Loss of SMYD1 Results in Perinatal Lethality via Selective Defects within Myotonic Muscle Descendants

Abstract
:1. Introduction
2. Methods and Materials
2.1. Generation of Myog-Cre/Smyd1flox Mice
2.2. Mating Scheme
2.3. PCR Genotyping
2.4. Recombination Assays
2.5. In Situ Hybridization
2.6. Protein/Immunohistochemistry
2.7. End-Point RT-PCR
2.8. RT-qPCR
2.9. RNA In Situ Hybridization
3. Results and Discussion
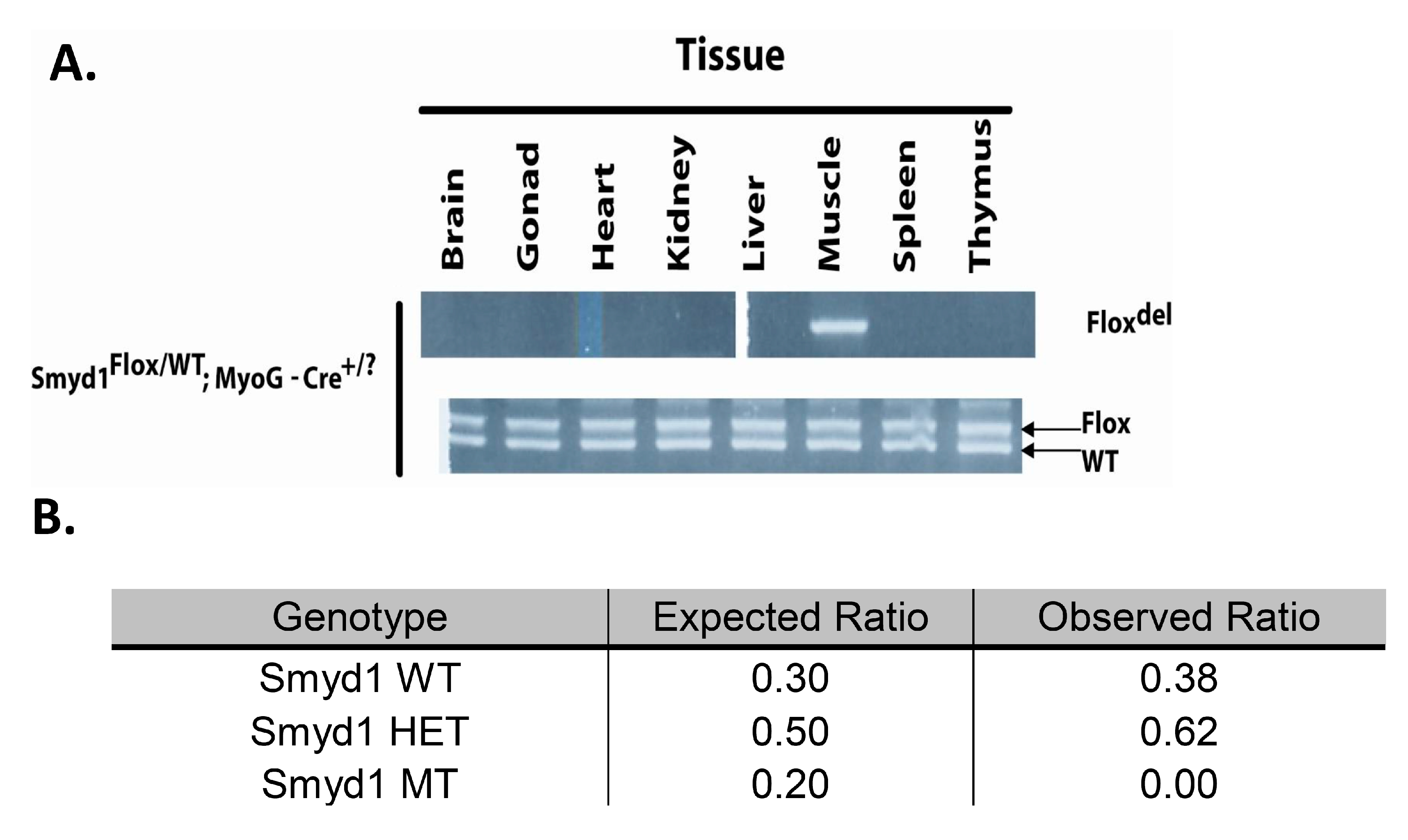
3.1. MyoG-Cre Deletion is Specific to Skeletal Muscle
3.2. MyoG-Cre-Mediated Smyd1 CKO Results in Perinatal Lethality
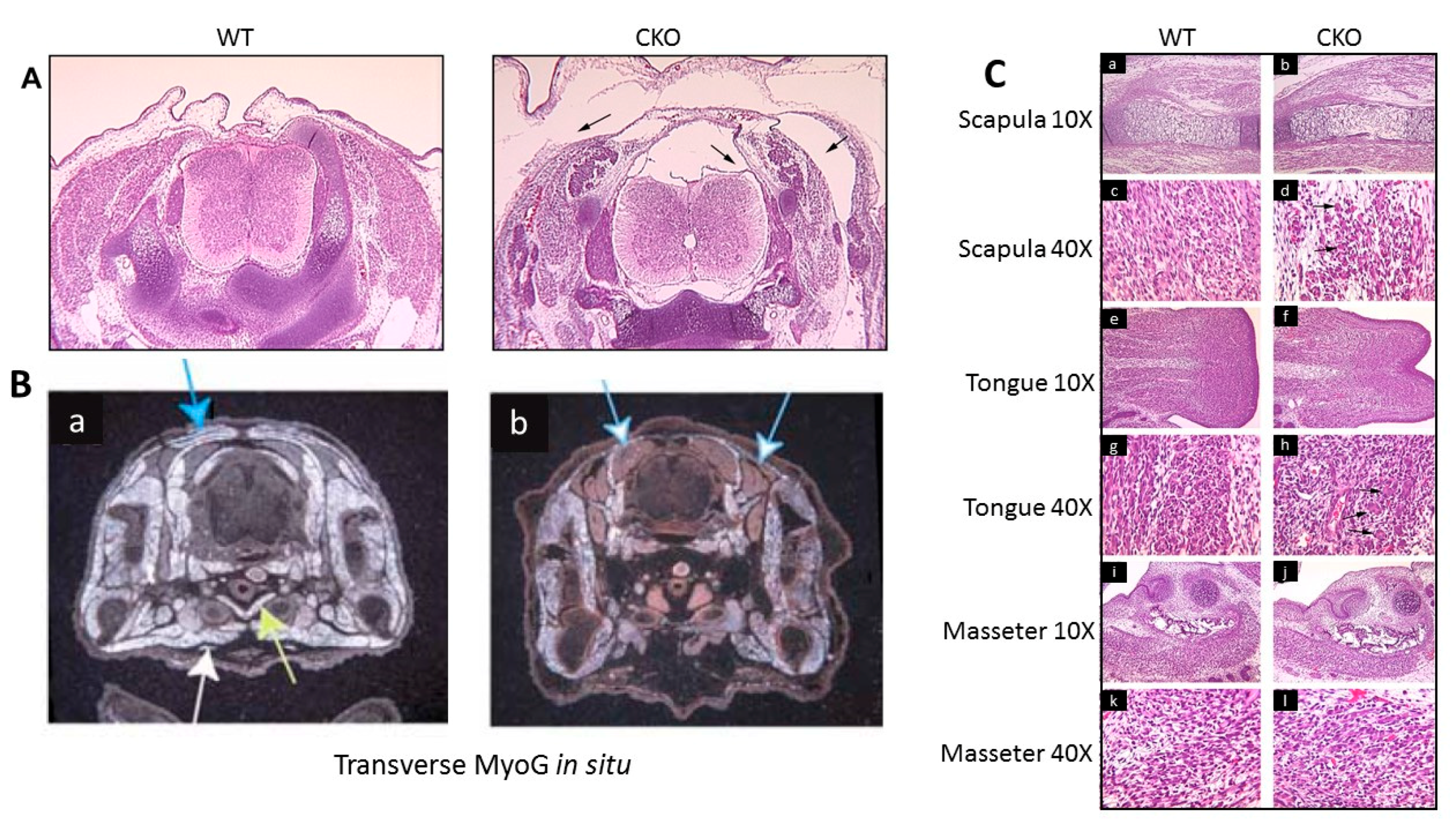
3.3. Skeletal Muscle Development in the MyoG-Cre-Induced Smyd1 CKO is Significantly Derailed
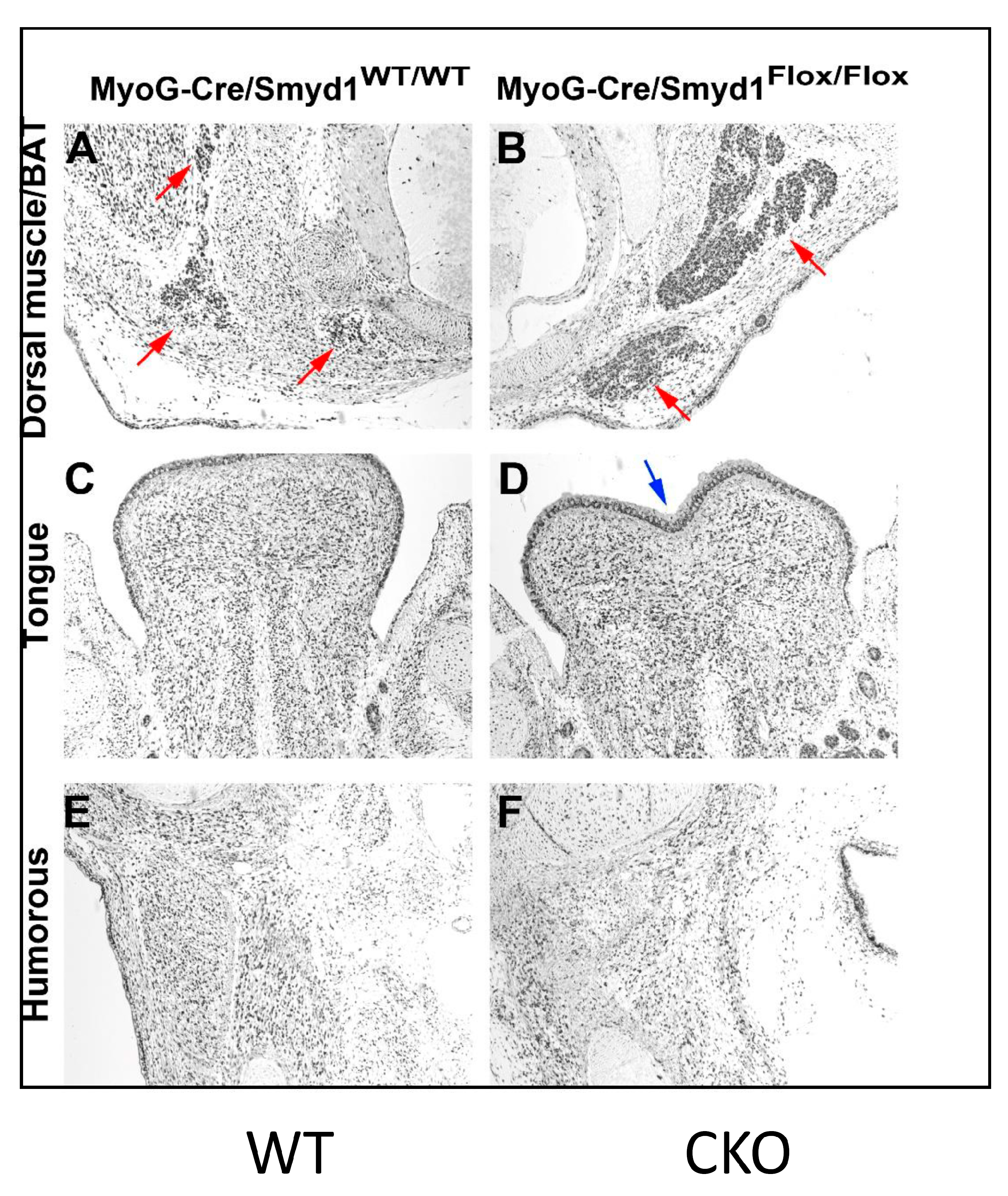
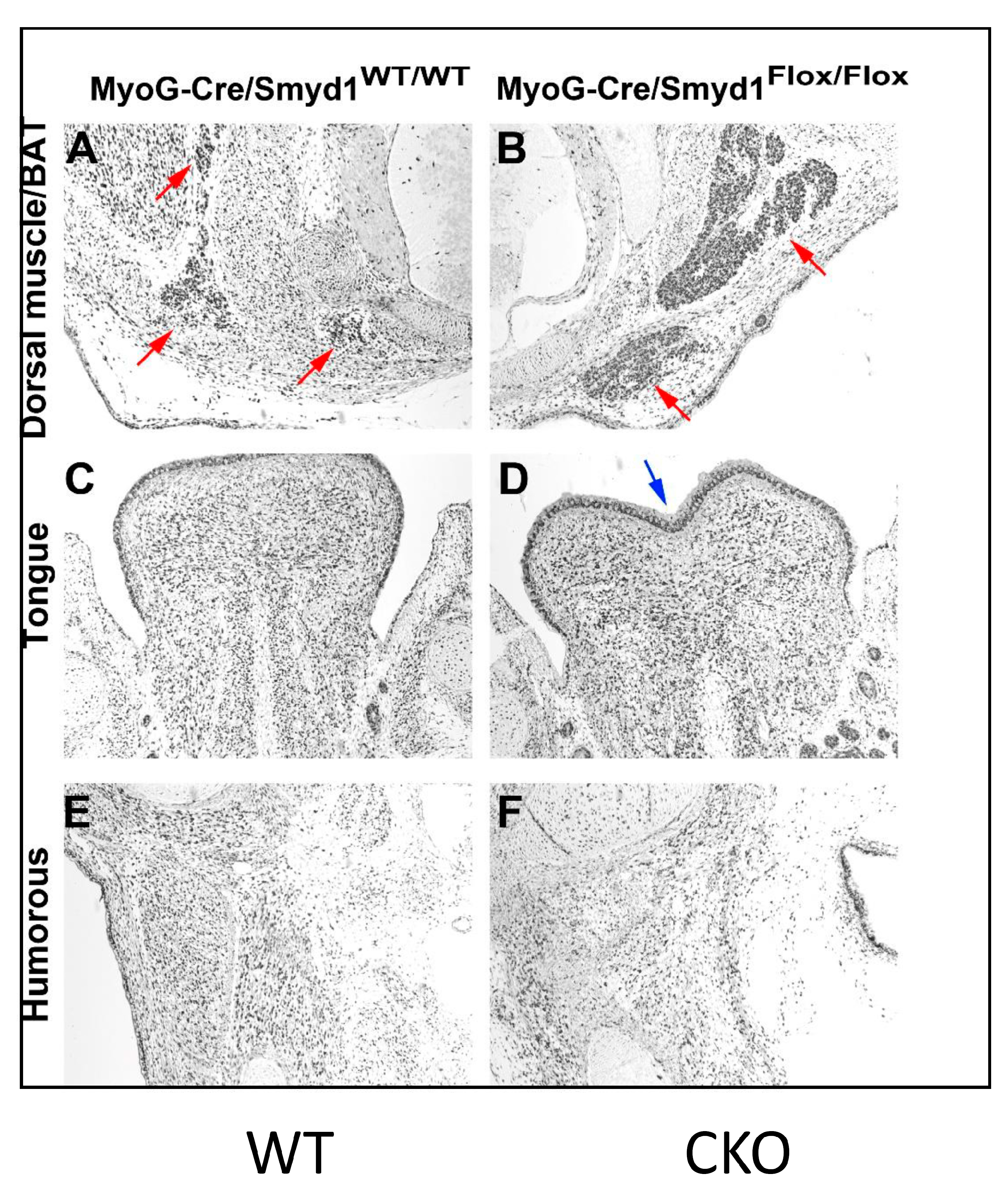
3.4. MyoG-Cre-Induced Smyd1 CKOs Exhibit Proliferative Defects and Non-Autonomous Deposits of Brown Adipose Tissue
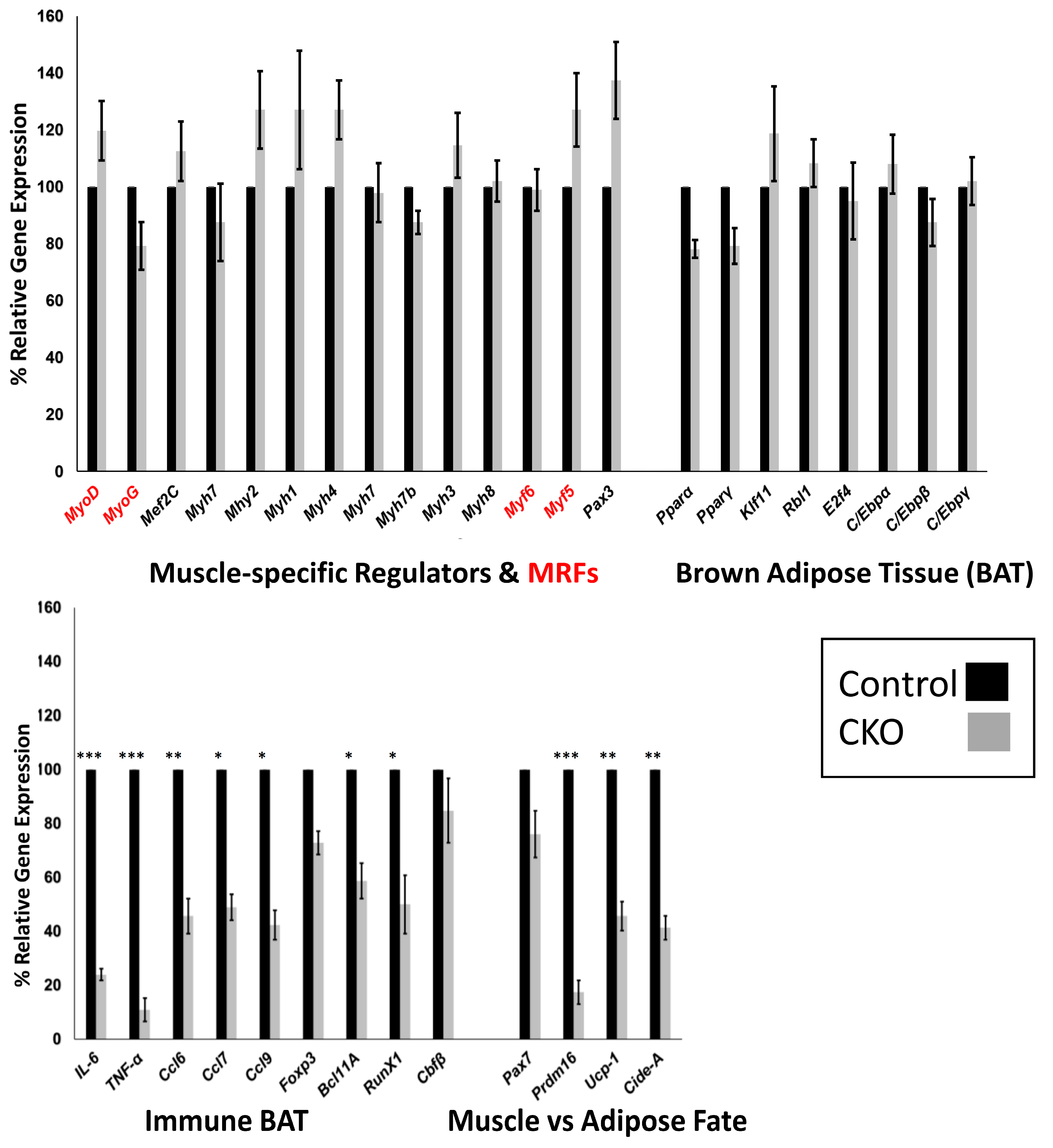
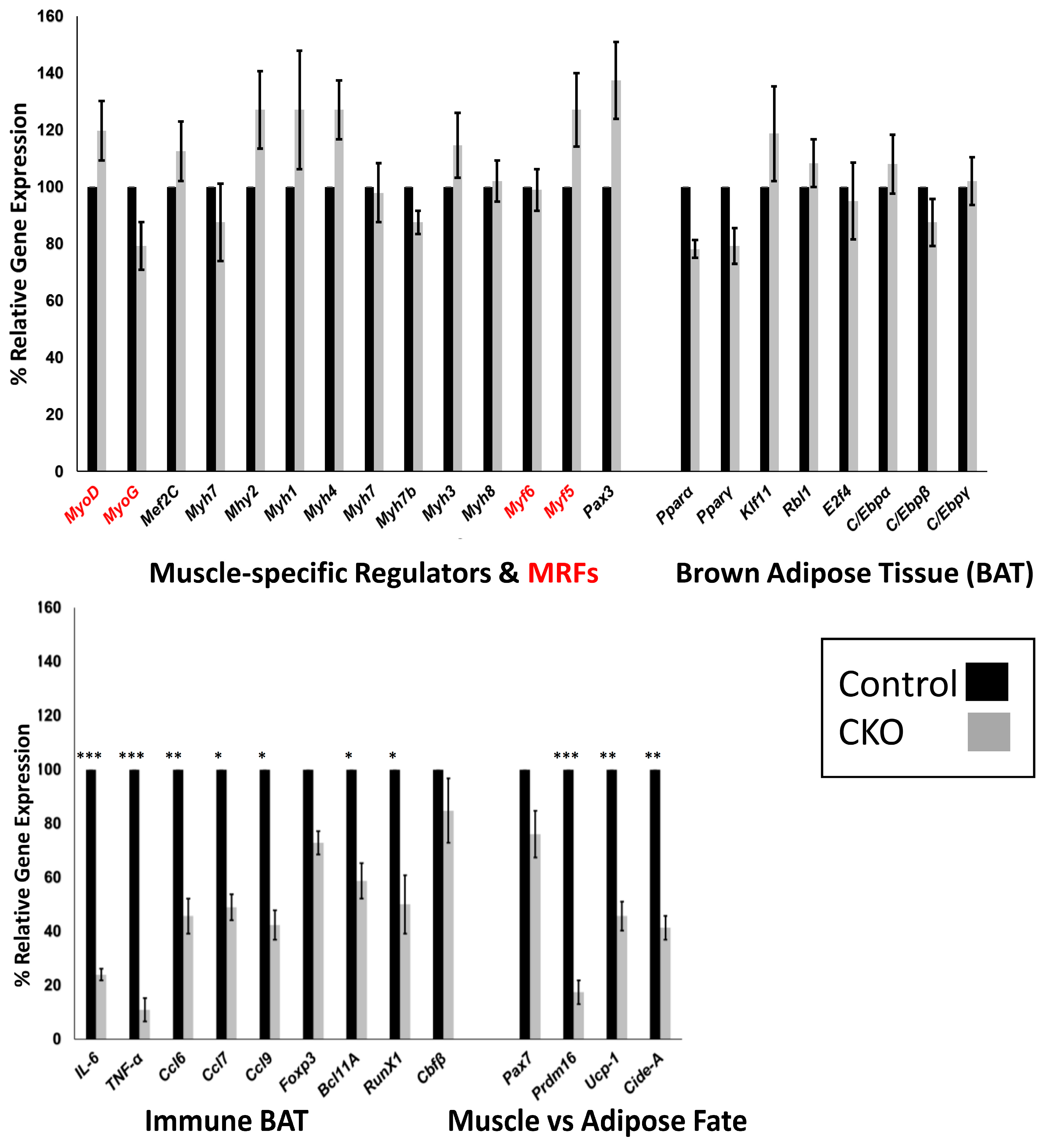
3.5. Skeletal Muscle SMYD1 Deficiency Does not Affect Transcript Levels of Myogenic Regulatory Factors
3.6. Central Regulators of BAT are Unaffected by SMYD1 CKO
3.7. SMYD1 Regulates Immune Factors Critical for BAT Development and Physiology
3.8. SMYD1 as a Potential Determinant of Muscle vs. Adipose Fate
3.9. SMYD1 Controls Specification of Muscle Fiber-Type in Rare Adult Survivors
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
References
- Buckingham, M.; Rigby, P.W. Gene regulatory networks and transcriptional mechanisms that control myogenesis. Dev. Cell 2014, 28, 225–238. [Google Scholar] [CrossRef]
- Chal, J.; Pourquié, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, P.D.; Pierce, S.A.; Sims, R.J.; Yamagishi, H.; Weihe, E.K.; Harriss, J.V.; Maika, S.D.; Kuziel, W.A.; King, H.L.; Olson, E.N.; et al. Bop encodes a muscle-restricted protein containing MYND and SET domains and is essential for cardiac differentiation and morphogenesis. Nat. Genet. 2002, 31, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Franklin, S.; Kimball, T.; Rasmussen, T.L.; Rosa-Garrido, M.; Chen, H.; Tran, T.; Miller, M.R.; Gray, R.; Jiang, S.; Ren, S.; et al. The chromatin-binding protein Smyd1 restricts adult mammalian heart growth. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1234–H1247. [Google Scholar] [CrossRef] [PubMed]
- Tracy, C.; Warren, J.S.; Szulik, M.; Wang, L.; Garcia, J.; Makaju, A.; Russell, K.; Miller, M.; Franklin, S. The Smyd family of methyltransferases: Role in cardiac and skeletal muscle physiology and pathology. Curr. Opin. Physiol. 2018, 1, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Garrido, M.; Chapski, D.J.; Vondriska, T.M. Epigenomes in Cardiovascular Disease. Circ. Res. 2018, 122, 1586–1607. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhong, Y.; Wang, Z.; Gao, J.; Xu, J.; Chu, W.; Zhang, J.; Fang, S.; Du, S.J. Smyd1b is required for skeletal and cardiac muscle function in zebrafish. Mol. Biol. Cell 2013, 24, 3511–3521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, X.; Rotllant, J.; Li, H.; De Deyne, P.; Du, S.J. SmyD1, a histone methyltransferase, is required for myofibril organization and muscle contraction in zebrafish embryos. Proc. Natl. Acad. Sci. USA 2006, 103, 2713–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Niu, Z.; Yu, W.; Qian, Y.; Wang, Q.; Li, Q.; Yi, Z.; Luo, J.; Wu, X.; Wang, Y.; et al. SMYD1, the myogenic activator, is a direct target of serum response factor and myogenin. Nucl. Acids Res. 2009, 37, 7059–7071. [Google Scholar] [CrossRef]
- Fishman, M.C.; Katus, H.A.; Strähle, U.; Rottbauer, W. The myosin-interacting protein SMYD1 is essential for sarcomere organization. J. Cell Sci. 2011, 124, 3127–3136. [Google Scholar] [Green Version]
- Rasmussen, T.L.; Ma, Y.; Park, C.Y.; Harriss, J.; Pierce, S.A.; Dekker, J.D.; Valenzuela, N.; Srivastava, D.; Schwartz, R.J.; Stewart, M.D.; et al. Smyd1 facilitates heart development by antagonizing oxidative and ER stress responses. PLoS ONE 2015, 10, e0121765. [Google Scholar] [CrossRef]
- Nagandla, H.; Lopez, S.; Yu, W.; Rasmussen, T.L.; Tucker, H.O.; Schwartz, R.J.; Stewart, M.D. Defective myogenesis in the absence of the muscle-specific lysine methyltransferase SMYD1. Dev. Biol. 2016, 410, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Stewart, M.D.; Lopez, S.; Nagandla, H.; Soibam, B.; Benham, A.; Nguyen, J.; Valenzuela, N.; Wu, H.J.; Burns, A.R.; Rasmussen, T.L.; et al. Mouse myofibers lacking the SMYD1 methyltransferase are susceptible to atrophy, internalization of nuclei and myofibrillar disarray. Dis. Model. Mech. 2016, 9, 347–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinterberger, T.J.; Sassoon, D.A.; Rhodes, S.J.; Konieczny, S.F. Expression of the muscle regulatory factor MRF4 during somite and skeletal myofiber development. Dev. Biol. 1991, 147, 144–156. [Google Scholar] [CrossRef]
- Southard, S.; Low, S.; Li, L.; Rozo, M.; Harvey, T.; Fan, C.M.; Lepper, C. A Series of Cre-ERT2 Drivers for Manipulation of the Skeletal Muscle Lineage. Genesis 2014, 52, 759–770. [Google Scholar] [CrossRef]
- Sassoon, D.; Lyons, G.; Wright, W.E.; Lin, V.; Lassar, A.; Weintraub, H.; Buckingham, M. Expression of two myogenic regulatory factors myogenin and MyoD1 during mouse embryogenesis. Nature 1989, 341, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Arnold, M.A.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Regulation of skeletal muscle sarcomere integrity and postnatal muscle function by Mef2c. Mol. Cell Biol. 2007, 27, 8143–8151. [Google Scholar] [CrossRef]
- Li, S.; Czubryt, M.P.; McAnally, J.; Bassel-Duby, R.; Richardson, J.A.; Wiebel, F.F.; Nordheim, A.; Olson, E.N. Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue-specific gene deletion in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 1082–1087. [Google Scholar] [CrossRef] [Green Version]
- McFadden, D.G.; Charité, J.; Richardson, J.A.; Srivastava, D.; Firulli, A.B.; Olson, E.N. A GATA-dependent right ventricular enhancer controls dHAND transcription in the developing heart. Development 2000, 127, 5331–5341. [Google Scholar]
- Traut, W.; Endl, E.; Scholzen, T.; Gerdes, J.; Winking, H. The temporal and spatial distribution of the proliferation associated Ki-67 protein during female and male meiosis. Chromosoma 2002, 111, 156–164. [Google Scholar] [CrossRef]
- Rosen, B.E.; Spleigleman, B.M. What we are talking about when we talk about fat. Cell 2013, 156, 20–44. [Google Scholar] [CrossRef] [PubMed]
- Zammit, P.S. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin. Cell Dev. Biol. 2017, 72, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucl. Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Fortuny, A.; Bragg, L.; Cossu, G.; Roostalu, U. MCAM contributes to the establishment of cell autonomous polarity in myogenic and chondrogenic differentiation. Biol. Open 2017, 6, 1592–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Q.; Zhang, J.; Wang, X.; Liu, Y.; He, R.; Liu, X.; Wang, F.; Feng, J.; Yang, D.; Wang, Z.; et al. The signaling receptor MCAM coordinates apical-basal polarity and planar cell polarity during morphogenesis. Nat. Commun. 2017, 8, 15279–15284. [Google Scholar] [CrossRef]
- Du, S.J.; Tan, X.; Zhang, J. SMYD proteins: Key regulators in skeletal and cardiac muscle development and function. Anat. Rec. 2014, 297, 1650–1662. [Google Scholar] [CrossRef]
- Kajimura, S.; Seale, P.; Spiegelman, B.M. Transcriptional control of brown fat development. Cell Metab. 2010, 11, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid- activated transcription factor. Cell 1994, 79, 1147–1156. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Spiegelman, B.M. Transcriptional activation of adipogenesis. Curr. Opin. Cell Biol. 1999, 11, 689–694. [Google Scholar] [CrossRef]
- Kajimura, S.; Seale, P.; Kubota, K.; Lunsford, E.; Frangioni, J.V.; Gygi, S.P.; Spiegelman, B.M. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature 2009, 460, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Ehei, A.; Medrikova, D.; Herzig, S. Immune cells and metabolic dysfunction. Semin. Immunopathol. 2011, 36, 13–25. [Google Scholar]
- Nguyen, K.D.; Qiu, Y.; Cui, X.; Goh, Y.P.; Mwangi, J.; David, T.; Chawla, A. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 2011, 480, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Thacker, R.I.; Hall, B.E.; Kong, R.; Granneman, J.G. Exploring the activated adipogenic niche: Interactions of macrophages and adipocyte progenitors. Cell Cycle 2014, 13, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.; Nguyen, K.D.; Odegaard, J.I.; Cui, X.; Tian, X.; Locksley, R.M.; Chawla, A. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell 2014, 157, 1292–1308. [Google Scholar] [CrossRef]
- Kalin, S.; Becker, M.; Ott, V.B.; Serr, I.; Hosp, F.; Mollah, M.M.; Keipert, S.; Lamp, D.; Rohner-Jeanrenaud, F.; Flynn, V.K.; et al. A Stat6/Pten axis links regulatory T cells with adipose tissue function. Cell Metab. 2017, 26, 475–492. [Google Scholar] [CrossRef]
- Medrikova, D.; Sijmonsma, T.P.; Sowodniok, K.; Richards, D.M.; Delacher, M.; Sticht, C.; Gretz, N.; Schafmeier, T.; Feuerer, M.; Herzig, S. Brown adipose tissue harbors a distinct sub-population of regulatory T cells. PLoS ONE 2015, 10, E0118534. [Google Scholar] [CrossRef] [PubMed]
- Rudra, D.; Egawa, T.; Chong, M.M.W.; Treuting, P.; Littman, D.R.; Rudensky, A.Y. Runx-CBFβ complexes control Foxp3 expression in regulatory T cells. Nat. Immunol. 2009, 10, 1170–1177. [Google Scholar] [CrossRef]
- Bruce, C.R.; Dyck, D.J. Cytokine regulation of skeletal muscle fatty acid metabolism: Effect of interleukin-6 and tumor necrosis factor-alpha. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E616–E621. [Google Scholar] [CrossRef]
- Chen, S.E.; Jin, B.; Li, Y.P. TNF-alpha regulates myogenesis and muscle regeneration by activating p38 MAPK. Am. J. Physiol. Cell Physiol. 2007, 292, 1660–1671. [Google Scholar] [CrossRef]
- Kandarian, S.C.; Jackman, R.W. Intracellular signaling during skeletal muscle atrophy. Muscle Nerve 2006, 33, 155–165. [Google Scholar] [CrossRef]
- Baeza-Raja, B.; Munoz-Canoves, P. p38 MAPK-induced nuclear factor-kappa B activity is required for skeletal muscle differentiation: Role of interleukin-6. Mol. Biol. Cell 2004, 15, 2013–2026. [Google Scholar] [CrossRef]
- Peirce, V.; Carobbio, S.; Vidal-Puig, A. The different shades of fat. Nature 2014, 510, 76–83. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef]
- Atit, R.; Sgaier, S.K.; Mohamed, O.A.; Taketo, M.M.; Dufort, D.; Joyner, A.L.; Niswander, L.; Conlon, R.A. Beta-catenin activation is necessary and sufficient to specify the dorsal dermal fate in the mouse. Dev. Biol. 2006, 296, 164–176. [Google Scholar] [CrossRef]
- Ben-Yair, R.; Kalcheim, C. Lineage analysis of the avian dermomyotome sheet reveals the existence of single cells with both dermal and muscle progenitor fates. Development 2005, 132, 689–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepper, C.; Fan, C.M. Inducible lineage tracing of Pax7-descendant cells reveals embryonic origin of adult satellite cells. Genesis 2010, 48, 424–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, G.; Wang, G.; Diao, Y.; Long, Y.; Fu, X.; Weng, M.; Zhou, L.; Sun, K.; Cheung, T.H.; Ip, N.Y.; et al. A molecular switch regulating cell fate choice between muscle progenitor cells and Brown adipocytes. Dev. Cell 2017, 41, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Relaix, F. PAX3 and PAX7 as upstream regulators of myogenesis. Semin. Cell Dev. Biol. 2015, 44, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, H.M.; Golozoubova, V.; Cannon, B.; Nedergaard, J. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 2009, 9, 203–209. [Google Scholar] [CrossRef]
- Abreu-Vieira, G.; Fischer, A.W.; Mattsson, C.; De Jong, J.M.; Shabalina, I.G.; Rydén, M.; Laurencikiene, J.; Arner, P.; Cannon, B.; Nedergaard, J.; et al. Cidea improves the metabolic profile through expansion of adipose tissue. Nat. Commun. 2015, 6, 7433–7439. [Google Scholar] [CrossRef] [PubMed]
- Kalcheim, C. Epithelial-mesenchymal transitions during neural crest and somite development. J. Clin. Med. 2016, 5, 1. [Google Scholar] [CrossRef]
- Borensztein, M.; Viengchareun, S.; Montarras, D.; Journot, L.; Binart, N.; Lombès, M.; Dandolo, L. Double Myod and Igf2 inactivation promotes brown adipose tissue development by increasing Prdm16 expression. FASEB J. 2012, 26, 4584–4591. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Rathbun, G.; Tucker, H. Smyd1C Mediates CD8 T Cell Death via Regulation of Bcl2-Mediated Restriction of outer Mitochondrial Membrane Integrity. J. Cell Signal. 2017, 3, 163–173. [Google Scholar] [CrossRef]
- Agbulut, O.; Noirez, P.; Beaumont, F.; Butler-Browne, G. Myosin heavy chain isoforms in postnatal muscle development of mice. Biol. Cell 2003, 95, 399–406. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Okutsu, M.; Akhtar, Y.N.; Lira, V.A. Regulation of exercise-induced fiber type transformation, mitochondrial biogenesis, and angiogenesis in skeletal muscle. J. Appl. Physiol. 2011, 110, 264–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuliani, C.; Sazzini, M.; Bacalini, M.G.; Pirazzini, C.; Marasco, E.; Fontanesi, E.; Franceschi, C.; Luiselli, D.; Garagnani, P. Epigenetic variability across human populations: A focus on DNA methylation profiles of the KRTCAP3, MAD1L1 and BRSK2 genes. Genome Biol. Evol. 2016, 8, 2760–2773. [Google Scholar] [CrossRef] [PubMed]
- Hawley, J.A.; Holloszy, J.O. Exercise: It’s the real thing! Nutr. Rev. 2009, 67, 172–178. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age (DPC) | Smyd1 MT | Smyd1 HET | Smyd1 WT | Total |
|---|---|---|---|---|
| 9.5 | 5 | 11 | 6 | 22 |
| 12.5 | 4 | 9 | 10 | 23 |
| 13.5 | 2 | 3 | 3 | 8 |
| 14.5 | 0 | 4 | 0 | 4 |
| 15.5 | 6 | 11 | 8 | 25 |
| 16.5 | 6 | 5 | 2 | 13 |
| 17.5 | 2 | 4 | 3 | 9 |
| Total | 25 | 47 | 32 | 104 |
| Expected Ratio | 0.25 | 0.5 | 0.25 | |
| Observed Ratio | 0.24 | 0.45 | 0.31 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasmussen, T.L.; Tucker, H.O. Loss of SMYD1 Results in Perinatal Lethality via Selective Defects within Myotonic Muscle Descendants. Diseases 2019, 7, 1. https://doi.org/10.3390/diseases7010001
Rasmussen TL, Tucker HO. Loss of SMYD1 Results in Perinatal Lethality via Selective Defects within Myotonic Muscle Descendants. Diseases. 2019; 7(1):1. https://doi.org/10.3390/diseases7010001
Chicago/Turabian StyleRasmussen, Tara L., and Haley O. Tucker. 2019. "Loss of SMYD1 Results in Perinatal Lethality via Selective Defects within Myotonic Muscle Descendants" Diseases 7, no. 1: 1. https://doi.org/10.3390/diseases7010001