
Gaucher-like Cells in Thalassemia Intermedia: Is It a Challenge?

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
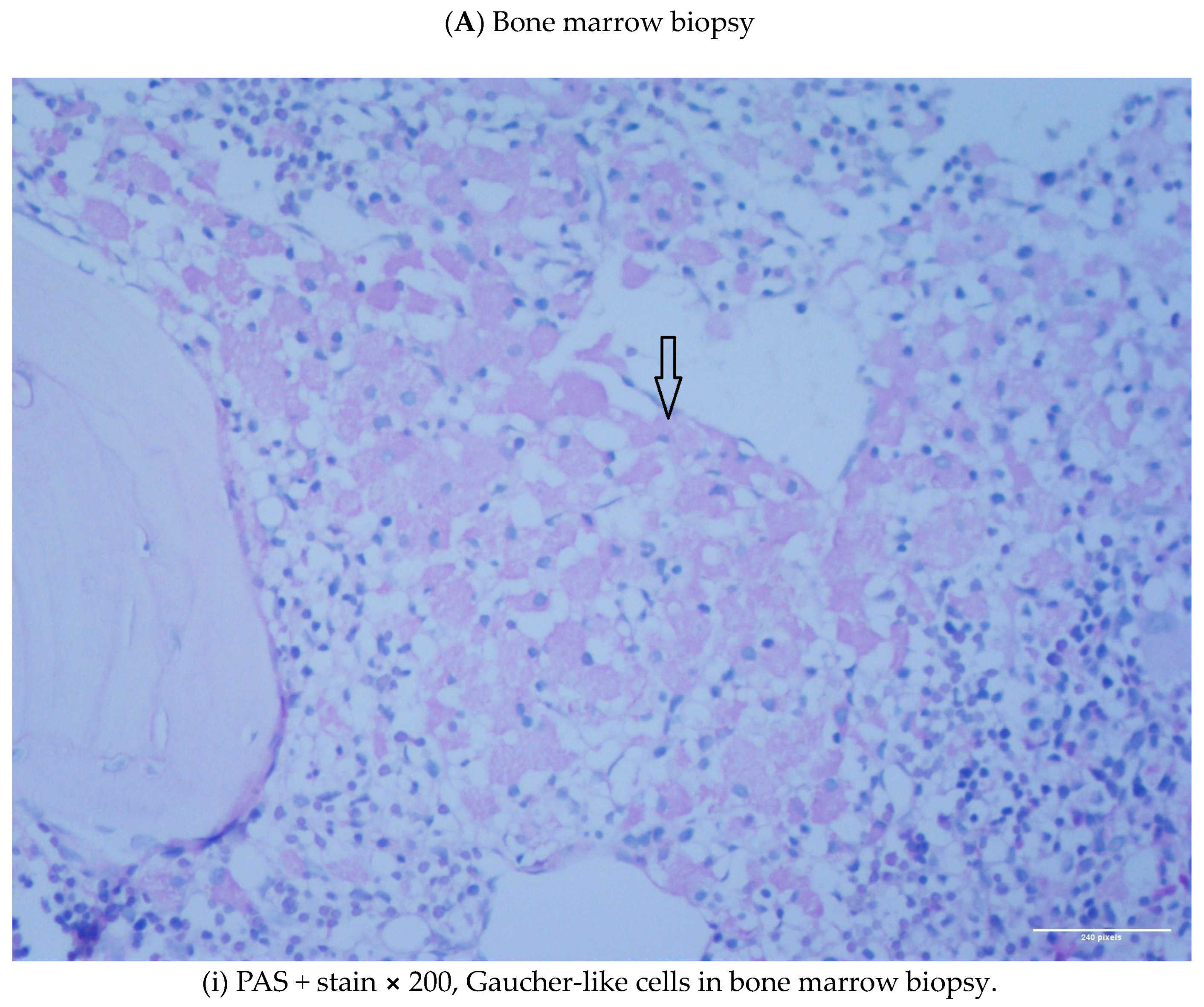
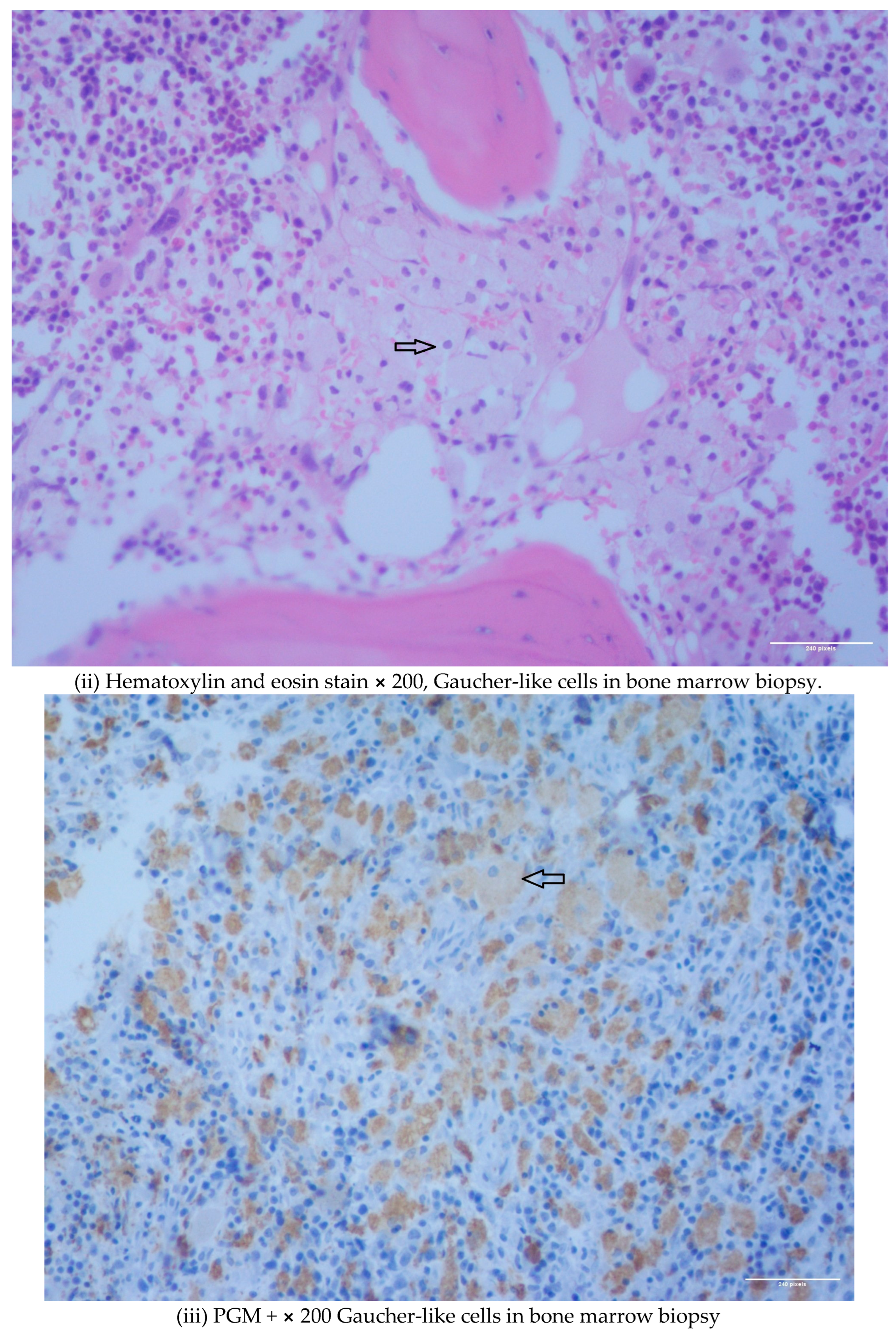
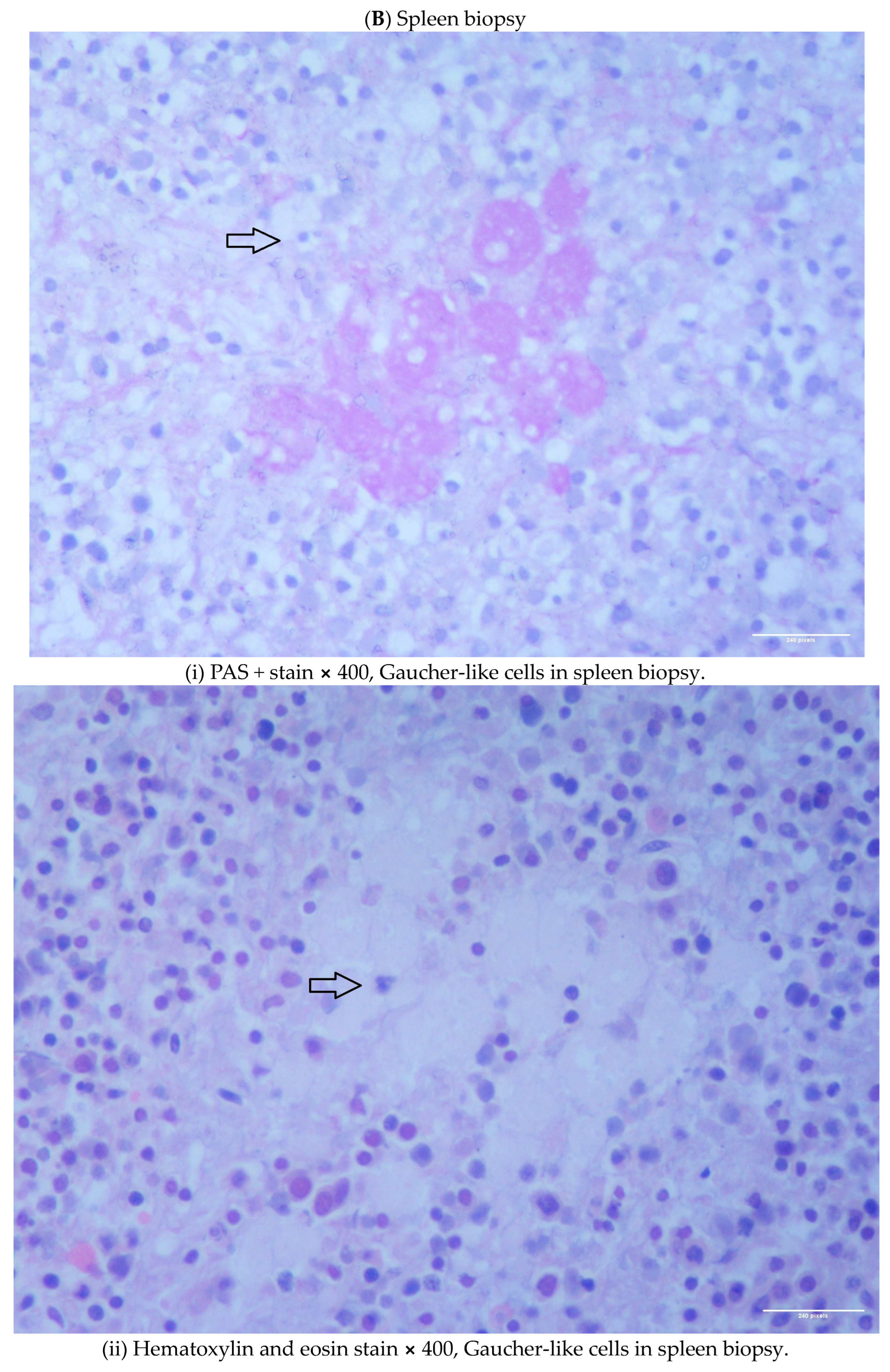
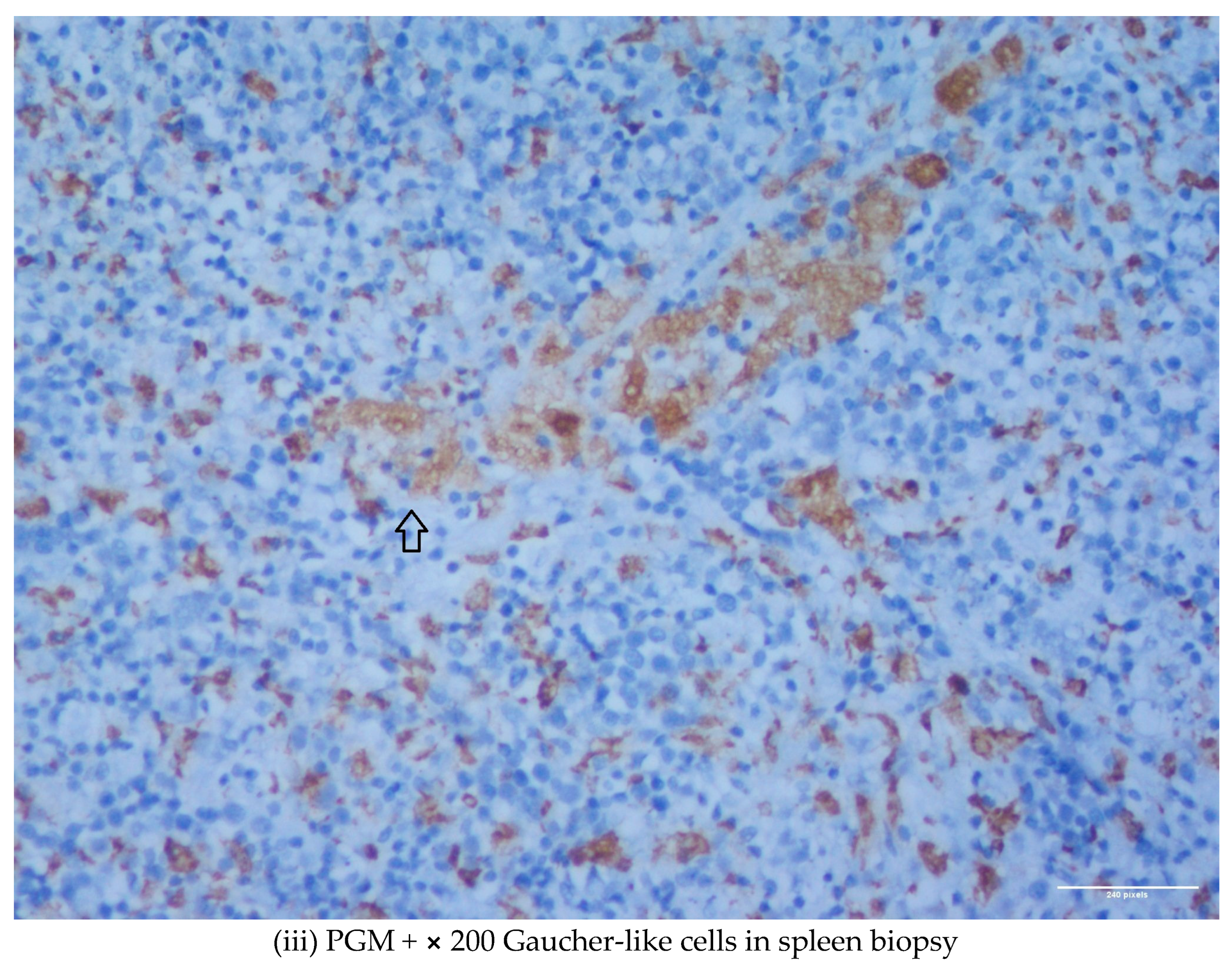
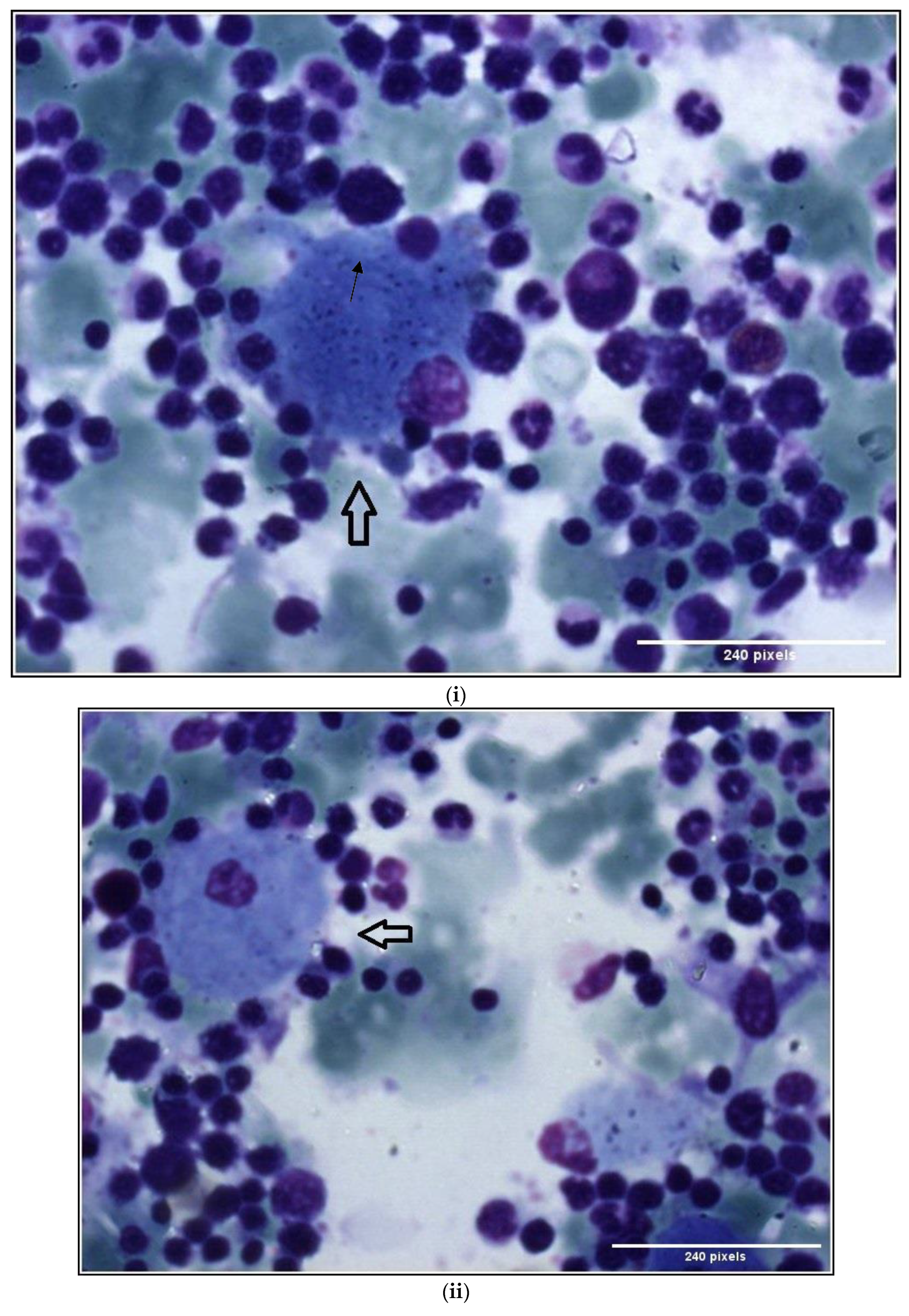
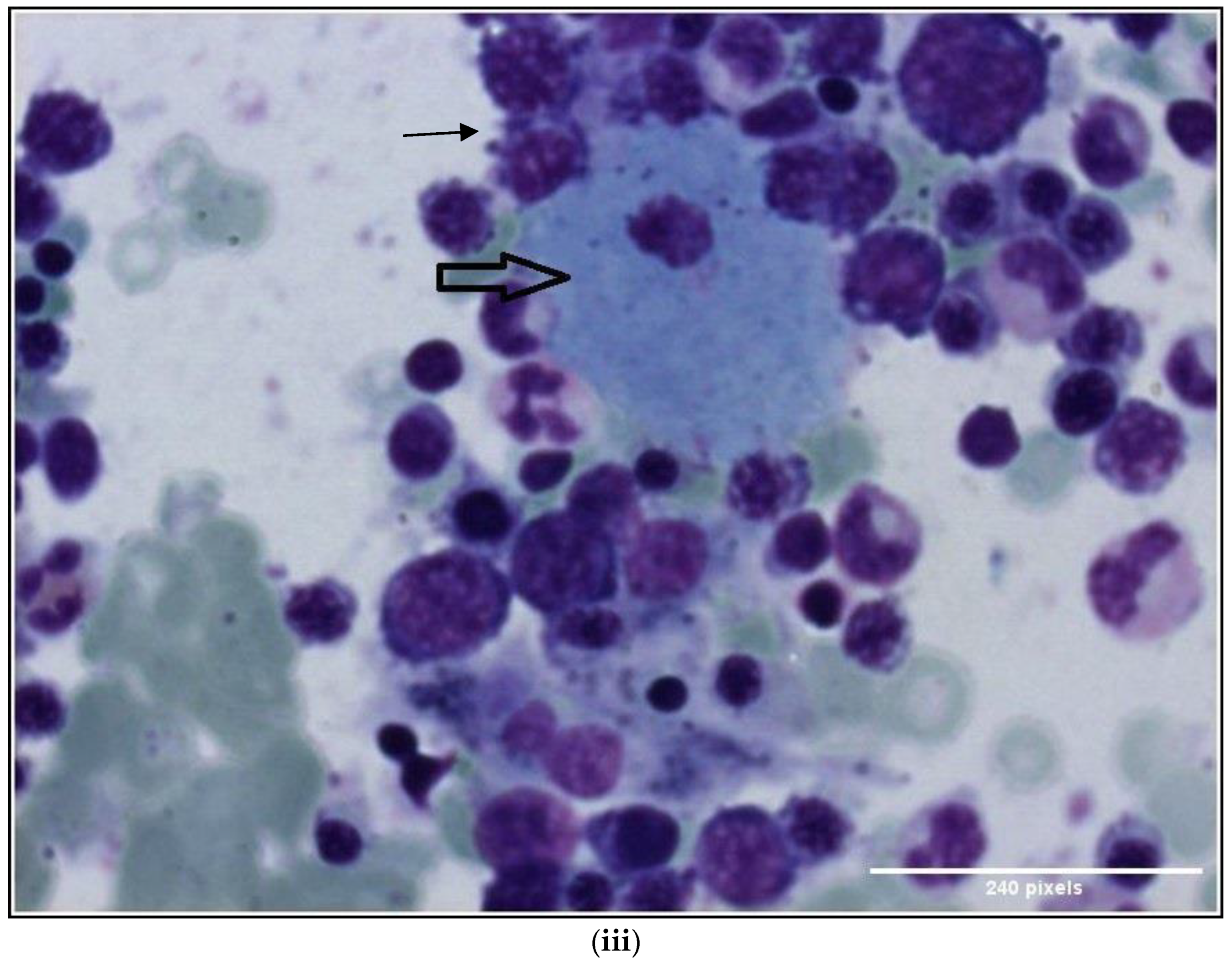
1.1. Case 1
1.2. Case 2
2. Discussion
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beutler, E. Gaucher’s disease. N. Engl. J. Med. 1991, 325, 1354–1360. [Google Scholar]
- Baris, H.N.; Cohen, I.J.; Mistry, P.K. Gaucher disease: The metabolic defect, pathophysiology, phenotypes and natural history. Pediatr. Endocrinol. Rev. 2014, 12 (Suppl. 1), 72–81. [Google Scholar]
- Lee, R.E.; Peters, S.P.; Glew, R.H. Gaucher’s disease: Clinical, morphologic, and pathogenetic considerations. Pathol. Annu. 1977, 12, 309–339. [Google Scholar]
- Beltrami, C.A.; Bearzi, I.; Fabris, G. Storage Cells of Spleen and Bone Marrow in Thalassemia: An Ultrastructural Study. Blood 1973, 41, 901–912. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, C.; Farhat, H. Gaucher-like cells in myelodysplastic syndrome with ring sideroblasts. Hematol. Transfus. Cell Ther. 2022, 44, 136–137. [Google Scholar] [CrossRef] [PubMed]
- Bain, B.J.; Lee, L. Pseudo-Gaucher cells in sickle cell anemia. Am. J. Hematol. 2010, 85, 435. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Khurana, N.; Singh, T. Pseudo-Gaucher cells in Hb E disease and thalassemia intermedia. Hematology 2007, 12, 457–459. [Google Scholar] [CrossRef] [PubMed]
- Dorpe, A.V.; Orshoven, A.B.; Desmet, V.; Verwilghen, R.L. Gaucher-like cells and congenital dyserythropoieticanaemia, type II (HEMPAS). Br. J. Haematol. 1973, 25, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Praveen, S.; Narender, K.; Neelam, V. Multifocal large aggregates of pseudo Gaucher-cells in chronic myeloid leukemia. Blood Res. 2018, 53, 187. [Google Scholar]
- Ash Image Bank. Waldenström macroglobulinemia with pseudo-Gaucher cells. Blood 2010, 116, 3388. [Google Scholar] [CrossRef] [PubMed]
- Saroha, V.; Gupta, P.; Singh, M.; Singh, T. Pseudogaucher cells obscuring multiple myeloma: A case report. Cases J. 2009, 2, 9147. [Google Scholar] [CrossRef] [PubMed]
- Scullin, D.C., Jr.; Shelburne, J.D.; Cohen, H.J. Pseudo-Gaucher cells in multiple myeloma. Am. J. Med. 1979, 67, 347–352. [Google Scholar] [CrossRef]
- Zidar, B.L.; Hartsock, R.J.; Lee, R.E.; Glew, R.H.; LaMarco, K.L.; Pugh, R.P.; Raju, R.N.; Shackney, S.E. Pseudo-Gaucher cells in the bone marrow of a patient with Hodgkin’s disease. Am. J. Clin. Pathol. 1987, 87, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Palta, A.; Dhiman, P.; Ram, J. Excess of Plasma Cells with Pseudo-Gaucher Cells in a Case of Tuberculosis—A Case Report. SAARC J. Tuberc. Lung Dis. HIV/AIDS 2013, 9, 30–32. [Google Scholar] [CrossRef]
- Lee, J.S.; Im, K.; Park, S.N.; Park, H.S.; Kim, J.A.; Choi, Q.; Kim, S.Y.; Cha, C.H.; Oh, H.S.; Kim, I.H. A challenging diagnosis: Crystal-storing histiocytosis in plasma cell myeloma. Am. J. Clin. Pathol. 2015, 143, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Saad Eldeen Bakheet, O.; Yusof, N.; Raja Zahratul, A.; Ithnin, A.; Abdul Aziz, S.; Alias, H. Secondary Sea-Blue Histiocytosis in a Patient with Transfusion Dependent HbE-Beta Thalassaemia and Osteosarcoma. Indian J. Hematol. Blood Transfus. 2016, 32 (Suppl. 1), 262–266. [Google Scholar] [CrossRef]
- Ishihara, T.; Yamashita, Y.; Okuzono, Y.; Yokota, T.; Takahashi, M.; Kamei, T.; Uchino, F.; Matsumoto, N.; Miwa, S.; Fuji, H. Three kinds of foamy cells in the spleen: Comparative histochemical and ultrastructural studies. Ultrastruct. Pathol. 1985, 8, 13–23. [Google Scholar] [CrossRef]
- Zaino, E.C.; Rossi, M.B.; Pham, T.D.; Azar, H.A. Gaucher’s cells in thalassemia. Blood 1971, 38, 457–462. [Google Scholar] [CrossRef]
- Gajendra, S.; Sachdev, R. Pseudo-Gaucher Cells in Thalassemia Intermedia. Int. J. Surg. Pathol. 2015, 23, 370–371. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.C.; Wu, P.C.; Ormiston, I.W.; Tideman, H. Periosteal Gaucher-like cells in beta-thalassemia major. J. Oral Pathol. Med. 1993, 22, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, I.M.; Canon, A.M.; Barcenas, W.; Groden, C.; Goker-Alpan, O.; Resnik, C.S.; Sidransky, E. Bilateral symmetrical cortical osteolytic lesions in two patients with Gaucher disease. Skelet. Radiol. 2011, 40, 1611–1615. [Google Scholar] [CrossRef] [PubMed]
- Mikosch, P.; Hughes, D. An overview on bone manifestations in Gaucher disease. Wien Med. Wochenschr. 2010, 160, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Wasserstein, M.P.; Martignetti, J.A.; Zeitlin, R.; Lumerman, H.; Solomon, M.; Grace, M.E.; Desnick, R.J. Type 1 Gaucher disease presenting with extensive mandibular lytic lesions: Identification and expression of a novel acid beta-glucosidase mutation. Am. J. Med. Genet. 1999, 84, 334–339. [Google Scholar] [CrossRef]
- Morabito, N.; Russo, G.T.; Gaudio, A.; Lasco, A.; Catalano, A.; Morini, E.; Franchina, F.; Maisano, D.; La Rosa, M.; Plota, M. The “lively” cytokines network in beta-Thalassemia Major-related osteoporosis. Bone 2007, 40, 1588–1594. [Google Scholar] [CrossRef] [PubMed]
- Shamshirsaz, A.A.; Bekheirnia, M.R.; Kamgar, M.; Pourzahedgilani, N.; Bouzari, N.; Habibzadeh, M.; Hashemi, R.; Shamshirsaz, A.A.; Aghakhani, S. Metabolic and endocrinologic complications in beta-thalassemia major: A multicenter study in Tehran. BMC Endocr. Disord. 2003, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Gaudio, A.; Morabito, N.; Catalano, A.; Rapisarda, R.; Xourafa, A.; Lasco, A. Pathogenesis of Thalassemia Major-associated Osteoporosis: A Review with Insights from Clinical Experience. J. Clin. Res. Pediatr. Endocrinol. 2019, 11, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Morabito, N.; Gaudio, A.; Lasco, A.; Atteritano, M.; Pizzoleo, M.A.; Cincotta, M.; La Rosa, M.; Guarino, R.; Meo, A.; Frisina, N. Osteoprotegerin and RANKL in the pathogenesis of thalassemia-induced osteoporosis: New pieces of the puzzle. J. Bone Miner. Res. 2004, 19, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Anastasilakis, A.D.; Tsoli, M.; Kaltsas, G.; Makras, P. Bone metabolism in Langerhans cell histiocytosis. Endocr. Connect. 2018, 7, R246–R253. [Google Scholar] [CrossRef] [PubMed]
- Gören Şahin, D. Gaucher Cells or Pseudo-Gaucher Cells: That’s the Question. Gaucher Hücreleri ya da Pseudo-Gaucher Hücreleri: İşte Soru Bu. Turk. J. Haematol. 2014, 31, 428–429. [Google Scholar] [CrossRef] [PubMed]
- Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Fotiou, D.; Kanellias, N.; Migkou, M.; Eleutherakis-Papaiakovou, E.; Kastritis, E.; Dimopoulos, M.A.; Terpos, E. Screening for Gaucher disease among patients with plasma cell dyscrasias. Leuk. Lymphoma 2021, 62, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Arends, M.; van Dussen, L.; Biegstraaten, M.; Hollak, C.E. Malignancies and monoclonal gammopathy in Gaucher disease; a systematic review of the literature. Br. J. Haematol. 2013, 161, 832–842. [Google Scholar] [CrossRef]
- Shagun, A.; Karthik, A. Ganapathi; Prominent pseudo-Gaucher cells in a patient with β-thalassemia intermedia and plasma cell myeloma. Blood 2020, 136, 2839. [Google Scholar] [CrossRef]
- Ricchi, P.; Costantini, S.; Spasiano, A.; De Dominicis, G.; Di Matola, T.; Cinque, P.; Ammirabile, M.; Marsella, M.; Filosa, A. The long-term and extensive efficacy of low dose thalidomide in a case of an untransfusable patient with Non-Transfusion-Dependent Thalassemia. Blood Cells Mol. Dis. 2016, 57, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Gooda, R.; Marouf, R. Pseudo-Gaucher cells in a splenectomised Beta-Thalassemia patient. Br. J. Haematol. 2023, 202, 911. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komninaka, V.; Flevari, P.; Karkaletsis, G.; Androutsakos, T.; Karkaletsi, T.; Ntanasis-Stathopoulos, I.; Ntelaki, E.-E.; Terpos, E. Gaucher-like Cells in Thalassemia Intermedia: Is It a Challenge? Diseases 2023, 11, 161. https://doi.org/10.3390/diseases11040161
Komninaka V, Flevari P, Karkaletsis G, Androutsakos T, Karkaletsi T, Ntanasis-Stathopoulos I, Ntelaki E-E, Terpos E. Gaucher-like Cells in Thalassemia Intermedia: Is It a Challenge? Diseases. 2023; 11(4):161. https://doi.org/10.3390/diseases11040161
Chicago/Turabian StyleKomninaka, Veroniki, Pagona Flevari, Georgios Karkaletsis, Theodoros Androutsakos, Theofili Karkaletsi, Ioannis Ntanasis-Stathopoulos, Evaggelia-Eleni Ntelaki, and Evangelos Terpos. 2023. "Gaucher-like Cells in Thalassemia Intermedia: Is It a Challenge?" Diseases 11, no. 4: 161. https://doi.org/10.3390/diseases11040161