McArdle Disease and Exercise Physiology

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Compensatory Energy Transfer Pathways
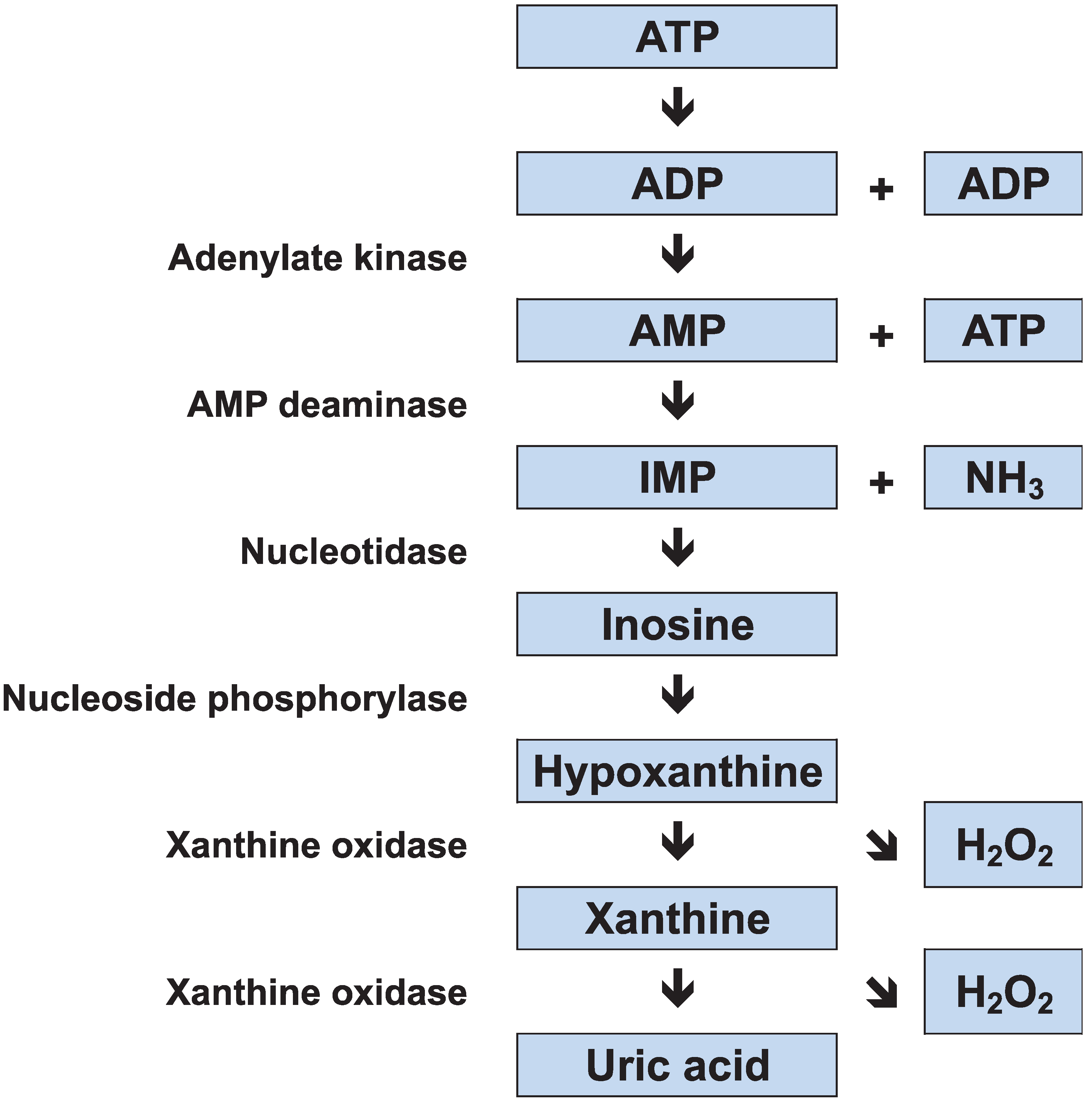
2.1. Adenine Nucleotide Degradation
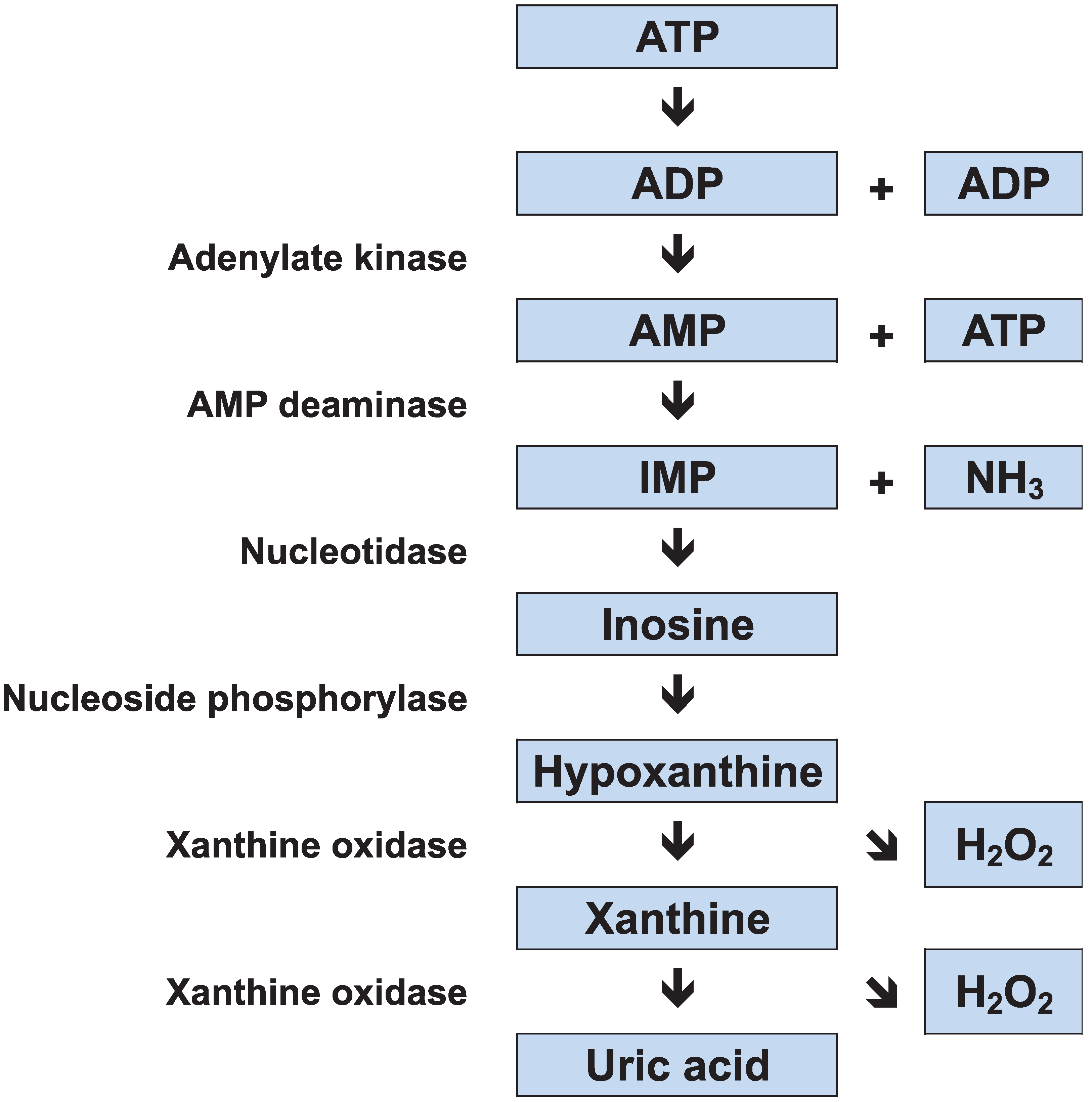
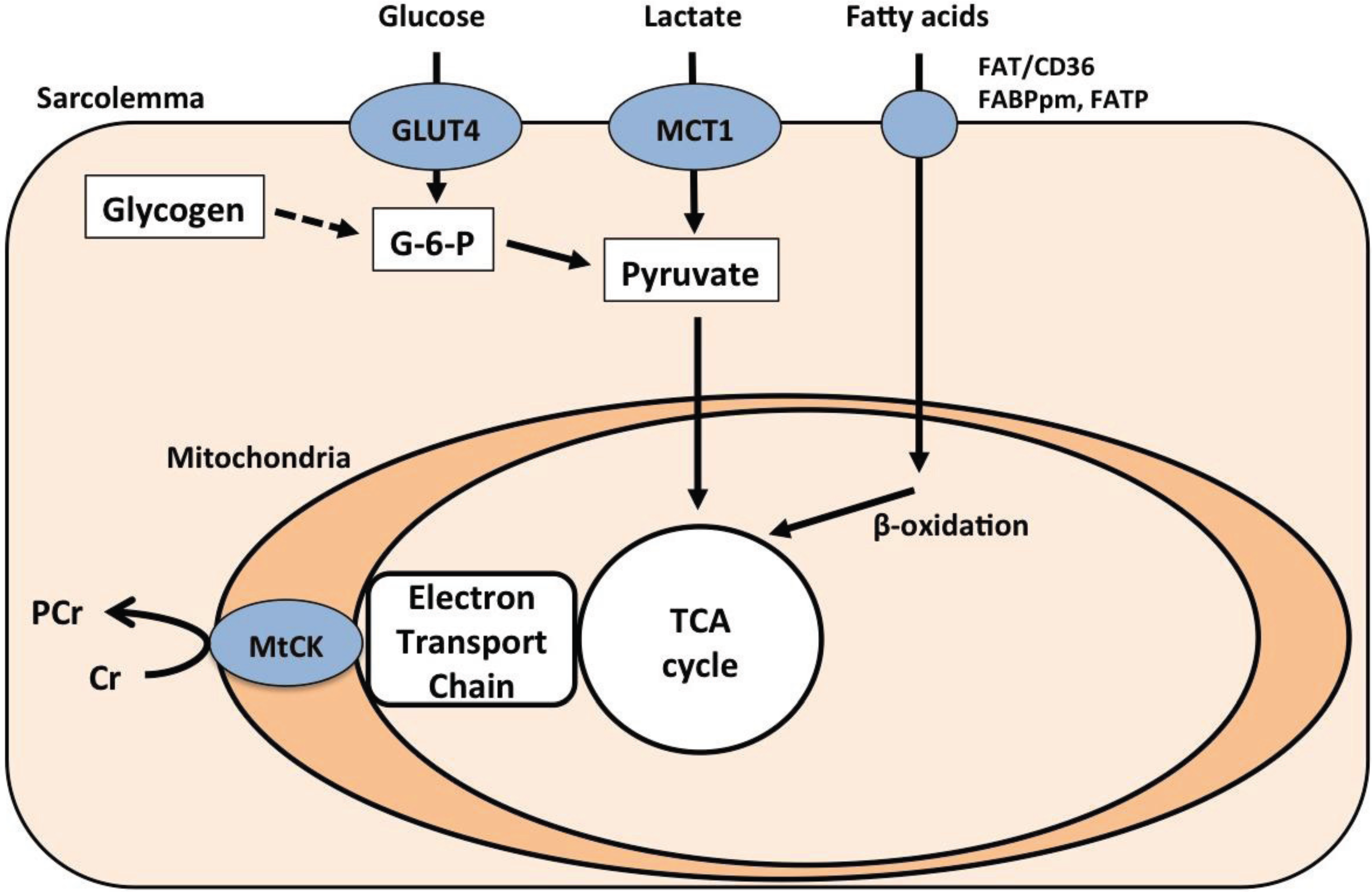
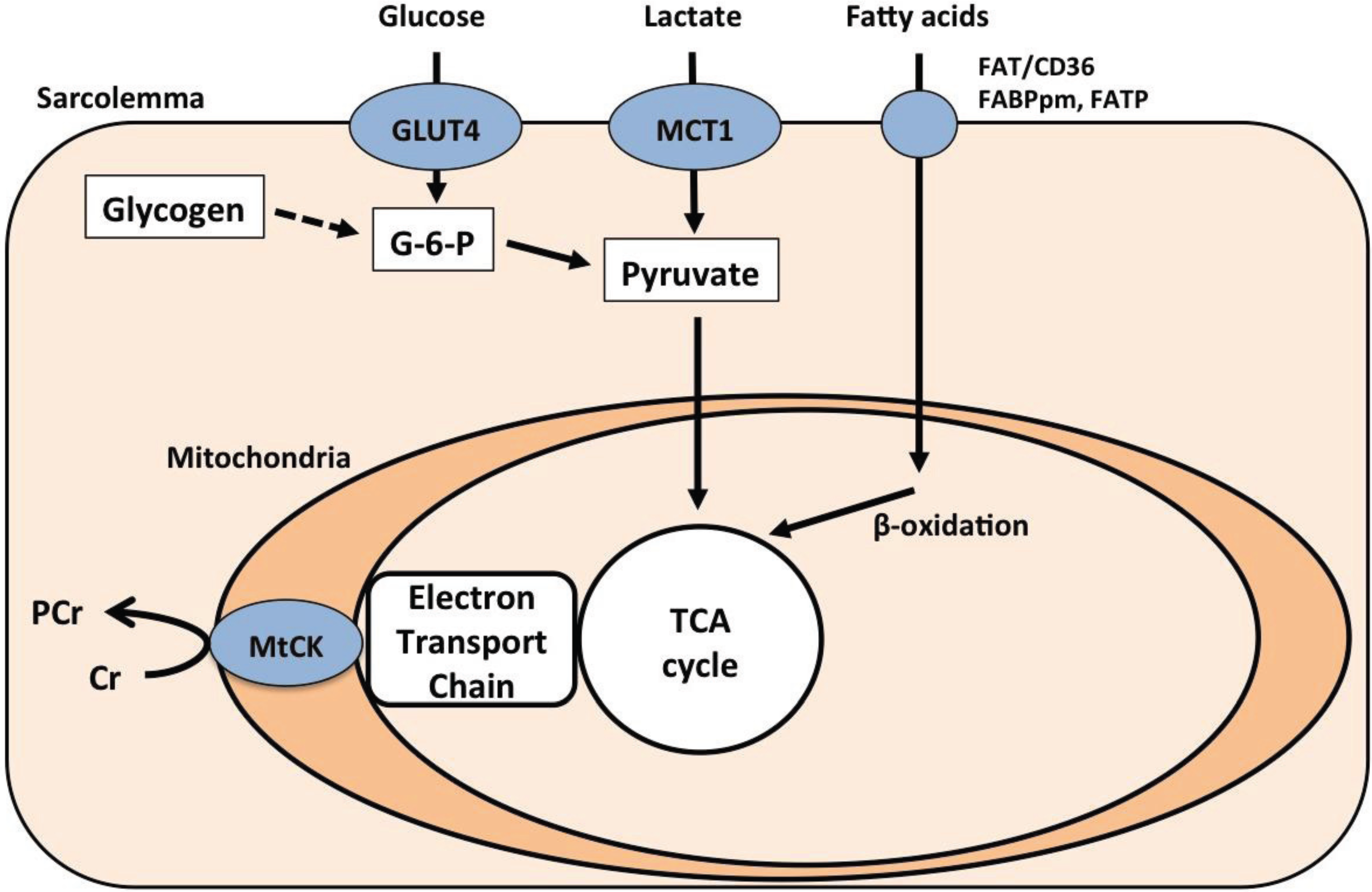
2.2. Carbohydrate Metabolism
2.3. Fat Metabolism
2.4. Creatine-Phosphocreatine Shuttle
3. Exercise Training as a Possible Treatment
4. Conclusions
Acknowledgment
Conflicts of Interest
References and Notes
- McArdle, B. Myopathy due to a defect in muscle glycogen breakdown. Clin. Sci. 1951, 10, 13–33. [Google Scholar]
- Lebo, R.V.; Gorin, F.; Fletterick, R.J.; Kao, F.T.; Cheung, M.C.; Bruce, B.D.; Kan, Y.W. High-resolution chromosome sorting and DNA spot-blot analysis assign McArdle's syndrome to chromosome 11. Science 1984, 225, 57–59. [Google Scholar]
- Burke, J.; Hwang, P.; Anderson, L.; Lebo, R.; Gorin, F.; Fletterick, R. Intron/exon structure of the human gene for the muscle isozyme of glycogen phosphorylase. Proteins 1987, 2, 177–187. [Google Scholar] [CrossRef]
- Krawczak, M.; Ball, E.V.; Fenton, I.; Stenson, P.D.; Abeysinghe, S.; Thomas, N.; Cooper, D.N. Human gene mutation database-a biomedical information and research resource. Hum. Mutat. 2000, 15, 45–51. [Google Scholar] [CrossRef]
- Tsujino, S.; Shanske, S.; DiMauro, S. Molecular genetic heterogeneity of myophosphorylase deficiency (McArdle's disease). N. Engl. J. Med. 1993, 329, 241–245. [Google Scholar] [CrossRef]
- Bartram, C.; Edwards, R.H.; Clague, J.; Beynon, R.J. McArdle's disease: A nonsense mutation in exon 1 of the muscle glycogen phosphorylase gene explains some but not all cases. Hum. Mol. Genet. 1993, 2, 1291–1293. [Google Scholar] [CrossRef]
- Vieitez, I.; Teijeira, S.; Fernandez, J.M.; San Millan, B.; Miranda, S.; Ortolano, S.; Louis, S.; Laforet, P.; Navarro, C. Molecular and clinical study of McArdle's disease in a cohort of 123 European patients. Identification of 20 novel mutations. Neuromuscul. Disord. 2011, 21, 817–823. [Google Scholar] [CrossRef]
- Vorgerd, M.; Kubisch, C.; Burwinkel, B.; Reichmann, H.; Mortier, W.; Tettenborn, B.; Pongratz, D.; Lindemuth, R.; Tegenthoff, M.; Malin, J.P.; et al. Mutation analysis in myophosphorylase deficiency (McArdle's disease). Ann. Neurol. 1998, 43, 326–331. [Google Scholar] [CrossRef]
- Bruno, C.; Cassandrini, D.; Martinuzzi, A.; Toscano, A.; Moggio, M.; Morandi, L.; Servidei, S.; Mongini, T.; Angelini, C.; Musumeci, O.; et al. McArdle disease: The mutation spectrum of PYGM in a large Italian cohort. Hum. Mutat. 2006, 27, 718. [Google Scholar]
- Martin, M.A.; Rubio, J.C.; Wevers, R.A.; van Engelen, B.G.; Steenbergen, G.C.; van Diggelen, O.P.; de Visser, M.; de Die-Smulders, C.; Blazquez, A.; Andreu, A.L.; et al. Molecular analysis of myophosphorylase deficiency in Dutch patients with McArdle's disease. Ann. Hum. Genet. 2004, 68, 17–22. [Google Scholar] [CrossRef]
- Sugie, H.; Sugie, Y.; Ito, M.; Fukuda, T.; Nonaka, I.; Igarashi, Y. Genetic analysis of Japanese patients with myophosphorylase deficiency (McArdle's disease): Single-codon deletion in exon 17 is the predominant mutation. Clin. Chim. Acta 1995, 236, 81–86. [Google Scholar] [CrossRef]
- Tsujino, S.; Shanske, S.; Nonaka, I.; Eto, Y.; Mendell, J.R.; Fenichel, G.M.; DiMauro, S. Three new mutations in patients with myophosphorylase deficiency (McArdle disease). Am. J. Hum. Genet. 1994, 54, 44–52. [Google Scholar] [CrossRef]
- Vissing, J.; Duno, M.; Schwartz, M.; Haller, R.G. Splice mutations preserve myophosphorylase activity that ameliorates the phenotype in McArdle disease. Brain 2009, 132, 1545–1552. [Google Scholar] [CrossRef]
- Schmid, R.; Mahler, R. Chronic progressive myopathy with myoglobinuria: Demonstration of a glycogenolytic defect in the muscle. J. Clin. Invest. 1959, 38, 2044–2058. [Google Scholar] [CrossRef]
- Mommaerts, W.F.; Illingworth, B.; Pearson, C.M.; Guillory, R.J.; Seraydarian, K. A Functional Disorder of Muscle Associated with the Absence of Phosphorylase. Proc. Natl. Acad. Sci. USA 1959, 45, 791–797. [Google Scholar] [CrossRef]
- Pearson, C.M.; Rimer, D.G.; Mommaerts, W.F. A metabolic myopathy due to absence of muscle phosphorylase. Am. J. Med. 1961, 30, 502–517. [Google Scholar] [CrossRef]
- Nadaj-Pakleza, A.A.; Vincitorio, C.M.; Laforet, P.; Eymard, B.; Dion, E.; Teijeira, S.; Vietez, I.; Jeanpierre, M.; Navarro, C.; Stojkovic, T. Permanent muscle weakness in McArdle disease. Muscle Nerve 2009, 40, 350–357. [Google Scholar] [CrossRef]
- Zange, J.; Grehl, T.; Disselhorst-Klug, C.; Rau, G.; Muller, K.; Schroder, R.; Tegenthoff, M.; Malin, J.P.; Vorgerd, M. Breakdown of adenine nucleotide pool in fatiguing skeletal muscle in McArdle's disease: A noninvasive 31P-MRS and EMG study. Muscle Nerve 2003, 27, 728–736. [Google Scholar] [CrossRef]
- Mineo, I.; Kono, N.; Shimizu, T.; Hara, N.; Yamada, Y.; Sumi, S.; Nonaka, K.; Tarui, S. Excess purine degradation in exercising muscles of patients with glycogen storage disease types V and VII. J. Clin. Invest. 1985, 76, 556–560. [Google Scholar] [CrossRef]
- Brooke, M.H.; Patterson, V.H.; Kaiser, K.K. Hypoxanthine and Mcardle disease: A clue to metabolic stress in the working forearm. Muscle Nerve 1983, 6, 204–206. [Google Scholar] [CrossRef]
- Jinnai, K.; Kono, N.; Yamamoto, Y.; Kanda, F.; Ohno, S.; Tsutsumi, M.; Yamada, Y.; Kawachi, M.; Tarui, S.; Fujita, T. Glycogenosis type V (McArdle's disease) with hyperuricemia. A case report and clinical investigation. Eur. Neurol. 1993, 33, 204–207. [Google Scholar] [CrossRef]
- Mineo, I.; Kono, N.; Hara, N.; Shimizu, T.; Yamada, Y.; Kawachi, M.; Kiyokawa, H.; Wang, Y.L.; Tarui, S. Myogenic hyperuricemia. A common pathophysiologic feature of glycogenosis types III, V, and VII. N. Engl. J. Med. 1987, 317, 75–80. [Google Scholar] [CrossRef]
- Kitaoka, Y.; Ogborn, D.I.; Nilsson, M.I.; Mocellin, N.J.; Macneil, L.G.; Tarnopolsky, M.A. Oxidative stress and Nrf2 signaling in McArdle disease. Mol. Genet. Metab. 2013, 110, 297–302. [Google Scholar] [CrossRef]
- Vissing, J.; Lewis, S.F.; Galbo, H.; Haller, R.G. Effect of deficient muscular glycogenolysis on extramuscular fuel production in exercise. J. Appl. Physiol. 1992, 72, 1773–1779. [Google Scholar]
- Vissing, J.; Haller, R.G. The effect of oral sucrose on exercise tolerance in patients with McArdle's disease. N. Engl. J. Med. 2003, 349, 2503–2509. [Google Scholar] [CrossRef]
- Nielsen, J.N.; Wojtaszewski, J.F.; Haller, R.G.; Hardie, D.G.; Kemp, B.E.; Richter, E.A.; Vissing, J. Role of 5'AMP-activated protein kinase in glycogen synthase activity and glucose utilization: insights from patients with McArdle's disease. J. Physiol. 2002, 541, 979–989. [Google Scholar] [CrossRef]
- Robertshaw, H.A.; Raha, S.; Kaczor, J.J.; Tarnopolsky, M.A. Increased PFK activity and GLUT4 protein content in McArdle's disease. Muscle Nerve 2008, 37, 431–437. [Google Scholar] [CrossRef]
- Kitaoka, Y.; Ogborn, D.I.; Mocellin, N.J.; Schlattner, U.; Tarnopolsky, M.A. Monocarboxylate transporters and mitochondrial creatine kinase protein content in McArdle disease. Mol. Genet. Metab. 2013, 108, 259–262. [Google Scholar] [CrossRef]
- Gladden, L.B. Lactate metabolism: A new paradigm for the third millennium. J. Physiol. 2004, 558, 5–30. [Google Scholar] [CrossRef]
- Kitaoka, Y.; Hoshino, D.; Hatta, H. Monocarboxylate transporter and lactate metabolism. J. Phys. Fitness Sports Med. 2012, 1, 247–252. [Google Scholar] [CrossRef]
- Azevedo, J.L.; Tietz, E.; Two-Feathers, T.; Paull, J.; Chapman, K. Lactate, fructose and glucose oxidation profiles in sports drinks and the effect on exercise performance. PLoS One 2007, 2, e927. [Google Scholar] [CrossRef]
- Turk, W.R.; Heller, S.L.; Norris, B.J.; Nemeth, P.M. Increased muscular beta-hydroxyacyl CoA dehydrogenase with McArdle's disease. Muscle Nerve 1990, 13, 607–612. [Google Scholar] [CrossRef]
- Orngreen, M.C.; Jeppesen, T.D.; Andersen, S.T.; Taivassalo, T.; Hauerslev, S.; Preisler, N.; Haller, R.G.; van Hall, G.; Vissing, J. Fat metabolism during exercise in patients with McArdle disease. Neurology 2009, 72, 718–724. [Google Scholar] [CrossRef]
- Andersen, S.T.; Jeppesen, T.D.; Taivassalo, T.; Sveen, M.L.; Heinicke, K.; Haller, R.G.; Vissing, J. Effect of changes in fat availability on exercise capacity in McArdle disease. Arch. Neurol. 2009, 66, 762–766. [Google Scholar]
- Glatz, J.F.; Luiken, J.J.; Bonen, A. Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol. Rev. 2010, 90, 367–417. [Google Scholar] [CrossRef]
- Smith, B.K.; Jain, S.S.; Rimbaud, S.; Dam, A.; Quadrilatero, J.; Ventura-Clapier, R.; Bonen, A.; Holloway, G.P. FAT/CD36 is located on the outer mitochondrial membrane, upstream of long-chain acyl-CoA synthetase, and regulates palmitate oxidation. Biochem. J. 2011, 437, 125–134. [Google Scholar] [CrossRef]
- Wallimann, T.; Tokarska-Schlattner, M.; Schlattner, U. The creatine kinase system and pleiotropic effects of creatine. Amino Acids 2011, 40, 1271–1296. [Google Scholar] [CrossRef]
- Schlattner, U.; Tokarska-Schlattner, M.; Wallimann, T. Mitochondrial creatine kinase in human health and disease. Biochim. Biophys. Acta 2006, 1762, 164–180. [Google Scholar] [CrossRef]
- Vorgerd, M.; Grehl, T.; Jager, M.; Muller, K.; Freitag, G.; Patzold, T.; Bruns, N.; Fabian, K.; Tegenthoff, M.; Mortier, W.; et al. Creatine therapy in myophosphorylase deficiency (McArdle disease): a placebo-controlled crossover trial. Arch. Neurol. 2000, 57, 956–963. [Google Scholar] [CrossRef]
- Vorgerd, M.; Zange, J.; Kley, R.; Grehl, T.; Husing, A.; Jager, M.; Muller, K.; Schroder, R.; Mortier, W.; Fabian, K.; et al. Effect of high-dose creatine therapy on symptoms of exercise intolerance in McArdle disease: Double-blind, placebo-controlled crossover study. Arch. Neurol. 2002, 59, 97–101. [Google Scholar] [CrossRef]
- Lucia, A.; Quinlivan, R.; Wakelin, A.; Martin, M.A.; Andreu, A.L. The 'McArdle paradox': exercise is a good advice for the exercise intolerant. Br. J. Sports Med. 2013, 47, 728–729. [Google Scholar] [CrossRef]
- Quinlivan, R.; Vissing, J.; Hilton-Jones, D.; Buckley, J. Physical training for McArdle disease. Cochrane Database Syst. Rev. 2011, CD007931. [Google Scholar]
- Haller, R.G.; Wyrick, P.; Taivassalo, T.; Vissing, J. Aerobic conditioning: an effective therapy in McArdle's disease. Ann. Neurol. 2006, 59, 922–928. [Google Scholar] [CrossRef]
- Richter, E.A.; Hargreaves, M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol. Rev. 2013, 93, 993–1017. [Google Scholar] [CrossRef]
- Burgomaster, K.A.; Cermak, N.M.; Phillips, S.M.; Benton, C.R.; Bonen, A.; Gibala, M.J. Divergent response of metabolite transport proteins in human skeletal muscle after sprint interval training and detraining. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1970–R1976. [Google Scholar] [CrossRef]
- Thomas, C.; Bishop, D.J.; Lambert, K.; Mercier, J.; Brooks, G.A. Effects of acute and chronic exercise on sarcolemmal MCT1 and MCT4 contents in human skeletal muscles: Current status. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R1–R14. [Google Scholar] [CrossRef]
- Perry, C.G.; Heigenhauser, G.J.; Bonen, A.; Spriet, L.L. High-intensity aerobic interval training increases fat and carbohydrate metabolic capacities in human skeletal muscle. Appl. Physiol. Nutr. Metab. 2008, 33, 1112–1123. [Google Scholar] [CrossRef]
- Yoshida, Y.; Jain, S.S.; McFarlan, J.T.; Snook, L.A.; Chabowski, A.; Bonen, A. Exercise- and training-induced upregulation of skeletal muscle fatty acid oxidation are not solely dependent on mitochondrial machinery and biogenesis. J. Physiol. 2013, 591, 4415–4426. [Google Scholar] [CrossRef]
- Talanian, J.L.; Holloway, G.P.; Snook, L.A.; Heigenhauser, G.J.; Bonen, A.; Spriet, L.L. Exercise training increases sarcolemmal and mitochondrial fatty acid transport proteins in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E180–E188. [Google Scholar]
- Block, N.E.; Menick, D.R.; Robinson, K.A.; Buse, M.G. Effect of denervation on the expression of two glucose transporter isoforms in rat hindlimb muscle. J. Clin. Invest. 1991, 88, 1546–1552. [Google Scholar] [CrossRef]
- Koonen, D.P.; Benton, C.R.; Arumugam, Y.; Tandon, N.N.; Calles-Escandon, J.; Glatz, J.F.; Luiken, J.J.; Bonen, A. Different mechanisms can alter fatty acid transport when muscle contractile activity is chronically altered. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E1042–E1049. [Google Scholar] [CrossRef]
- Kitaoka, Y.; Takahashi, Y.; Machida, M.; Takeda, K.; Takemasa, T.; Hatta, H. Effect of AMPK activation on monocarboxylate transporter (MCT)1 and MCT4 in denervated muscle. J. Physiol. Sci. 2014, 64, 59–64. [Google Scholar] [CrossRef]
- Mate-Munoz, J.L.; Moran, M.; Perez, M.; Chamorro-Vina, C.; Gomez-Gallego, F.; Santiago, C.; Chicharro, L.; Foster, C.; Nogales-Gadea, G.; Rubio, J.C.; Andreu, A.L.; Martin, M.A.; Arenas, J.; Lucia, A. Favorable responses to acute and chronic exercise in McArdle patients. Clin. J. Sport Med. 2007, 17, 297–303. [Google Scholar] [CrossRef]
- Perez, M.; Moran, M.; Cardona, C.; Mate-Munoz, J.L.; Rubio, J.C.; Andreu, A.L.; Martin, M.A.; Arenas, J.; Lucia, A. Can patients with McArdle's disease run? Br. J. Sports Med. 2007, 41, 53–54. [Google Scholar]
- Garcia-Benitez, S.; Fleck, S.J.; Naclerio, F.; Martin, M.A.; Lucia, A. Resistance (weight lifting) training in an adolescent with McArdle disease. J. Child. Neurol. 2013, 28, 805–808. [Google Scholar] [CrossRef]
- Nogales-Gadea, G.; Pinos, T.; Lucia, A.; Arenas, J.; Camara, Y.; Brull, A.; de Luna, N.; Martin, M.A.; Garcia-Arumi, E.; Marti, R.; et al. Knock-in mice for the R50X mutation in the PYGM gene present with McArdle disease. Brain 2012, 135, 2048–2057. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kitaoka, Y. McArdle Disease and Exercise Physiology. Biology 2014, 3, 157-166. https://doi.org/10.3390/biology3010157
Kitaoka Y. McArdle Disease and Exercise Physiology. Biology. 2014; 3(1):157-166. https://doi.org/10.3390/biology3010157
Chicago/Turabian StyleKitaoka, Yu. 2014. "McArdle Disease and Exercise Physiology" Biology 3, no. 1: 157-166. https://doi.org/10.3390/biology3010157
APA StyleKitaoka, Y. (2014). McArdle Disease and Exercise Physiology. Biology, 3(1), 157-166. https://doi.org/10.3390/biology3010157