Exploiting a Reference Genome in Terms of Duplications: The Network of Paralogs and Single Copy Genes in Arabidopsis thaliana



Abstract
:
1. Introduction
2. Methods
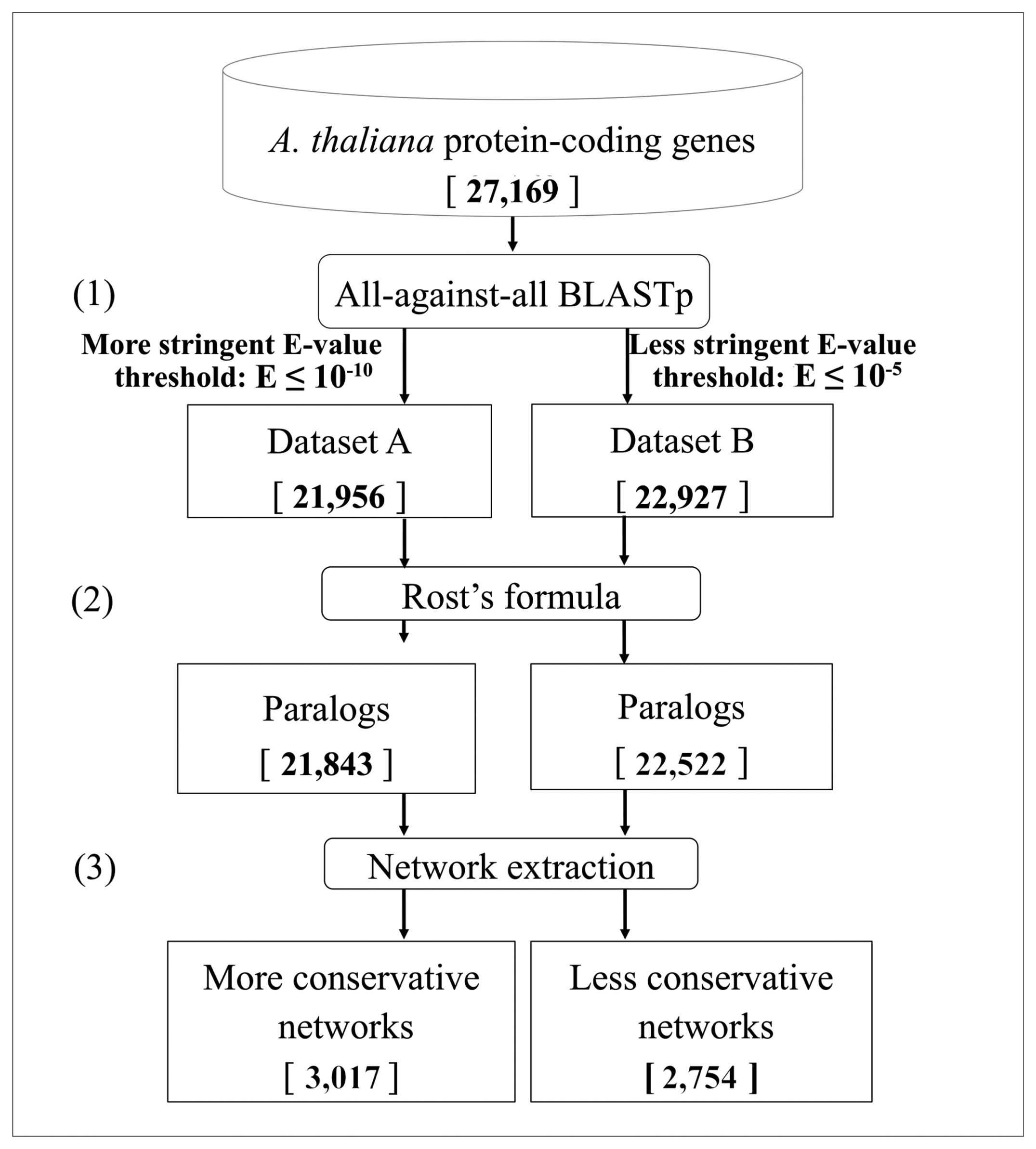
2.1. Data Retrieval
2.2. BLASTp Analysis
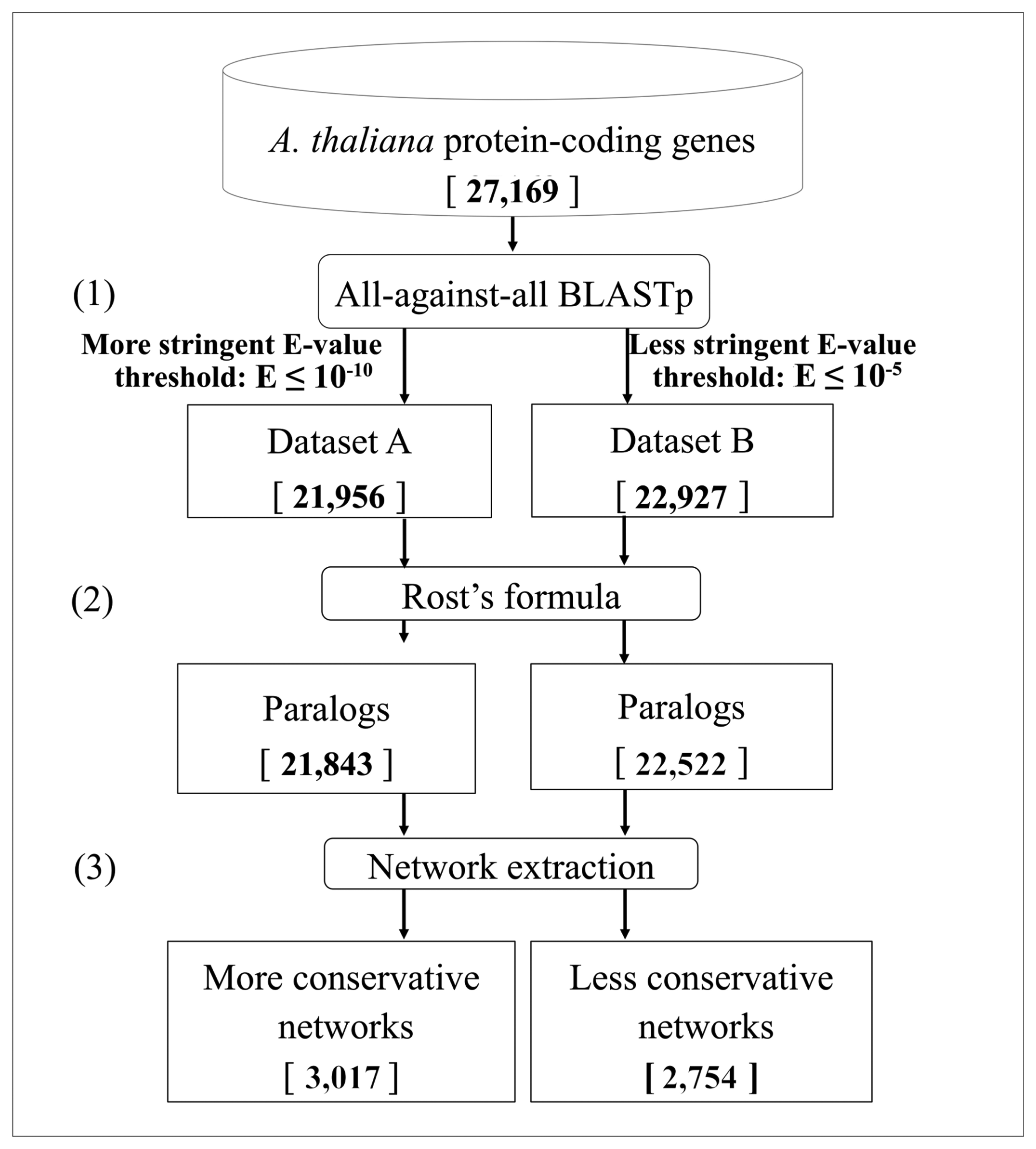
- The Expect-value cut-off. To detect different levels of sequence similarity among protein-coding genes, two different Expect-value (E-value, E) cutoffs were used: a more stringent threshold (E ≤ 10−10) and a less stringent one (E ≤ 10−5) [64,65]. To ensure the correct definition of singleton genes as those not having similar copies in the genome, we considered an even looser E-value threshold (E ≤ 10−3).
- The Rost's formula. To determine whether two proteins in a genome are paralogs when the similarity between them is in the so-called twilight zone (20%–30% of identity/length ratio), the Rost's formula [56,58] was applied with the cut-off threshold set to n = 5, according to Li et al., [59]. The use of this formula implies that all the alignments with a length shorter than 150 amino acids and with an identity score lower than 30% were discarded. These genes were removed both from the networks and from the singleton analysis, due to the intrinsic ambiguity of their paralogy relationships.
- The low-complexity BLAST filter. The BLAST analysis applies by default a masking of low-complexity regions in the query sequence [66]. Indeed, because of their repetitive nature, low-complexity regions, which are very abundant in biological sequences, may result in biologically meaningless high scoring hits [67]. However, often, sequence similarities can be missed using the masking of low complexity regions, in particular in the case of small sequences. Therefore, after a first BLASTp analyses with the default parameters, both using the more stringent cut-off and the less stringent one, another BLASTp analysis (E ≤ 10−5) was performed without the masking filter, to identify genes that could have been masked hiding similarities to other genes. Sequences showing similarity when unmasked were not included in the subsequent analyses, since they cannot be considered as singletons and their similarity with other proteins may not be necessarily associated with a paralogy relationship.
2.3. Detection of Differences in ORF Assignments
2.4. Identification of the Similarities of Protein Coding vs. Non-Protein-Coding Regions
2.5. EST-Based Functional Validation of Singleton Genes
2.6. Searching for Orthologs
2.7. Network Definition
2.8. The Web-Accessible Database
3. Results
3.1. Identification of Duplicated Genes
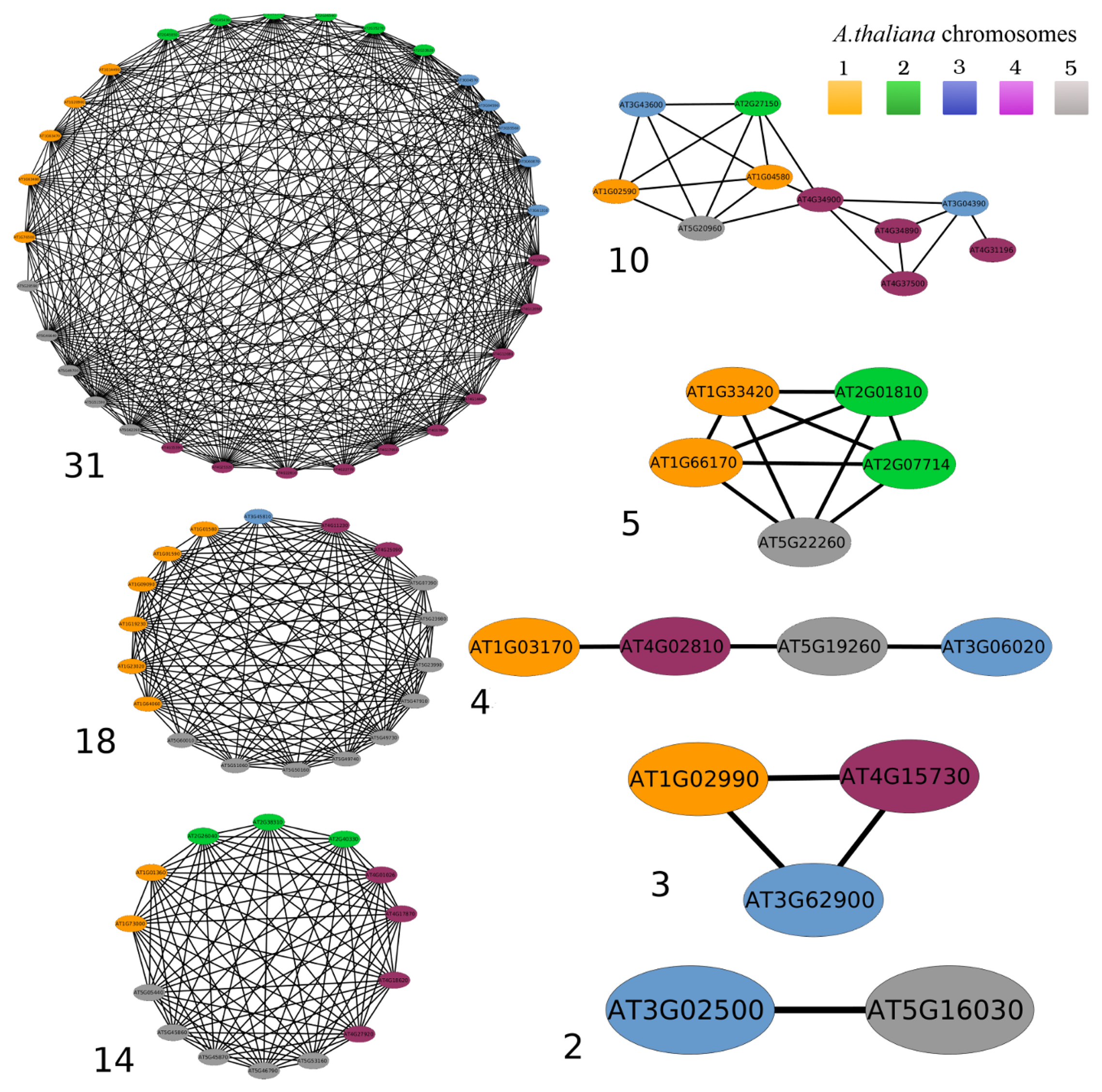
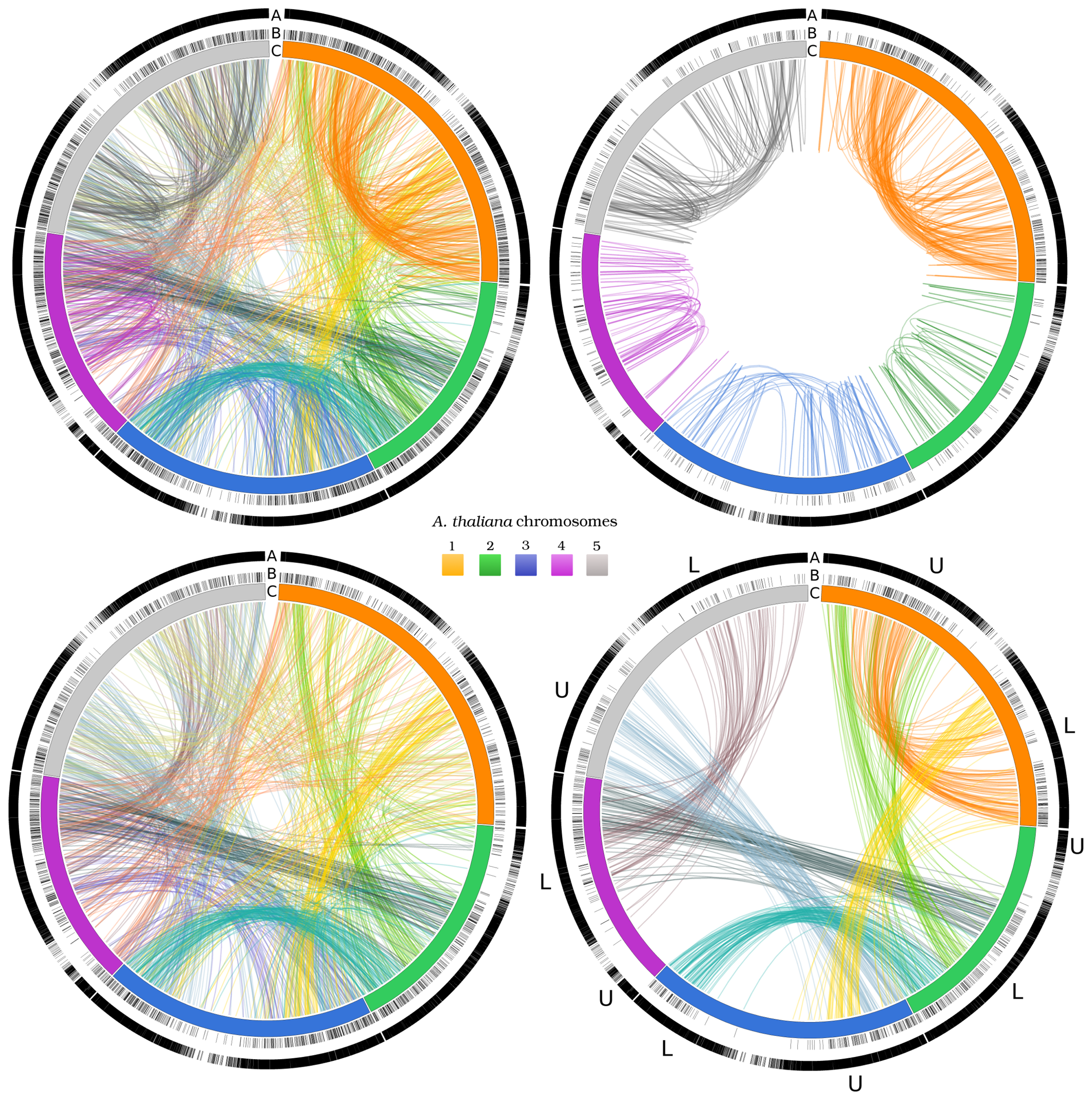
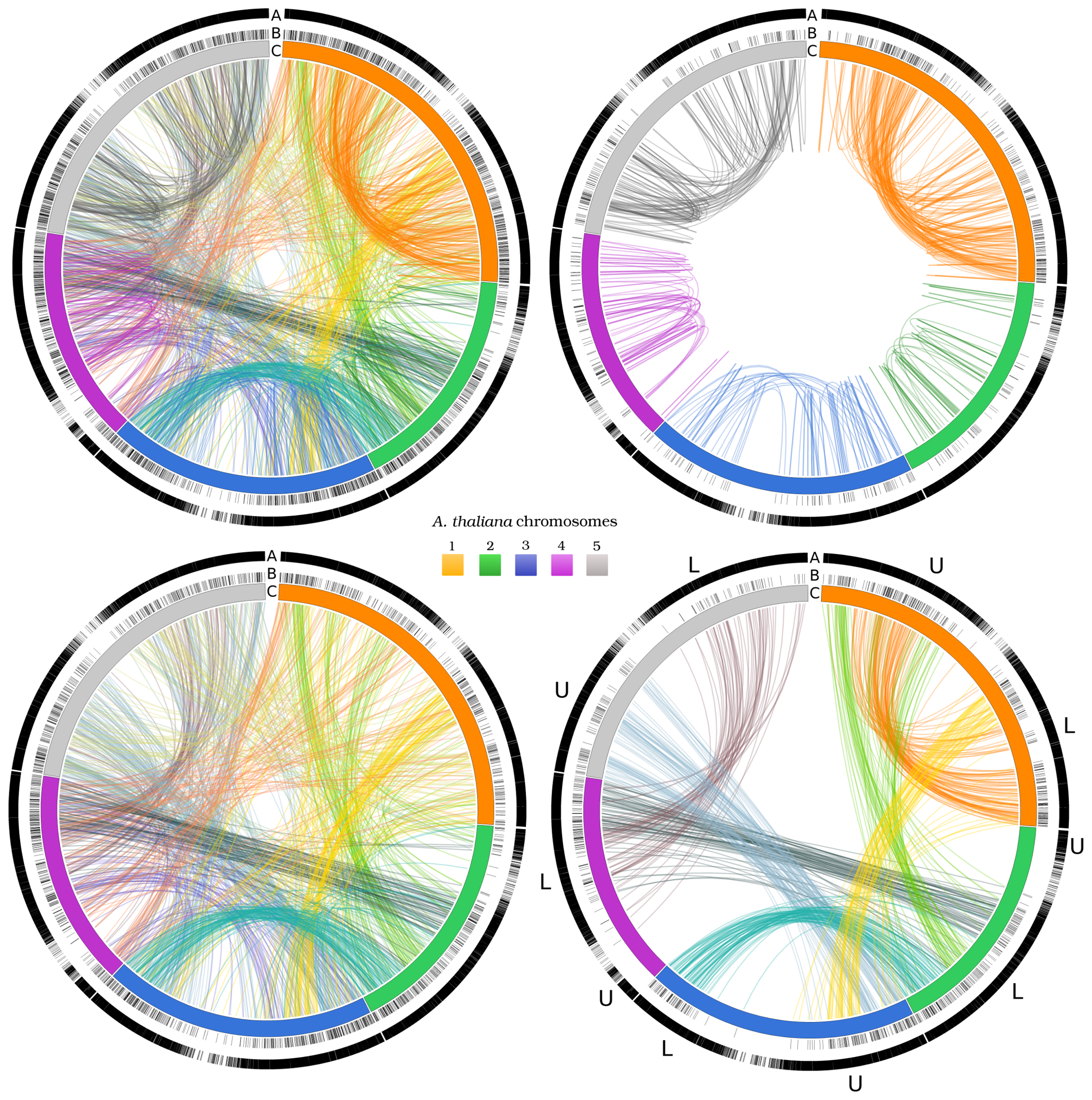
3.2. Networks of Paralogs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Network Size | Less conservative networks (E ≤ 10−5) | More conservative networks (E ≤ 10−10) | ||
|---|---|---|---|---|
| Network Number | Gene Number | Network Number | Gene Number | |
| 2 | 1, 215 | 2, 430 | 1, 347 | 2, 694 |
| 3 to 9 | 1, 232 | 5, 966 | 1, 370 | 6, 217 |
| 10 to 30 | 241 | 3, 421 | 216 | 3, 603 |
| 31 to 209 | 65 | 3, 871 | 83 | 4, 161 |
| > 210 | 1 | 6, 834 | 1 | 5, 168 |
| Number of paralogies | Number of genes |
|---|---|
| 1 | 92 |
| 2 | 118 |
| 3 | 118 |
| 4 | 120 |
| 5 to 10 | 491 |
| 11 to 50 | 1, 292 |
| 51 to 200 | 1, 804 |
| >200 | 1, 133 |
| Removed paralogies | Up to 2 | Up to 3 | Up to 4 |
|---|---|---|---|
| Removed genes | 211 | 328 | 448 |
| 4, 766 | 4, 658 | 4, 197 | |
| 169 | 138 | 157 | |
| 13 | 28 | 134 | |
| 5 | 13 | 95 | |
| Resulting network sizes | 90 | ||
| 28 | |||
| 18 | |||
| 9 | |||
| 7 |
3.3. Two-Gene Networks
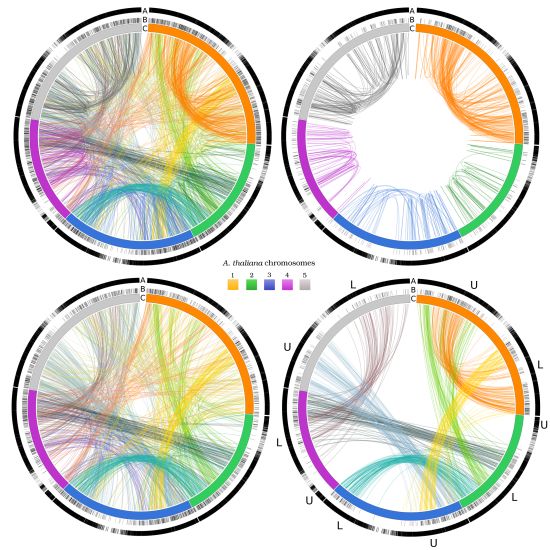
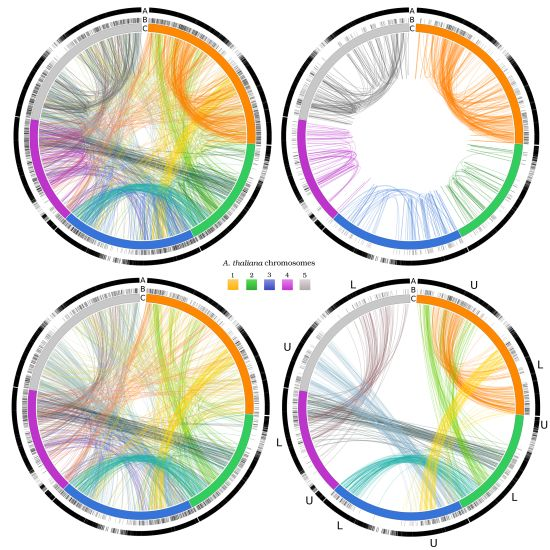
| CHR1 | CHR 2 | CHR 3 | CHR 4 | CHR 5 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 714/7,054 | 394/4,237 | 561/5,436 | 401/4,124 | 624/6,318 | |||||||
| CHR 1 | U | 23 | 107 | 5 | 62 | 21 | 18 | 14 | 34 | 18 | 28 |
| L | 107 | 33 | 4 | 21 | 61 | 17 | 4 | 25 | 19 | 37 | |
| CHR 2 | U | 5 | 4 | 5 | 1 | 3 | 0 | 0 | 3 | 2 | 6 |
| L | 62 | 21 | 1 | 30 | 21 | 81 | 2 | 68 | 24 | 20 | |
| CHR 3 | U | 21 | 61 | 3 | 21 | 28 | 16 | 7 | 37 | 86 | 29 |
| L | 18 | 17 | 0 | 81 | 16 | 8 | 5 | 12 | 22 | 37 | |
| CHR 4 | U | 14 | 4 | 0 | 2 | 7 | 5 | 2 | 6 | 7 | 5 |
| L | 34 | 25 | 3 | 68 | 37 | 12 | 6 | 43 | 19 | 57 | |
| CHR 5 | U | 18 | 19 | 2 | 24 | 86 | 22 | 7 | 19 | 35 | 39 |
| L | 28 | 37 | 6 | 20 | 29 | 37 | 5 | 57 | 39 | 30 | |
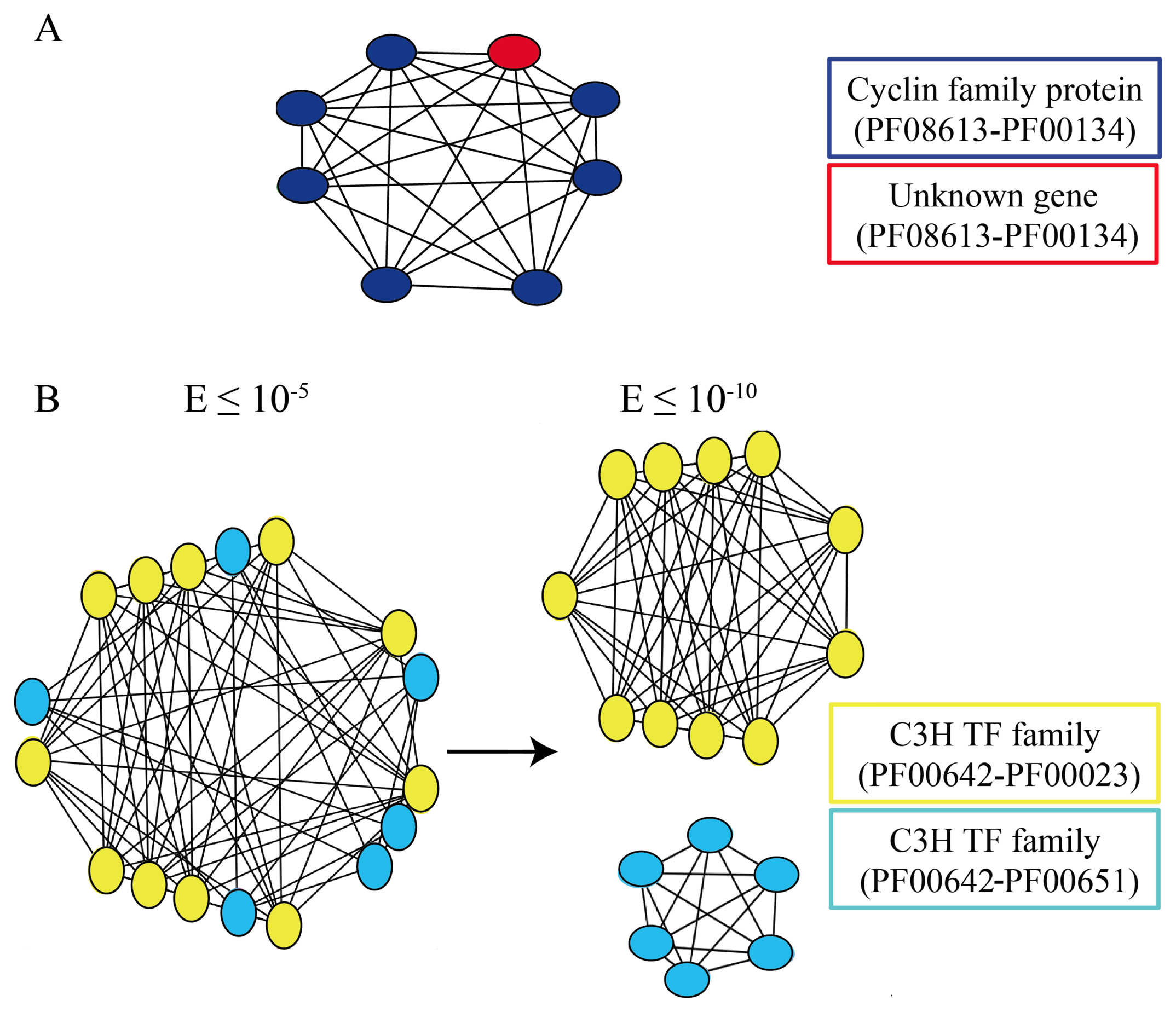
3.4. Exploiting Networks: Analysis of Arabidopsis Gene Families
| Less conservative networks (E ≤ 10−5) | More conservative networks (E ≤ 10−10) | |
|---|---|---|
| Exclusive families in exclusive networks | 52 | 58 |
| Exclusive families in mixed networks | 227 | 212 |
| Split families in exclusive networks | 20 | 29 |
| Split families in mixed networks | 81 | 81 |
3.5. Classification of Non-Paralog Genes and Identification of True Singletons
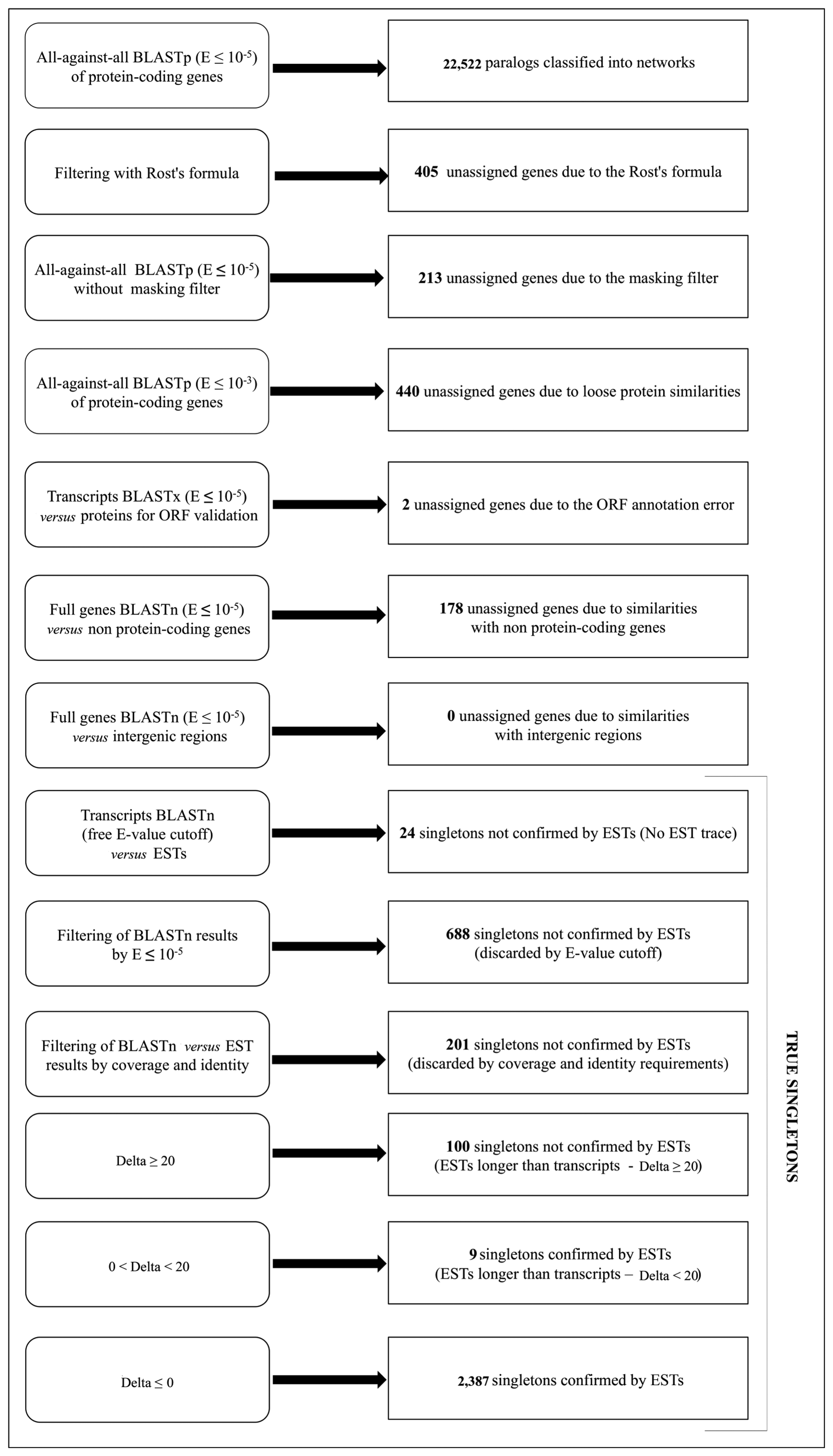
3.6. True Singletons Validation Based on Expressed Sequence Tags
3.7. Orthologs of True Singleton Genes in Other Plant Species
| Singletons with orthologs in all the considered species | Singletons with orthologs in at least one species+ | Singletons without orthologs | Total | ||
|---|---|---|---|---|---|
| EST confirmed | 374 | 1,126 | 896 | 2,396 | |
| A. Lyrata | No ESTs | 1 | 55 | 957 | 1,013 |
| B. Rapa | Totals | 375 | 1,181 | 1,853 | 3,409 |
| O. sativa | EST confirmed | 1,449 | 498 | 449 | 2,396 |
| P. trichocarpa | No ESTs | 0 | 0 | 1,013 | 1,013 |
| S. bicolor | Totals | 1,449 | 498 | 1,462 | 3,409 |
| V. vinifera | |||||
3.8. The Web-Accessible Database
4. Discussion
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Meyerowitz, E.; Somerville, C. Arabidopsis, Cold Spring Harbor Monograph Series; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1994. [Google Scholar]
- Somerville, C. Arabidopsis blooms. Plant Cell 1989, 1, 1131. [Google Scholar]
- Somerville, C.; Koornneef, M. A fortunate choice: The history of Arabidopsis as a model plant. Nat. Rev. Genet. 2002, 3, 883–889. [Google Scholar]
- Meinke, D.W.; Cherry, J.M.; Dean, C.; Rounsley, S.D.; Koornneef, M. Arabidopsis thaliana: A model plant for genome analysis. Science 1998, 282, 679–682. [Google Scholar]
- The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815.
- Hall, A.E.; Fiebig, A.; Preuss, D. Beyond the Arabidopsis genome: Opportunities for comparative genomics. Plant Physiol. 2002, 129, 1439–1447. [Google Scholar]
- Yogeeswaran, K.; Frary, A.; York, T.L.; Amenta, A.; Lesser, A.H.; Nasrallah, J.B.; Tanksley, S.D.; Nasrallah, M.E. Comparative genome analyses of Arabidopsis spp.: Inferring chromosomal rearrangement events in the evolutionary history of A. thaliana. Genome Res. 2005, 15, 505–515. [Google Scholar]
- Taji, T.; Seki, M.; Satou, M.; Sakurai, T.; Kobayashi, M.; Ishiyama, K.; Narusaka, Y.; Narusaka, M.; Zhu, J.K.; Shinozaki, K. Comparative genomics in salt tolerance between Arabidopsis and Arabidopsis-related halophyte salt cress using Arabidopsis microarray. Plant Physiol. 2004, 135, 1697–1709. [Google Scholar]
- Nelson, D.R.; Schuler, M.A.; Paquette, S.M.; Werck-Reichhart, D.; Bak, S. Comparative genomics of rice and Arabidopsis. Analysis of 727 cytochrome P450 genes and pseudogenes from a monocot and a dicot. Plant Physiol. 2004, 135, 756–772. [Google Scholar]
- Town, C.D.; Cheung, F.; Maiti, R.; Crabtree, J.; Haas, B.J.; Wortman, J.R.; Hine, E.E.; Althoff, R.; Arbogast, T.S.; Tallon, L.J.; et al. Comparative genomics of Brassica oleracea and Arabidopsis thaliana reveal gene loss, fragmentation, and dispersal after polyploidy. Plant Cell 2006, 18, 1348–1359. [Google Scholar]
- Ku, H.M.; Vision, T.; Liu, J.; Tanksley, S.D. Comparing sequenced segments of the tomato and Arabidopsis genomes: large-scale duplication followed by selective gene loss creates a network of synteny. Proc. Natl. Acad. Sci. USA 2000, 97, 9121–9126. [Google Scholar]
- Boivin, K.; Acarkan, A.; Mbulu, R.S.; Clarenz, O.; Schmidt, R. The Arabidopsis genome sequence as a tool for genome analysis in Brassicaceae. A comparison of the Arabidopsis and Capsella rubella genomes. Plant Physiol. 2004, 135, 735–744. [Google Scholar]
- International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature 2005, 436, 793–800.
- Ming, R.; Hou, S.; Feng, Y.; Yu, Q.; Dionne-Laporte, A.; Saw, J.H.; Senin, P.; Wang, W.; Ly, B.V.; Lewis, K.L.; et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 2008, 452, 991–996. [Google Scholar]
- Somers, D.A.; Langridge, P.; Gustafson, J.P. Plant Genomics: Methods And Protocols; Humana Press: New York, NY, USA, 2009. [Google Scholar]
- Koch, M.A.; Kiefer, M. Genome evolution among cruciferous plants: A lecture from the comparison of the genetic maps of three diploid species—Capsella rubella, Arabidopsis lyrata subsp. petraea, and A. thaliana. Am. J. Bot. 2005, 92, 761–767. [Google Scholar]
- Koornneef, M.; Meinke, D. The development of Arabidopsis as a model plant. Plant J. 2010, 61, 909–921. [Google Scholar]
- Simillion, C.; Vandepoele, K.; van Montagu, M.C.; Zabeau, M.; van de Peer, Y. The hidden duplication past of Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2002, 99, 13627–13632. [Google Scholar]
- Debodt, S.; Maere, S.; Vandepeer, Y. Genome duplication and the origin of angiosperms. Trends Ecol. Evol. 2005, 20, 591–597. [Google Scholar]
- Vision, T.J.; Brown, D.G.; Tanksley, S.D. The origins of genomic duplications in Arabidopsis. Science 2000, 290, 2114–2117. [Google Scholar]
- Blanc, G.; Barakat, A.; Guyot, R.; Cooke, R.; Delseny, M. Extensive duplication and reshuffling in the Arabidopsis genome. Plant Cell 2000, 12, 1093–1101. [Google Scholar]
- Simillion, C.; Vandepoele, K.; van Montagu, M.C.; Zabeau, M.; van de Peer, Y. The hidden duplication past of Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2002, 99, 13627–13632. [Google Scholar]
- Wolfe, K.H. Yesterday's polyploids and the mystery of diploidization. Nat. Rev. Genet. 2001, 2, 333–341. [Google Scholar]
- Cui, L.; Wall, P.K.; Leebens-Mack, J.H.; Lindsay, B.G.; Soltis, D.E.; Doyle, J.J.; Soltis, P.S.; Carlson, J.E.; Arumuganathan, K.; Barakat, A.; et al. Widespread genome duplications throughout the history of flowering plants. Genome Res. 2006, 16, 738–749. [Google Scholar]
- Blanc, G.; Hokamp, K.; Wolfe, K.H. A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res. 2003, 13, 137–144. [Google Scholar]
- Blanc, G.; Wolfe, K.H. Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell 2004, 16, 1679–1691. [Google Scholar]
- Van de Peer, Y.; Meyer, A. Large-scale gene and ancient genome duplications. In The Evolution of The Genome; Elsevier Academic Press: San Diego, CA, USA, 2005; pp. 328–368. [Google Scholar]
- Jiao, Y.; Leebens-Mack, J.; Ayyampalayam, S.; Bowers, J.E.; McKain, M.R.; McNeal, J.; Rolf, M.; Ruzicka, D.R.; Wafula, E.; Wickett, N.J.; et al. A genome triplication associated with early diversification of the core eudicots. Genome Biol. 2012, 13, R3. [Google Scholar]
- Jiao, Y.; Wickett, N.J.; Norman, J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar]
- Van de Peer, Y. A mystery unveiled. Genome Biol. 2011, 12, 113. [Google Scholar]
- Lysak, M.A.; Fransz, P.F.; Ali, H.B.; Schubert, I. Chromosome painting in Arabidopsis thaliana. Plant J. 2001, 28, 689–697. [Google Scholar]
- Lysak, M.A.; Koch, M.A.; Pecinka, A.; Schubert, I. Chromosome triplication found across the tribe Brassiceae. Genome Res. 2005, 15, 516–525. [Google Scholar]
- Tang, H.; Bowers, J.E.; Wang, X.; Ming, R.; Alam, M.; Paterson, A.H. Synteny and collinearity in plant genomes. Science 2008, 320, 486–488. [Google Scholar]
- Conner, J.A.; Conner, P.; Nasrallah, M.E.; Nasrallah, J.B. Comparative mapping of the Brassica S locus region and its homeolog in Arabidopsis: Implications for the evolution of mating systems in the Brassicaceae. Plant Cell Online 1998, 10, 801–812. [Google Scholar]
- Johnston, J.; Pepper, A.; Hall, A.; Chen, Z.; Hodnett, G.; Drabek, J.; Lopez, R.; Price, H. Evolution of genome size in Brassicaceae. Ann. Bot. 2005, 95, 229–235. [Google Scholar]
- Rong, J.; Bowers, J.E.; Schulze, S.R.; Waghmare, V.N.; Rogers, C.J.; Pierce, G.J.; Zhang, H.; Estill, J.C.; Paterson, A.H. Comparative genomics of Gossypium and Arabidopsis: Unraveling the consequences of both ancient and recent polyploidy. Genome Res. 2005, 15, 1198–1210. [Google Scholar]
- Jaillon, O.; Aury, J.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar]
- Adams, K.L.; Wendel, J.F. Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 2005, 8, 135–141. [Google Scholar]
- Bowers, J.E.; Chapman, B.A.; Rong, J.; Paterson, A.H. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 2003, 422, 433–438. [Google Scholar]
- Ermolaeva, M.; Wu, M.; Eisen, J.; Salzberg, S. The age of the Arabidopsis thaliana genome duplication. Plant Mol. Biol. 2003, 51, 859–866. [Google Scholar]
- Raes, J.; Klaas, V.; Klaas, V.; Simillion, C.; Saeys, Y.; van de Peer, Y. Investigating ancient duplication events in the Arabidopsis genome. J. Struct. Funct. Genomics 2003, 3, 117–129. [Google Scholar]
- Seoighe, C.; Gehring, C. Genome duplication led to highly selective expansion of the Arabidopsis thaliana proteome. Trends Genet. 2004, 20, 461–464. [Google Scholar]
- Taylor, J.S.; Raes, J. Duplication and divergence: The evolution of new genes and old ideas. Annu. Rev. Genet. 2004, 38, 615–643. [Google Scholar]
- Duarte, J.; Wall, P.K.; Edger, P.; Landherr, L.; Ma, H.; Pires, J.C.; Leebens-Mack, J.; dePamphilis, C. Identification of shared single copy nuclear genes in Arabidopsis, populus, vitis and oryza and their phylogenetic utility across various taxonomic levels. BMC Evol. Biol. 2010, 10, 61. [Google Scholar]
- Proost, S.; van Bel, M.; Sterck, L.; Billiau, K.; van Parys, T.; van de Peer, Y.; Vandepoele, K. PLAZA: A comparative genomics resource to study gene and genome evolution in plants. Plant Cell 2009, 21, 3718–3731. [Google Scholar]
- Rouard, M.; Guignon, V.; Aluome, C.; Laporte, M.A.; Droc, G.; Walde, C.; Zmasek, C.M.; Prin, C.; Conte, M.G. GreenPhylDB v2.0: Comparative and functional genomics in plants. Nucleic Acids Res. 2011, 39, 1095–1102. [Google Scholar]
- Duvick, J.; Fu, A.; Muppirala, U.; Sabharwal, M.; Wilkerson, M.D.; Lawrence, C.J.; Lushbough, C.; Brendel, V. PlantGDB: A resource for comparative plant genomics. Nucleic Acids Res. 2008, 36, 959–965. [Google Scholar]
- Zhang, P.; Gu, Z.; Li, W. Different evolutionary patterns between young duplicate genes in the human genome. Genome Biol. 2003, 4, R56. [Google Scholar]
- Gu, Z.; Nicolae, D.; Lu, H.; Li, W. Rapid divergence in expression between duplicate genes inferred from microarray data. Trends Genet. 2002, 18, 609–613. [Google Scholar]
- Gu, Z.; Steinmetz, L.M.; Gu, X.; Scharfe, C.; Davis, R.W.; Li, W.H.H. Role of duplicate genes in genetic robustness against null mutations. Nature 2003, 421, 63–66. [Google Scholar]
- Friedman, R.; Hughes, A. Gene duplication and the structure of eukaryotic genomes. Genome Res. 2001, 11, 373–381. [Google Scholar]
- Hsiao, T.; Vitkup, D. Role of duplicate genes in robustness against deleterious human mutations. PLoS Genet. 2008, 4, e1000014. [Google Scholar]
- Li, Q.; Lee, B.; Zhang, L. Genome-scale analysis of positional clustering of mouse testis-specific genes. BMC Genomics 2005, 6, 7. [Google Scholar]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar]
- Östlund, G.; Schmitt, T.; Forslund, K.; Köstler, T.; Messina, D.N.; Roopra, S.; Frings, O.; Sonnhammer, E.L. InParanoid 7: New algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 2010, 38, D196–D203. [Google Scholar]
- Yang, L.; Gaut, B.S. Factors that contribute to variation in evolutionary rate among Arabidopsis genes. Mol. Biol. Evol. 2011, 28, 2359–2369. [Google Scholar]
- Vilella, A.; Severin, J.; Ureta-Vidal, A.; Durbin, R.; Heng, L.; Birney, E. EnsemblCompara GeneTrees: Analysis of complete, duplication aware phylogenetic trees in vertebrates. Genome Res. 2008, 14, 327–335. [Google Scholar]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. 1999, 12, 85–94. [Google Scholar]
- Li, W.H.; Gu, Z.; Wang, H.; Nekrutenko, A. Evolutionary analyses of the human genome. Nature 2001, 409, 847–849. [Google Scholar]
- The Arabidopsis Information Resource (TAIR). Available online: http://www.arabidopsis.org/ (accessed on 27 November 2013).
- Zhang, H.; Jin, J.; Tang, L.; Zhao, Y.; Gu, X.; Gao, G.; Luo, J. PlantTFDB 2.0: Update and improvement of the comprehensive plant transcription factor database. Nucleic Acids Res. 2011, 39, 1114–1117. [Google Scholar]
- Yilmaz, A.; Mejia-Guerra, M.K.; Kurz, K.; Liang, X.; Welch, L.R.; Grotewold, E. AGRIS: The Arabidopsis Gene Regulatory Information Server, an update. Nucleic Acids Res. 2011, 39, 1118–1122. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
- Rubin, G.M.; Yandell, M.D.; Wortman, J.R.; Gabor Miklos, G.L.; Nelson, C.R.; Hariharan, I.K.; Fortini, M.E.; Li, P.W.; Apweiler, R.; Fleischmann, W.; et al. Comparative genomics of the eukaryotes. Science 2000, 287, 2204–2215. [Google Scholar]
- He, X.; Zhang, J. Gene complexity and gene duplicability. Curr. Biol. 2005, 15, 1016–1021. [Google Scholar]
- Wootton, J.C.; Federhen, S. Analysis of compositionally biased regions in sequence databases. Methods Enzymol. 1996, 266, 554–571. [Google Scholar]
- Sharon, I.; Birkland, A.; Chang, K.; El-Yaniv, R.; Yona, G. Correcting blast e-Values for low-complexity segments. J. Comput. Biol. 2005, 12, 980–1003. [Google Scholar]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar]
- Loraine, A.E.; McCormick, S.; Estrada, A.; Patel, K.; Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant Physiol. 2013, 162, 1092–1109. [Google Scholar]
- Smedley, D.; Haider, S.; Ballester, B.; Holland, R.; London, D.; Thorisson, G.; Kasprzyk, A. BioMart-biological queries made easy. BMC Genomics 2009, 10, 22. [Google Scholar]
- The PHP scripting language network. Available online: http://www.php.net/ (accessed on 27 November 2013).
- The MySQL open source database. Available online: http://www.mysql.com/ (accessed on 27 November 2013).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar]
- Moreno-Hagelsieb, G.; Latimer, K. Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics 2008, 24, 319–324. [Google Scholar]
- Kawabe, A.; Hansson, B.; Hagenblad, J.; Forrest, A.; Charlesworth, D. Centromere locations and associated chromosome rearrangements in Arabidopsis lyrata and A. thaliana. Genetics 2006, 173, 1613–1619. [Google Scholar]
- Pérez-Rodríguez, P.; Riaño-Pachón, D.M.; Corrêa, L.G.G.; Rensing, S.A.; Kersten, B.; Mueller-Roeber, B. PlnTFDB: Updated content and new features of the plant transcription factor database. Nucleic Acids Res. 2010, 38, D822–D827. [Google Scholar]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar]
- Rosenfeld, J.; DeSalle, R. E value cutoff and eukaryotic genome content phylogenetics. Mol. Phylogenet. Evol. 2012, 63, 342–350. [Google Scholar]
- Van de Peer, Y.; Fawcett, J.; Proost, S.; Sterck, L.; Vandepoele, K. The flowering world: A tale of duplications. Trends Plant Sci. 2009, 14, 680–688. [Google Scholar]
- pARsi: paralogs and singleton genes browser for Arabidopsis. Available online: http://biosrv.cab.unina.it/athparalogs/main/index/ (accessed on 27 November 2013).
- Fransz, P.; Armstrong, S.; Alonso-Blanco, C.; Fischer, T.C.; Torres-Ruiz, R.A.; Jones, G. Cytogenetics for the model system Arabidopsis thaliana. Plant J. 1998, 13, 867–876. [Google Scholar]
- Van de Peer, Y. Computational approaches to unveiling ancient genome duplications. Nat. Rev. Genet. 2004, 5, 752–763. [Google Scholar]
- Wootton, J.C.; Federhen, S. Statistics of local complexity in amino acid sequences and sequence databases. Comput. Chem. 1993, 17, 149–163. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sangiovanni, M.; Vigilante, A.; Chiusano, M.L. Exploiting a Reference Genome in Terms of Duplications: The Network of Paralogs and Single Copy Genes in Arabidopsis thaliana. Biology 2013, 2, 1465-1487. https://doi.org/10.3390/biology2041465
Sangiovanni M, Vigilante A, Chiusano ML. Exploiting a Reference Genome in Terms of Duplications: The Network of Paralogs and Single Copy Genes in Arabidopsis thaliana. Biology. 2013; 2(4):1465-1487. https://doi.org/10.3390/biology2041465
Chicago/Turabian StyleSangiovanni, Mara, Alessandra Vigilante, and Maria Luisa Chiusano. 2013. "Exploiting a Reference Genome in Terms of Duplications: The Network of Paralogs and Single Copy Genes in Arabidopsis thaliana" Biology 2, no. 4: 1465-1487. https://doi.org/10.3390/biology2041465