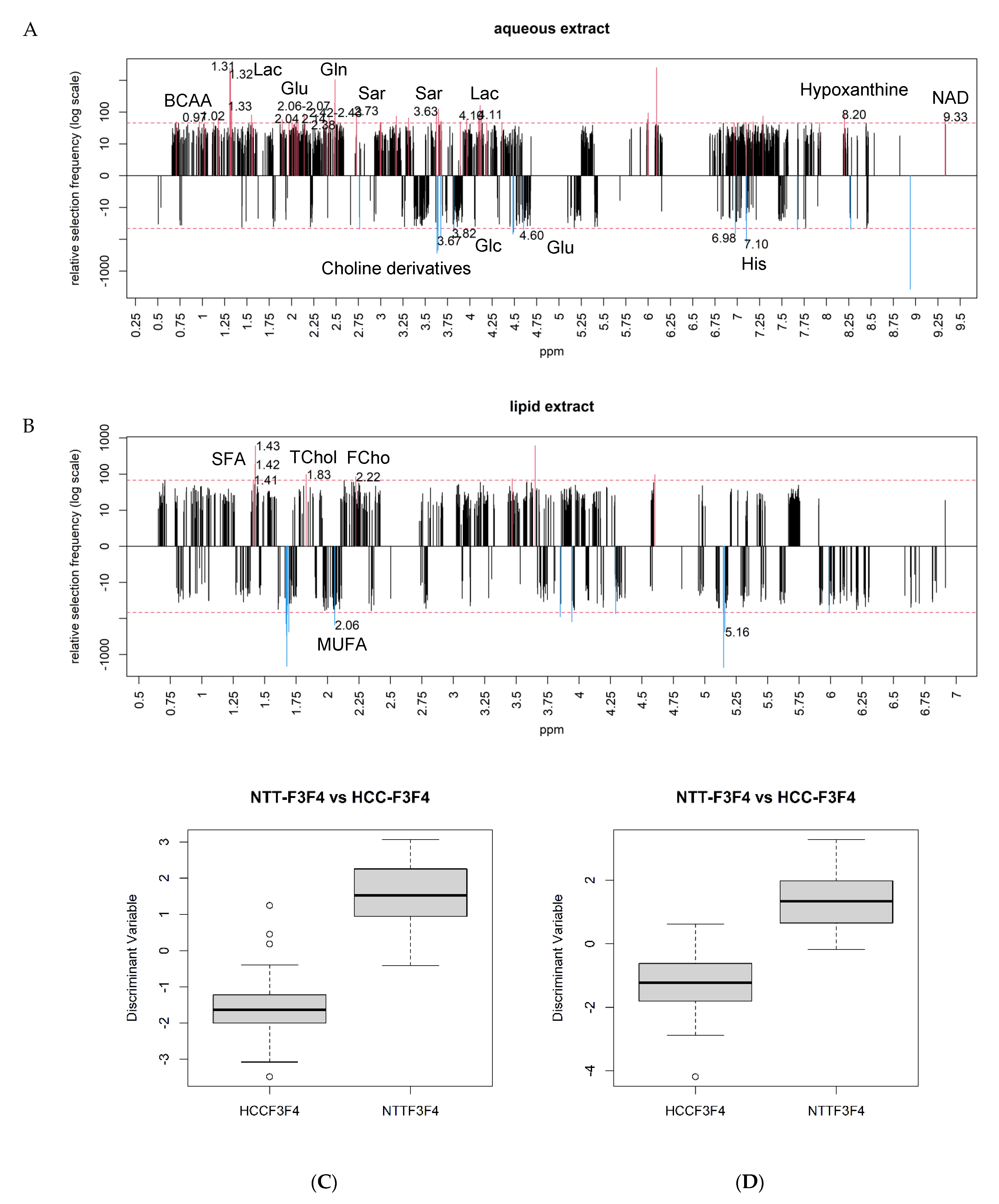
2.4. Differential Metabolites between NAFLD-HCC-F3F4 vs. NTT- F3F4
2.5. Differential Metabolites between NAFLD-HCCs according the Severity of Fibrosis
3. Discussion
3.1. Carbohydrate Metabolism in NAFLD-HCC: A Common Warburg Effect but Enhanced Neoglucogenesis in Severe Fibrosis
3.2. Preserved Antioxidant Defenses in HCC-F0F1
3.3. Enhanced Glutamine Synthetase Activity in HCC-F0F1 and Putative Involvement of the Beta-Catenin Pathway in NAFLD
3.4. BCAA Content and Possible Activation of the mTOR Pathway in HCC-F3F4
3.5. Methylation Disorders in HCC-F3F4
3.6. NAFLD-HCC in Non-Severe Fibrosis Displays a Cholinic Phenotype
3.7. Different Lipid Metabolism Reprogramming in NAFLD-HCC according to Fibrosis Severity
4. Patients and Methods
4.1. Patients and Collection of Specimens
4.2. Histology
4.3. Sample Preparation for NMR-Spectroscopy
4.4. 1H-NMR Spectroscopy
4.5. Data Processing
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HCC | hepatocellular carcinoma |
| NAFLD | non-alcoholic fatty liver disease |
| MS | metabolic syndrome |
| MAFLD | metabolic associated fatty liver disease |
| NAFLD-HCC | NAFLD associated HCC |
| NMR | nuclear magnetic resonance |
| NTT | non-tumoral tissue |
| ppm | parts per million |
| FA | fatty acids |
| AA | Aminoacids |
| Plp | Phospholipids |
| Lac | Lactate |
| Glc | glucose |
| Gln | glutamine |
| GS | glutamine synthase |
| His | histidine |
| GSx | glutathione |
| Asc A | ascorbic acid |
| PC | phosphocholine |
| PtdCho | Phosphatidylcholine |
| CK | choline kinase |
| MUFA | monounsaturated fatty acids |
| TCho | total cholesterol |
| AUC | area under curve |
| BCAA | branched chain aminoacids |
| Val | Valine |
| Leu | Leucine |
| IsoLeu | Isoleucine |
| Sar | Sarcosine |
| GNMT | Glycine N-methyltransferase |
| NAD | nicotinamide adenine dinucleotide |
| SFA | saturated fatty acids |
| DNL | de novo lipogenesis |
| FASN | fatty acid synthase |
| SCD | stearoyl coA desaturase |
| FCho | free cholesterol |
| TAG | Triacylglycerol |
| PE | phosphoethanolamine |
| PET | positron-emission tomography |
| FDG | fluorodeoxyglucose |
| ROS | reactive oxygen species |
| TCA | tricarboxylic acid |
| AAA | aromatic aminoacid |
| Gly | Glycine |
| THF | tetrahydrofolate |
| CDP PtdEth | cytidine diphosphate phosphatidylethanolamine |
| PEMT | phosphatidylethanolamine N-methyltransferase |
| MestReNova | Mestrelab Research chemistry software solutions |
| GA LDA ROC | Genetic Algorithm Linear Discriminant Analysis Receiver Operating Characteristic |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, A.; Sandhu, S.; Lai, J.-P.; Sandhu, D.S. Hepatocellular carcinoma in non-cirrhotic liver: A comprehensive review. World J. Hepatol. 2019, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J.; Neuschwander-Tetri, B.; Tiribelli, C.; Kleiner, D.E.; Brunt, E.; Bugineusi, E.; Yilmaz, Y.; Younossi, Z.; et al. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014. [Google Scholar] [CrossRef] [PubMed]
- Bedossa, P.; Poynard, T. An algorithm for the grading of activity in chronic hepatitis C. The METAVIR Cooperative Study Group. Hepatology 1996, 24, 289–293. [Google Scholar] [CrossRef]
- Ertle, J.; Dechêne, A.; Sowa, J.-P.; Penndorf, V.; Herzer, K.; Kaiser, G.; Schlaak, J.F.; Gerken, G.; Syn, W.K.; Canbay, A. Non-alcoholic fatty liver disease progresses to hepatocellular carcinoma in the absence of apparent cirrhosis. Int. J. Cancer. 2011, 128, 2436–2443. [Google Scholar] [CrossRef]
- Piscaglia, F.; Svegliati-Baroni, G.; Barchetti, A.; Pecorelli, A.; Marinelli, S.; Tiribelli, C.; Bellentani, S.; HCC-NAFLD Italian Study Group. Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: A multicenter prospective study. Hepatology 2016, 63, 827–838. [Google Scholar] [CrossRef]
- Paradis, V.; Zalinski, S.; Chelbi, E.; Guedj, N.; Degos, F.; Vilgrain, V.; Bedossa, P.; Belghiti, J. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: A pathological analysis. Hepatology 2009, 49, 851–859. [Google Scholar] [CrossRef]
- Sircana, A.; Paschetta, E.; Saba, F.; Molinaro, F.; Musso, G. Recent Insight into the Role of Fibrosis in Nonalcoholic Steatohepatitis-Related Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 1745. [Google Scholar] [CrossRef] [Green Version]
- Calderaro, J.; Couchy, G.; Imbeaud, S.; Amaddeo, G.; Letouzé, E.; Blanc, J.-F.; Laurent, C.; Hajji, Y.; Azoulay, D.; Bioulac-Sage, P.; et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J. Hepatol. 2017, 67, 727–738. [Google Scholar] [CrossRef]
- Wong, C.R.; Nguyen, M.H.; Lim, J.K. Hepatocellular carcinoma in patients with non-alcoholic fatty liver disease. World J. Gastroenterol. 2016, 37, 8294–8303. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Teilhet, C.; Morvan, D.; Joubert-Zakeyh, J.; Biesse, A.-S.; Pereira, B.; Massoulier, S.; Dechelotte, P.; Pezet, D.; Buc, E.; Lamblin, G.; et al. Specificities of Human Hepatocellular Carcinoma Developed on Non-Alcoholic Fatty Liver Disease in Absence of Cirrhosis Revealed by Tissue Extracts 1H-NMR Spectroscopy. Metabolites 2017, 7, 49. [Google Scholar] [CrossRef]
- Beyoğlu, D.; Idle, J.R. The metabolomic window into hepatobiliary disease. J. Hepatol. 2013, 59, 842–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyoğlu, D.; Imbeaud, S.; Maurhofer, O.; Bioulac-Sage, P.; Zucman-Rossi, J.; Dufour, J.-F.; Idle, J.R. Tissue metabolomics of hepatocellular carcinoma: Tumor energy metabolism and the role of transcriptomic classification. Hepatology 2013, 58, 229–238. [Google Scholar]
- Wong, C.C.-L.; Au, S.L.-K.; Tse, A.P.-W.; Xu, I.M.-J.; Lai, R.K.-H.; Chiu, D.K.-C.; Wei, L.L.; Fan, D.N.-Y.; Lo, R.C.-L.; Tsang, F.H.-C.; et al. Switching of pyruvate kinase isoform L to M2 promotes metabolic reprogramming in hepatocarcinogenesis. PLoS ONE 2014, 9, e115036. [Google Scholar] [CrossRef] [Green Version]
- Kwee, S.A.; Hernandez, B.; Chan, O.; Wong, L. Choline Kinase Alpha and Hexokinase-2 Protein Expression in Hepatocellular Carcinoma: Association with Survival. PLoS ONE 2012, 7, e46591. [Google Scholar] [CrossRef]
- Wang, D.; Moothart, D.R.; Lowy, D.R.; Qian, X. The Expression of Glyceraldehyde-3-Phosphate Dehydrogenase Associated Cell Cycle (GACC) Genes Correlates with Cancer Stage and Poor Survival in Patients with Solid Tumors. PLoS ONE 2013, 8, e61262. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.-C.; She, B.; Gao, W.-T.; Ji, Y.-H.; Xu, D.-D.; Wang, Q.-S.; Wang, S.-B. Positron-emission tomography for hepatocellular carcinoma: Current status and future prospects. World J. Gastroenterol. 2019, 25, 4682–4695. [Google Scholar] [CrossRef]
- García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial Oxidative Stress and Antioxidants Balance in Fatty Liver Disease. Hepatol. Commun. 2018, 2, 1425–1439. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Granados, B.; Morales, J.M.; Rodrigo, J.M.; Del Omo, J.; Serra, M.A.; Ferrandez, A.; Celda, B.; Monleon, D. Metabolic profile of chronic liver disease by NMR spectroscopy of human biopsies. Int. J. Mol. Med. 2011, 27, 111–117. [Google Scholar] [PubMed]
- Christa, L.; Simon, M.T.; Flinois, J.P.; Gebhadt, R.; Brechot, C.; Lasserre, C. Overexpression of glutamine synthetase in human primary liver cancer. Gastroenterology 1994, 106, 1312–1320. [Google Scholar] [CrossRef]
- Di Tommaso, L.; Franchi, G.; Park, Y.N.; Destro, A.; Morenghi, E.; Montorsi, M.; Torzilli, G.; Tommasini, M.; Terracciono, L.; Tornillo, L.; et al. Diagnostic value of HSP70, glypican 3, and glutamine synthetase in hepatocellular nodules in cirrhosis. Hepatology 2007, 45, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Osada, T.; Nagashima, I.; Tsuno, N.H.; Kitayama, J.; Nagawa, H. Prognostic significance of glutamine synthetase expression in unifocal advanced hepatocellular carcinoma. J. Hepatol. 2000, 33, 247–253. [Google Scholar] [CrossRef]
- Cadoret, A.; Ovejero, C.; Terris, B.; Souil, E.; Lévy, L.; Lamers, W.H.; Kitajewski, J.; Kahn, A.; Perret, C. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002, 1, 8293–8301. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-T.; Tsai, S.-M.; Wang, S.-N.; Lin, S.-K.; Wu, S.-H.; Chuang, S.-C.; Wu, S.-H.; Ma, H.; Tsai, L.-Y. Glutathione status in the blood and tissues of patients with virus-originated hepatocellular carcinoma. Clin. Biochem. 2007, 40, 1157–1162. [Google Scholar] [CrossRef]
- Björnson, E.; Mukhopadhyay, B.; Asplund, A.; Pristovsek, N.; Cinar, R.; Romeo, S.; Uhlen, M.; Kunos, G.; Nielsen, J.; Mardinoglu, A. Stratification of Hepatocellular Carcinoma Patients Based on Acetate Utilization. Cell Rep. 2015, 13, 2014–2026. [Google Scholar]
- Holeček, M. Branched-chain amino acids in health and disease: Metabolism, alterations in blood plasma, and as supplements. Nutr. Metab. 2018, 15, 33. [Google Scholar] [CrossRef] [Green Version]
- Ericksen, R.E.; Lim, S.L.; McDonnell, E.; Shuen, W.H.; Vadiveloo, M.; Phillip, J.W.; Ding, Z.; Kwok, R.; Lee, P.; Radda, G.K. Loss of BCAA Catabolism during Carcinogenesis Enhances mTORC1 Activity and Promotes Tumor Development and Progression. Cell Metab. 2019, 29, 1151–1165. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Li, C.; Nie, X.; Feng, X.; Chang, W.; Yue, Y.; Tang, H.; Deng, F. Metabonomic studies of human hepatocellular carcinoma using high-resolution magic-angle spinning 1H NMR spectroscopy in conjunction with multivariate data analysis. J. Proteom. Res. 2007, 6, 2605–2614. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, S.; Yan, W.; Xia, Y.; Chen, X.; Wang, W.; Zhang, J.; Gao, C.; Peng, C.; Yan, F.; et al. Branched Chain Amino Acids Cause Liver Injury in Obese/Diabetic Mice by Promoting Adipocyte Lipolysis and Inhibiting Hepatic Autophagy. EBioMedicine 2016, 13, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Miyanishi, K.; Kobune, M.; Kawano, Y.; Hoki, T.; Kubo, T.; Hayashi, T.; Sato, T.; Sato, Y.; Takimoto, R.; et al. Increased Hepatic Oxidative DNA Damage in Patients with Nonalcoholic Steatohepatitis Who Develop Hepatocellular Carcinoma. J. Gastroenterol. 2013, 48, 1249–1258. [Google Scholar] [CrossRef]
- Simile, M.M.; Latte, G.; Feo, C.F.; Feo, C.F.; Feo, F.; Calvisi, D.F.; Pascale, R.M. Alterations of methionine metabolism in hepatocarcinogenesis: The emergent role of glycine N-methyltransferase in liver injury. Ann. Gastroenterol. 2018, 31, 552–560. [Google Scholar] [CrossRef]
- Lin, W.-C.; Chakraborty, A.; Huang, S.-C.; Wang, P.-Y.; Hsiek, Y.-J.; Chien, K.-Y.; Lee, Y.-H.; Chang, C.-C.; Tang, H.-Y.; Lin, Y.-T.; et al. Histidine-Dependent Protein Methylation Is Required for Compartmentalization of CTP Synthase. Cell Rep. 2018, 24, 2733–2745. [Google Scholar] [CrossRef] [Green Version]
- Glunde, K.; Bhujwalla, Z.M.; Ronen, S.M. Choline metabolism in malignant transformation. Nat. Rev. Cancer. 2011, 11, 835–848. [Google Scholar] [CrossRef] [Green Version]
- Kiss, Z. Regulation of Mitogenesis by Water-Soluble Phospholipid Intermediates. Cell. Signal. 1999, 11, 149–157. [Google Scholar] [CrossRef]
- Lin, X.-M.; Hu, L.; Gu, J.; Wang, R.-Y.; Li, L.; Tang, J.; Zhang, B.-H.; Yan, X.-Z.; Zhu, Y.-J.; Hu, C.-L.; et al. Choline Kinase α Mediates Interactions Between the Epidermal Growth Factor Receptor and Mechanistic Target of Rapamycin Complex 2 in Hepatocellular Carcinoma Cells to Promote Drug Resistance and Xenograft Tumor Progression. Gastroenterology 2017, 152, 1187–1202. [Google Scholar] [CrossRef]
- Kwee, S.A.; Wong, L.; Chan, O.T.M.; Kalathil, S.; Tsai, N. PET/CT with 18F Fluorocholine as an Imaging Biomarker for Chronic Liver Disease: A Preliminary Radiopathologic Correspondence Study in Patients with Liver Cancer. Radiology 2018, 287, 294–302. [Google Scholar] [CrossRef] [Green Version]
- Van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacob, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta. Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef]
- Tessitore, L.; Marengo, B.; Vance, D.E.; Papotti, M.; Mussa, A.; Daidone, M.G.; Costa, A. Expression of phosphatidylethanolamine N-methyltransferase in human hepatocellular carcinomas. Oncology 2003, 65, 152–158. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo Lipogenesis is a Distinct Characteristic of Individuals with Nonalcoholic Fatty Liver Disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Budhu, A.; Roessler, S.; Zhao, X.; Zu, X.; Forgues, M.; Ji, J.; Karoly, E.; Qin, L.-X.; Ye, Q.-H.; Jia, H.-L.; et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology 2013, 144, 1066–1075.e1. [Google Scholar] [CrossRef] [Green Version]
- Buechler, C.; Aslanidis, C. Role of lipids in pathophysiology, diagnosis and therapy of hepatocellular carcinoma. Biochim. Biophys. Acta. Mol. Cell. Biol. Lipids 2020, 1865, 158658. [Google Scholar] [CrossRef]
- Hall, Z.; Chiarugi, D.; Charidemou, E.; Lesli, J.; Scott, E.; Pellegrinet, L.; Allison, M.; Mocciaro, G.; Anstee, Q.M.; Evan, G.I.; et al. Lipid remodelling in hepatocyte proliferation and hepatocellular carcinoma. Hepatology 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Che, L.; Pilo, M.G.; Cigliano, A.; Latte, G.; Simile, M.M.; Ribback, R.; Dombrowski, F.; Evert, M.; Chan, X.; Calvisi, D.F. Oncogene dependent requirement of fatty acid synthase in hepatocellular carcinoma. Cell Cycle 2017, 16, 499–507. [Google Scholar] [CrossRef]
- Rousselet, M.C.; Michalak, S.; Dupré, F.; Croué, A.; Bedossa, P.; Saint-André, J.-P.; Calès, P. Sources of variability in histological scoring of chronic viral hepatitis. Hepatology 2005, 41, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.W.-M. Metabolite profiling by one- and two-dimensional NMR analysis of complex mixtures. Prog. Nucl. Magn. Reson. Spectrosc. 1996, 28, 161–219. [Google Scholar] [CrossRef]
- Human Metabolome Database: Browsing Metabolites. Available online: https://hmdb.ca/metabolites (accessed on 3 March 2018).
- Goldberg, D.E. Genetic Learning in Optimization, Search and Machine Learning; Addisson Wesley: Boston, MA, USA, 1994. [Google Scholar]
- Reeves, C.; Rowe, J.E. Genetic Algorithms: Principles and Perspectives: A Guide to GA Theory; Springer Science & Business Media: Berlin, Germany, 2002. [Google Scholar]
- Hastie, T.; Tibshirani, R.; Friedman, J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction, 2nd ed.; Springer: New York, NY, USA, 2009. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group F0F1 N = 26 | Group F3F4 N = 26 | p-Value | ||
| PARAMETERS | ||||
| Gender (M:F) | 21:5 | 24:2 | ns | |
| Age in years (mean ± SD) | 69.9 ± 10.7 | 70.5 ± 5.9 | ns | |
| CLINICAL AND BIOLOGICAL DATA | ||||
| Body Mass Index (missing data n = 3) | Normal | 4 | 4 | ns |
| Overweight | 11 | 10 | ns | |
| Obesity | 10 | 10 | ns | |
| Diabetes (missing data n = 5) | Yes | 16 | 19 | ns |
| No | 7 | 5 | ns | |
| Tobacco (missing data n = 6) | Yes | 7 | 11 | ns |
| No | 16 | 12 | ns | |
| Blood Alpha-Foeto-Protein (missing data n = 4) | <20ng/mL | 18 | 23 | ns |
| 20–200ng/mL | 2 | 0 | ns | |
| 200–1000ng/mL | 1 | 1 | ns | |
| >1000ng/mL | 2 | 1 | ns | |
| HISTOLOGICAL DATA | ||||
| Degree of steatosis in NTT | No | 5 | 1 | ns |
| Low (5–33%) | 5 | 6 | ns | |
| Moderate (33–66%) | 11 | 14 | ns | |
| Severe (>66%) | 5 | 5 | ns | |
| Tumor Differentiation (WHO) (missing data n = 1) | Well | 11 | 12 | ns |
| Moderate | 14 | 12 | ns | |
| Poor | 1 | 1 | ns | |
| Metabolites | Abbreviations | Chemical Shifts (ppm) | |
| AqueousPhase | |||
| Carbohydrates/TCA cycle derivatives | Lactate | Lac | 1.31–1.33/4.10–4.11 |
| Glucose | Glc | 3.39/3.46/3.51/3.75/4.63/5.22 | |
| Glycogen | Gly | 5.38–5.43 | |
| Amino Acids and derivatives | Glutamine | Gln | 2.14/2.44 |
| Glutamate | Glu | 2.04/2.34 | |
| Glutathione | GSx | 2.15/2.54/2.97/3.78 | |
| Leucine | Leu | 0.93–0.97 | |
| Isoleucine | Isoleu | 0.93–0.97 | |
| Valine | Val | 1.02-1.04/2.26 | |
| Histidine | His | 7.07–7.11 | |
| Sarcosine | Sar | 2.70–2.73 | |
| Nucleotides derivatives | Hypoxanthine | 8.20 | |
| Nicotinamide Adenine Dinucleotide | NAD | 9.33 | |
| Vitamins | Ascorbic acid | Asc A | 4.50 |
| Phospholipids Derivatives | Phosphocholine | PC | 3.22 |
| Choline derivatives | 3.62–3.68 | ||
| Lipid Phase | |||
| Phospholipids derivatives | Phosphoethanolamine | PE | 3.05–3.07/3.13 |
| Cholesterol | Total cholesterol | TChol | 0.69/0.93–0.94/1.01/1.52–1.54/2.19/2.25/3.49/3.57/3.89 |
| Free cholesterol | FChol | 0.94/1.07/1.50/1.79/2.22/3.45/3.48/3.57 | |
| Fatty acids | Saturated FA (CH2)n | SFA | 1.24–1.44 |
| Monounsaturated FA –CH2CH= | MUFA | 2.02–2.12 | |
| Triacylglycerides | TAG | TAG | 4.14–4.34 5.26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buchard, B.; Teilhet, C.; Abeywickrama Samarakoon, N.; Massoulier, S.; Joubert-Zakeyh, J.; Blouin, C.; Reynes, C.; Sabatier, R.; Biesse-Martin, A.-S.; Vasson, M.-P.; et al. Two Metabolomics Phenotypes of Human Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease According to Fibrosis Severity. Metabolites 2021, 11, 54. https://doi.org/10.3390/metabo11010054
Buchard B, Teilhet C, Abeywickrama Samarakoon N, Massoulier S, Joubert-Zakeyh J, Blouin C, Reynes C, Sabatier R, Biesse-Martin A-S, Vasson M-P, et al. Two Metabolomics Phenotypes of Human Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease According to Fibrosis Severity. Metabolites. 2021; 11(1):54. https://doi.org/10.3390/metabo11010054
Chicago/Turabian StyleBuchard, Benjamin, Camille Teilhet, Natali Abeywickrama Samarakoon, Sylvie Massoulier, Juliette Joubert-Zakeyh, Corinne Blouin, Christelle Reynes, Robert Sabatier, Anne-Sophie Biesse-Martin, Marie-Paule Vasson, and et al. 2021. "Two Metabolomics Phenotypes of Human Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease According to Fibrosis Severity" Metabolites 11, no. 1: 54. https://doi.org/10.3390/metabo11010054