Control of Glycosylation-Related Genes by DNA Methylation: the Intriguing Case of the B3GALT5 Gene and Its Distinct Promoters


Abstract
:1. Introduction
2. Methylation Control of Glycogenes
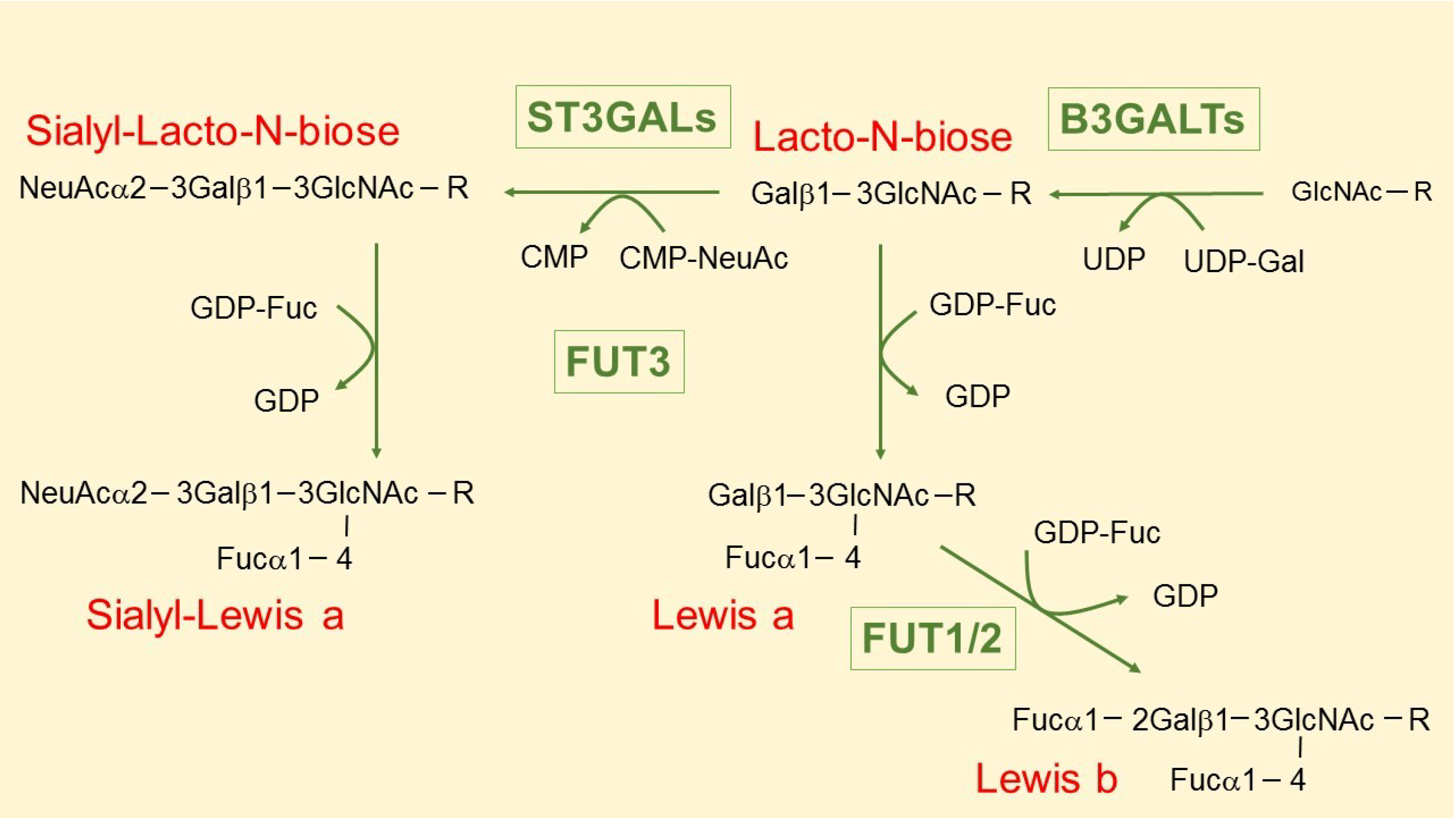
3. The Intriguing Case of the B3GALT5 Gene
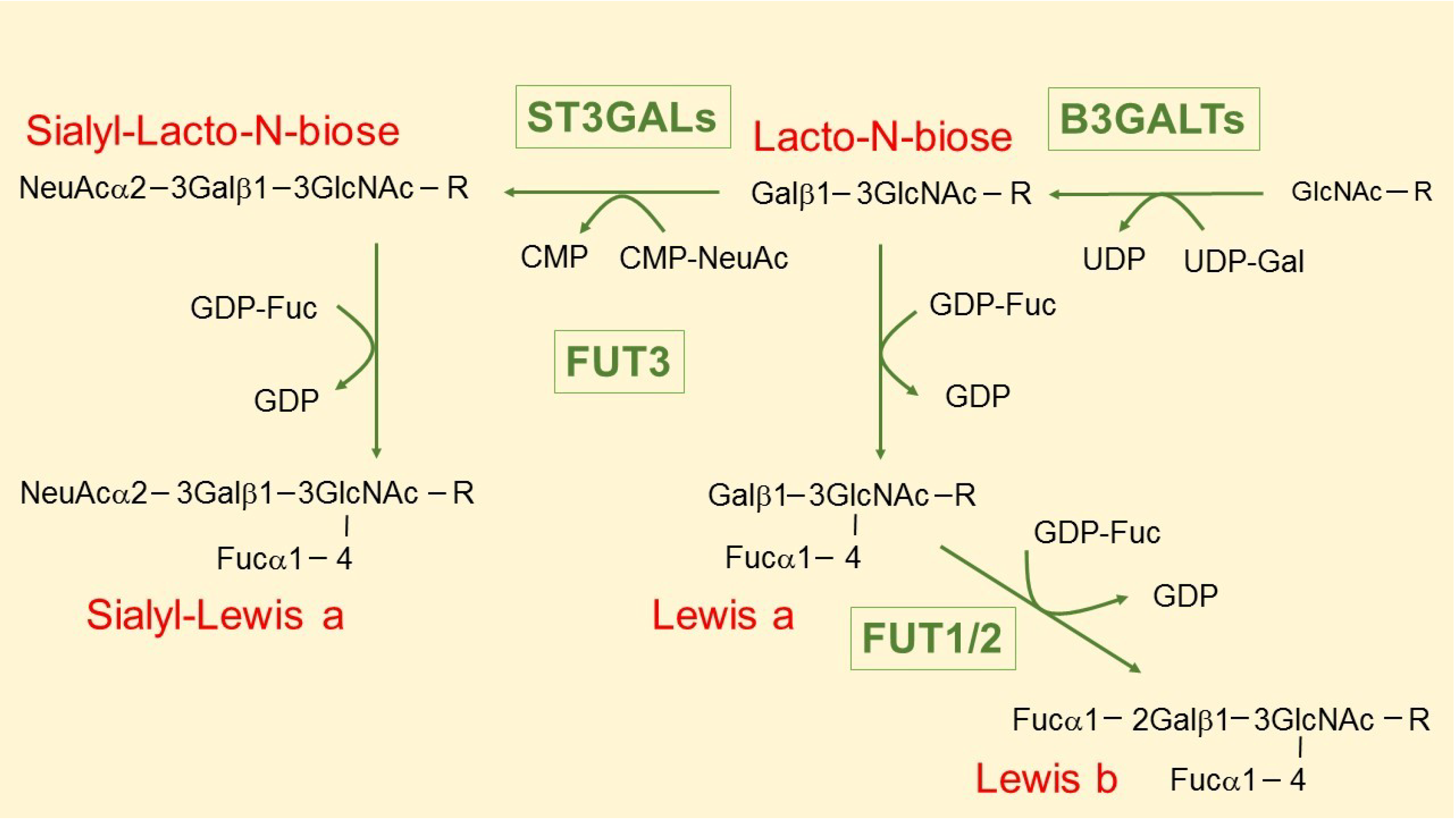
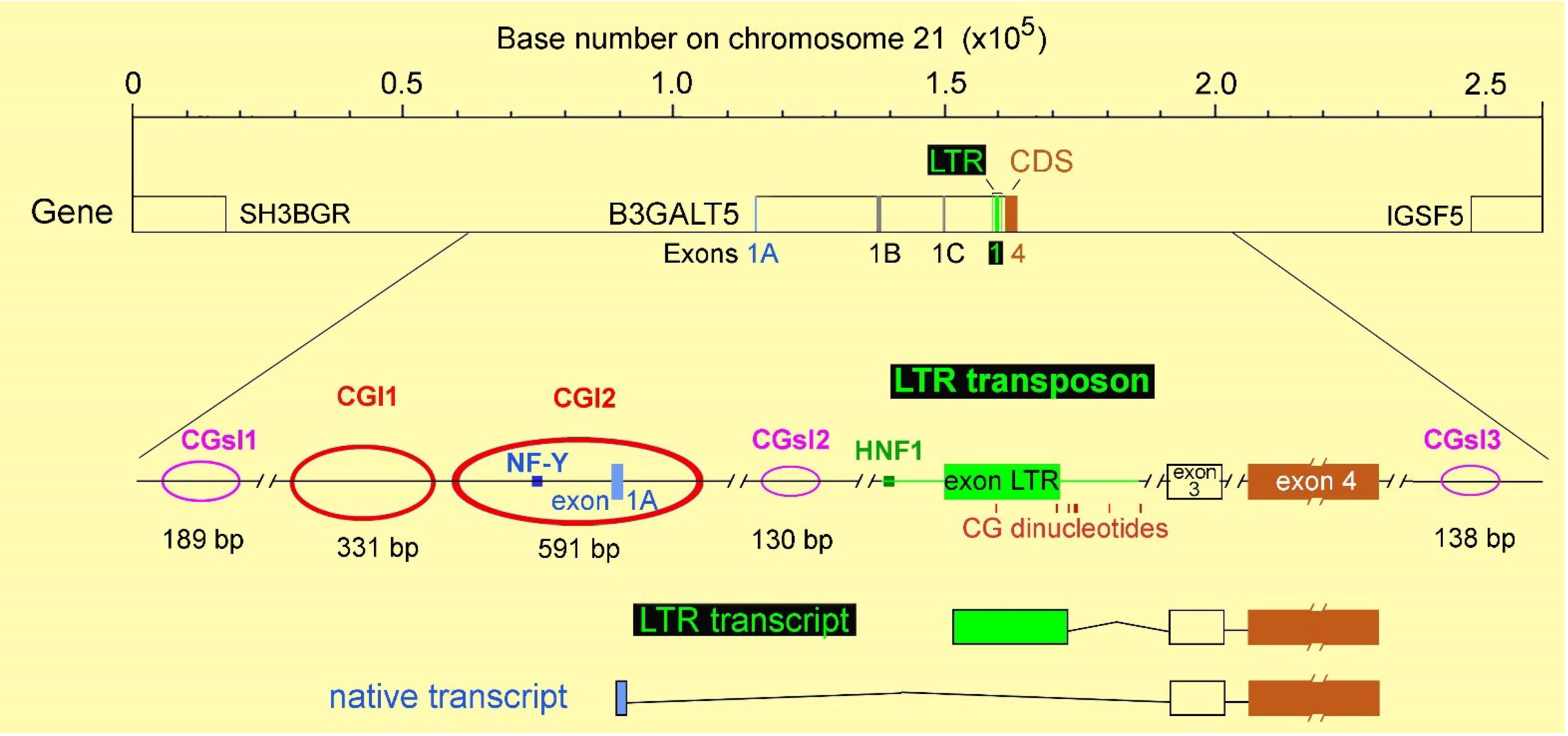
4. Regulation of B3GALT5 Native Promoter

{kind=link}
{kind=link}
| HuCC-T1 | COLO-205 | HCT-15 | MKN-45 | MCF-7 | MDA-MB-231 | |
|---|---|---|---|---|---|---|
| NATIVE TRANSCRIPT | ||||||
| Expression levels | ++ | ++ | - | + | + | - |
| Effect of 5AZA | = | ND | = | ↑ | ND | = |
| Effect of TSA | = | ND | = | ↑ | ND | = |
| Methylation status of CpG island 1/2 (%) | 20/5 | ND | 80/75 | 80/30 | 80/5 | 90/80 |
| H3K27me2, H4K20me3 | very low | ND | high | low | low | high |
| H3K4me3, H3K79me2, H3K9Ac, H3K9-14Ac | very high | ND | low | high | high | low |
| LTR TRANSCRIPT | ||||||
| Expression levels | - | ++++ | - | ++ | - | - |
| Effect of 5AZA | = | ↓↓ | = | ↓↓ | ND | ND |
| Effect of TSA | = | = | = | = | ND | ND |
5. Regulation of the B3GALT5 LTR Promoter
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Varki, A. Biological roles of oligosaccharides: All of the theories are correct. Glycobiology 1993, 3, 97–130. [Google Scholar] [CrossRef]
- Chase, S.D.; Magnani, J.L.; Simon, S.I. E-selectin ligands as mechano sensitive receptors on neutrophils in health and disease. Ann. Biomed. Eng. 2012, 40, 849–859. [Google Scholar] [CrossRef]
- Dall’Olio, F.; Vanhooren, C.V.; Chen, C.P.; Slagboom, E.; Wuhrer, M.; Franceschi, C. N-glycomic biomarkers of biological aging and longevity: A link with inflammaging. Ageing Res. Rev. 2013, 12, 685–698. [Google Scholar] [CrossRef]
- Zoldoš, V.; Novokmet, M.; Bečeheli, I.; Lauc, G. Genomics and epigenomics of the human glycome. Glycoconj. J. 2013, 30, 41–50. [Google Scholar] [CrossRef]
- Lauc, G.; Vojta, A.; Zoldoš, V. Epigenetic regulation of glycosylation is the quantum mechanics of biology. Biochim. Biophys. Acta 2014, 1840, 65–70. [Google Scholar] [CrossRef]
- Yamada, N.; Kitamoto, S.; Yokoyama, S.; Hamada, T.; Goto, M.; Tsutsumida, H.; Higashi, M.; Yonezawa, S. Epigenetic regulation of mucin genes in human cancers. Clin. Epigenet. 2011, 2, 85–96. [Google Scholar] [CrossRef]
- McLarty, J.L.; Marsh, S.A.; Chatham, J.C. Post-translational protein modification by O-linked N-acetyl-glucosamine: Its role in mediating the adverse effects of diabetes on the heart. Life Sci. 2013, 92, 621–627. [Google Scholar] [CrossRef]
- Hanover, J.A.; Krause, M.W.; Love, D.C. Bittersweet memories: Linking metabolism to epigenetics through O-GlcNAcylation. Nat. Rev. Mol. Cell. Biol. 2012, 13, 312–321. [Google Scholar] [CrossRef]
- Jablonka, E.; Raz, G. Transgenerational epigenetic inheritance: Prevalence, mechanisms, and implications for the study of heredity and evolution. Q. Rev. Biol. 2009, 84, 131–176. [Google Scholar] [CrossRef]
- Schmitz, R.J.; Schultz, M.D.; Lewsey, M.G.; O’Malley, R.C.; Urich, M.A.; Libiger, O.; Schork, N.J.; Ecker, J.R. Transgenerational epigenetic instability is a source of novel methylation variants. Science 2011, 334, 369–373. [Google Scholar] [CrossRef]
- Gama-Sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983, 11, 6883–6894. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Gehrke, C.W.; Kuo, K.C.; Ehrlich, M. Reduced genomic 5-methylcytosine content in human colonic neoplasia. Cancer Res. 1988, 48, 1159–1161. [Google Scholar]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef]
- Kulis, M.; Queirós, A.C.; Beekman, R.; Martín-Subero, J.I. Intragenic DNA methylation in transcriptional regulation, normal differentiation and cancer. Biochim. Biophys. Acta 2013, 1829, 1161–1174. [Google Scholar]
- Saldova, R.; Dempsey, E.; Perez-Garay, M.; Marino, K.; Watson, J.A.; Blanco-Fernandez, A.; Struwe, W.B.; Harvey, D.J.; Madden, S.F.; Peracaula, R.; et al. 5-AZA-2'-deoxycytidine induced demethylation influences N-glycosylation of secreted glycoproteins in ovarian cancer. Epigenetics 2011, 6, 1362–1372. [Google Scholar] [CrossRef]
- Syrbe, U.; Jennrich, S.; Schottelius, A.; Richter, A.; Radbruch, A.; Hamann, A. Differential regulation of P-selectin ligand expression in naive versus memory CD4+ T cells: Evidence for epigenetic regulation of involved glycosyltransferase genes. Blood 2004, 104, 3243–3248. [Google Scholar] [CrossRef]
- Chachadi, V.B.; Cheng, H.; Klinkebiel, D.; Christman, J.K.; Cheng, P.W. 5-Aza-2'-deoxycytidine increases sialyl Lewis X on MUC1 by stimulating β-galactoside: α2,3-sialyltransferase 6 gene. Int. J. Biochem. Cell Biol. 2011, 43, 586–593. [Google Scholar] [CrossRef]
- Serpa, J.; Mesquita, P.; Mendes, N.; Oliveira, C.; Almeida, R.; Santos-Silva, F.; Reis, C.A.; Lependu, J.; David, L. Expression of Lea in gastric cancer cell lines depends on FUT3 expression regulated by promoter methylation. Cancer Lett. 2006, 242, 191–197. [Google Scholar] [CrossRef]
- Miyazaki, K.; Ohmori, K.; Izawa, M.; Koike, T.; Kumamoto, K.; Furukawa, K.; Ando, T.; Kiso, M.; Yamaji, T.; Hashimoto, Y.; et al. Loss of disialyl Lewisa the ligand for lymphocyte inhibitory receptor sialic acid-binding immunoglobulin-like lectin-7 (Siglec-7) associated with increased sialyl Lewisa expression on human colon cancers. Cancer Res. 2004, 64, 4498–4505. [Google Scholar] [CrossRef]
- Yusa, A.; Miyazaki, K.; Kimura, N.; Izawa, M.; Kannagi, R. Epigenetic silencing of the sulfate transporter gene DTDST induces sialyl Lewisx expression and accelerates proliferation of colon cancer cells. Cancer Res. 2010, 70, 4064–4073. [Google Scholar] [CrossRef]
- Kawamura, Y.I.; Toyota, M.; Kawashima, R.; Hagiwara, T.; Suzuki, H.; Imai, K.; Shinomura, Y.; Tokino, T.; Kannagi, R.; Dohi, T. DNA hypermethylation contributes to incomplete synthesis of carbohydrate determinants in gastrointestinal cancer. Gastroenterology 2008, 135, 142–151. [Google Scholar] [CrossRef]
- Tong, W.G.; Wierda, W.G.; Lin, E.; Kuang, S.Q.; Bekele, B.N.; Estrov, Z.; Wei, Y.; Yang, H.; Keating, M.J.; Garcia-Manero, G. Genome-wide DNA methylation profiling of chronic lymphocytic leukemia allows identification of epigenetically repressed molecular pathways with clinical impact. Epigenetics 2010, 5, 499–508. [Google Scholar] [CrossRef]
- Wang, H.R.; Hsieh, C.Y.; Twu, Y.C.; Yu, L.C. Expression of the human Sda β-1,4-N-acetylgalactosaminyltransferase II gene is dependent on the promoter methylation status. Glycobiology 2008, 18, 104–113. [Google Scholar]
- Dall’Olio, F.; Malagolini, N.; Chiricolo, M.; Trinchera, M.; Harduin-Lepers, A. The expanding roles of the Sda/Cad carbohydrate antigen and its cognate glycosyltransferase B4GALNT2. Biochim. Biophys. Acta 2014, 1840, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Ide, Y.; Miyoshi, E.; Nakagawa, T.; Gu, J.; Tanemura, M.; Nishida, T.; Ito, T.; Yamamoto, H.; Kozutsumi, Y.; Taniguchi, N. Aberrant expression of N-acetylglucosaminyltransferase-IVa and IVb (GnT-IVa and b) in pancreatic cancer. Biochem. Biophys. Res. Commun. 2006, 341, 478–482. [Google Scholar] [CrossRef]
- Kizuka, Y.; Kitazume, S.; Yoshida, M.; Taniguchi, N. Brain-specific expression of N-acetylglucosaminyltransferase IX (GnT-IX) is regulated by epigenetic histone modifications. J. Biol. Chem. 2011, 286, 31875–31884. [Google Scholar] [CrossRef]
- Chakraborty, A.K.; Sousa, J.F.; Chakraborty, D.; Funasaka, Y.; Bhattacharya, M.; Chatterjee, A.; Pawelek, J. GnT-V expression and metastatic phenotypes in macrophage-melanoma fusion hybrids is down-regulated by 5-Aza-dC: Evidence for methylation sensitive, extragenic regulation of GnT-V transcription. Gene 2006, 374, 166–173. [Google Scholar] [CrossRef]
- Chihara, Y.; Sugano, K.; Kobayashi, A.; Kanai, Y.; Yamamoto, H.; Nakazono, M.; Fujimoto, H.; Kakizoe, T.; Fujimoto, K.; Hirohashi, S.; et al. Loss of blood group A antigen expression in bladder cancer caused by allelic loss and/or methylation of the ABO gene. Lab. Invest. 2005, 85, 895–907. [Google Scholar] [CrossRef]
- Dabelsteen, E.; Gao, S. ABO blood-group antigens in oral cancer. J. Dent. Res. 2005, 84, 21–28. [Google Scholar] [CrossRef]
- Gao, S.; Worm, J.; Guldberg, P.; Eiberg, H.; Krogdahl, A.; Liu, C.J.; Reibel, J.; Dabelsteen, E. Genetic and epigenetic alterations of the blood group ABO gene in oral squamous cell carcinoma. Int. J. Cancer 2004, 109, 230–237. [Google Scholar] [CrossRef]
- Giordanengo, V.; Ollier, L.; Lanteri, M.; Lesimple, J.; March, D.; Thyss, S.; Lefebvre, J.C. Epigenetic reprogramming of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) in HIV-1-infected CEM T cells. FASEB J. 2004, 18, 1961–1963. [Google Scholar]
- Oetke, C.; Hinderlich, S.; Reutter, W.; Pawlita, M. Epigenetically mediated loss of UDP-GlcNAc 2-epimerase/ManNAc kinase expression in hyposialylated cell lines. Biochem. Biophys. Res. Commun. 2003, 308, 892–898. [Google Scholar] [CrossRef]
- Boscher, C.; Dennis, J.W.; Nabi, I.R. Glycosylation, galectins and cellular signaling. Curr. Opin. Cell Biol. 2011, 23, 383–392. [Google Scholar] [CrossRef]
- Satelli, A.; Rao, U.S. Galectin-1 is silenced by promoter hypermethylation and its re-expression induces apoptosis in human colorectal cancer cells. Cancer Lett. 2011, 301, 38–46. [Google Scholar] [CrossRef]
- Juszczynski, P.; Rodig, S.J.; Ouyang, J.; O’Donnell, E.; Takeyama, K.; Mlynarski, W.; Mycko, K.; Szczepanski, T.; Gaworczyk, A.; Krivtsov, A.; et al. MLL-rearranged B lymphoblastic leukemias selectively express the immunoregulatory carbohydrate-binding protein galectin-1. Clin. Cancer Res. 2010, 16, 2122–2130. [Google Scholar] [CrossRef]
- Newlaczyl, A.U.; Yu, L.G. Galectin-3—A jack-of-all-trades in cancer. Cancer Lett. 2011, 313, 123–128. [Google Scholar] [CrossRef]
- Ahmed, H.; Banerjee, P.P.; Vasta, G.R. Differential expression of galectins in normal, benign and malignant prostate epithelial cells: Silencing of galectin-3 expression in prostate cancer by its promoter methylation. Biochem. Biophys. Res. Commun. 2007, 358, 241–246. [Google Scholar] [CrossRef]
- Ahmed, H.; Cappello, F.; Rodolico, V.; Vasta, G.R. Evidence of heavy methylation in the galectin 3 promoter in early stages of prostate adenocarcinoma: Development and validation of a methylated marker for early diagnosis of prostate cancer. Transl. Oncol. 2009, 2, 146–156. [Google Scholar] [CrossRef]
- Keller, S.; Angrisano, T.; Florio, E.; Pero, R.; Decaussin-Petrucci, M.; Troncone, G.; Capasso, M.; Lembo, F.; Fusco, A.; Chiariotti, L. DNA methylation state of the galectin-3 gene represents a potential new marker of thyroid malignancy. Oncol. Lett. 2013, 6, 86–90. [Google Scholar]
- Ben Mahmoud, L.K.; Arfaoui, A.; Khiari, M.; Chaar, I.; El Amine, O.; Ben Hmida, A.M.; Gharbi, L.; Mzabi, S.R.; Bouraoui, S. Loss of galectin-3 expression in mucinous colorectal carcinomas is associated with 5’CpG island methylation in Tunisian patients. Appl. Immunohistochem. Mol. Morphol. 2011, 19, 258–265. [Google Scholar]
- Ruebel, K.H.; Jin, L.; Qian, X.; Scheithauer, B.W.; Kovacs, K.; Nakamura, N.; Zhang, H.; Raz, A.; Lloyd, R.V. Effects of DNA methylation on galectin-3 expression in pituitary tumors. Cancer Res. 2005, 65, 1136–1140. [Google Scholar] [CrossRef]
- Kim, S.J.; Hwang, J.A.; Ro, J.Y.; Lee, Y.S.; Chun, K.H. Galectin-7 is epigenetically-regulated tumor suppressor in gastric cancer. Oncotarget 2013, 4, 1461–1471. [Google Scholar]
- Demers, M.; Couillard, J.; Giglia-Mari, G.; Magnaldo, T.; St Pierre, Y. Increased galectin-7 gene expression in lymphoma cells is under the control of DNA methylation. Biochem. Biophys. Res. Commun. 2009, 387, 425–429. [Google Scholar] [CrossRef]
- Isshiki, S.; Kudo, T.; Nishihara, S.; Ikehara, Y.; Togayachi, A.; Furuya, A.; Shitara, K.; Kubota, T.; Watanabe, M.; Kitajima, M.; et al. Cloning, expression, and characterization of a novel UDP-galactose: β-N-acetylglucosamine β1,3-galactosyltransferase (β3Gal-T5) responsible for synthesis of type 1 chain in colorectal and pancreatic epithelia and tumor cells derived therefrom. J. Biol. Chem. 1999, 274, 12499–12507. [Google Scholar] [CrossRef]
- Lin, C.H.; Fan, Y.Y.; Chen, Y.Y.; Wang, S.H.; Chen, C.I.; Yu, L.C.; Khoo, K.H. Enhanced expression of beta 3-galactosyltransferase 5 activity is sufficient to induce in vivo synthesis of extended type 1 chains on lactosylceramides of selected human colonic carcinoma cell lines. Glycobiology 2009, 19, 418–427. [Google Scholar]
- Mare, L.; Trinchera, M. Comparative analysis of retroviral and native promoters driving expression of beta1,3-galactosyltransferase beta3Gal-T5 in human and mouse tissues. J. Biol. Chem. 2007, 282, 49–57. [Google Scholar] [CrossRef]
- Isshiki, S.; Togayachi, A.; Kudo, T.; Nishihara, S.; Watanabe, M.; Kubota, T.; Kitajima, M.; Shiraishi, N.; Sasaki, K.; Andoh, T.; et al. Lewis type 1 antigen synthase (beta3Gal-T5) is transcriptionally regulated by homeoproteins. J. Biol. Chem. 2003, 278, 36611–36620. [Google Scholar] [CrossRef]
- Dunn, C.A.; Medstrand, P.; Mager, D.L. An endogenous retroviral long terminal repeat is the dominant promoter for human beta1,3-galactosyltransferase 5 in the colon. Proc. Natl. Acad. Sci. USA 2003, 100, 12841–12846. [Google Scholar] [CrossRef]
- Dunn, C.A.; van de Lagemaat, L.N.; Baillie, G.J.; Mager, D.L. Endogenous retrovirus long terminal repeats as ready-to-use mobile promoters: The case of primate beta3GAL-T5. Gene 2005, 364, 2–12. [Google Scholar] [CrossRef]
- Mare, L.; Trinchera, M. Suppression of β1,3galactosyltransferase β3Gal-T5 in cancer cells reduces sialyl-Lewis a and enhances poly N-acetyllactosamines and sialyl-Lewis x on O-glycans. Eur. J. Biochem. 2004, 271, 186–194. [Google Scholar] [CrossRef]
- Salvini, R.; Bardoni, A.; Valli, M.; Trinchera, M. Beta 1,3-Galactosyltransferase beta 3Gal-T5 acts on the GlcNAcbeta 1-->3Galbeta 1-->4GlcNAcbeta 1-->R sugar chains of carcinoembryonic antigen and other N-linked glycoproteins and is down-regulated in colon adenocarcinomas. J. Biol. Chem. 2001, 276, 3564–3573. [Google Scholar]
- Caretti, G.; Salsi, V.; Vecchi, C.; Imbriano, C.; Mantovani, R. Dynamic recruitment of NF-Y and histone acetyltransferases on cell-cycle promoters. J. Biol. Chem. 2003, 278, 30435–30440. [Google Scholar]
- Caretti, A.; Sirchia, S.M.; Tabano, S.; Zulueta, A.; Dall’Olio, F.; Trinchera, M. DNA methylation and histone modifications modulate the β1,3 galactosyltransferase β3Gal-T5 native promoter in cancer cells. Int. J. Biochem. Cell. Biol. 2012, 44, 84–90. [Google Scholar] [CrossRef]
- Zulueta, A.; Caretti, A.; Signorelli, P.; Dall’olio, F.; Trinchera, M. Transcriptional control of the B3GALT5 gene by a retroviral promoter and methylation of distant regulatory elements. FASEB J. 2014, 28, 946–955. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Sabato, S.; Hellman, A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013, 14, R21. [Google Scholar] [CrossRef]
- Terraneo, L.; Avagliano, L.; Caretti, A.; Bianciardi, P.; Tosi, D.; Bulfamante, G.P.; Samaja, M.; Trinchera, M. Expression of carbohydrate-antigen sialyl-Lewis a on colon cancer cells promotes xenograft growth and angiogenesis in nude mice. Int. J. Biochem. Cell. Biol. 2013, 45, 2796–2800. [Google Scholar] [CrossRef] [Green Version]
- Mare, L.; Caretti, A.; Albertini, R.; Trinchera, M. CA19.9 antigen circulating in the serum of colon cancer patients: Where is it from? Int. J. Biochem. Cell. Biol. 2013, 45, 792–797. [Google Scholar] [CrossRef] [Green Version]
- Chachadi, V.B.; Ali, M.F.; Cheng, P.W. Prostatic cell-specific regulation of the synthesis of MUC1-associated sialyl Lewis a. PLoS One 2013, 8, e57416. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Trinchera, M.; Zulueta, A.; Caretti, A.; Dall'Olio, F. Control of Glycosylation-Related Genes by DNA Methylation: the Intriguing Case of the B3GALT5 Gene and Its Distinct Promoters. Biology 2014, 3, 484-497. https://doi.org/10.3390/biology3030484
Trinchera M, Zulueta A, Caretti A, Dall'Olio F. Control of Glycosylation-Related Genes by DNA Methylation: the Intriguing Case of the B3GALT5 Gene and Its Distinct Promoters. Biology. 2014; 3(3):484-497. https://doi.org/10.3390/biology3030484
Chicago/Turabian StyleTrinchera, Marco, Aida Zulueta, Anna Caretti, and Fabio Dall'Olio. 2014. "Control of Glycosylation-Related Genes by DNA Methylation: the Intriguing Case of the B3GALT5 Gene and Its Distinct Promoters" Biology 3, no. 3: 484-497. https://doi.org/10.3390/biology3030484