Application of Fluorescence Two-Dimensional Difference In-Gel Electrophoresis as a Proteomic Biomarker Discovery Tool in Muscular Dystrophy Research

Abstract
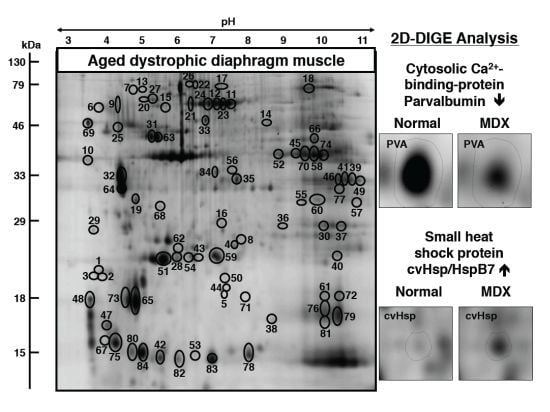
:
1. Introduction
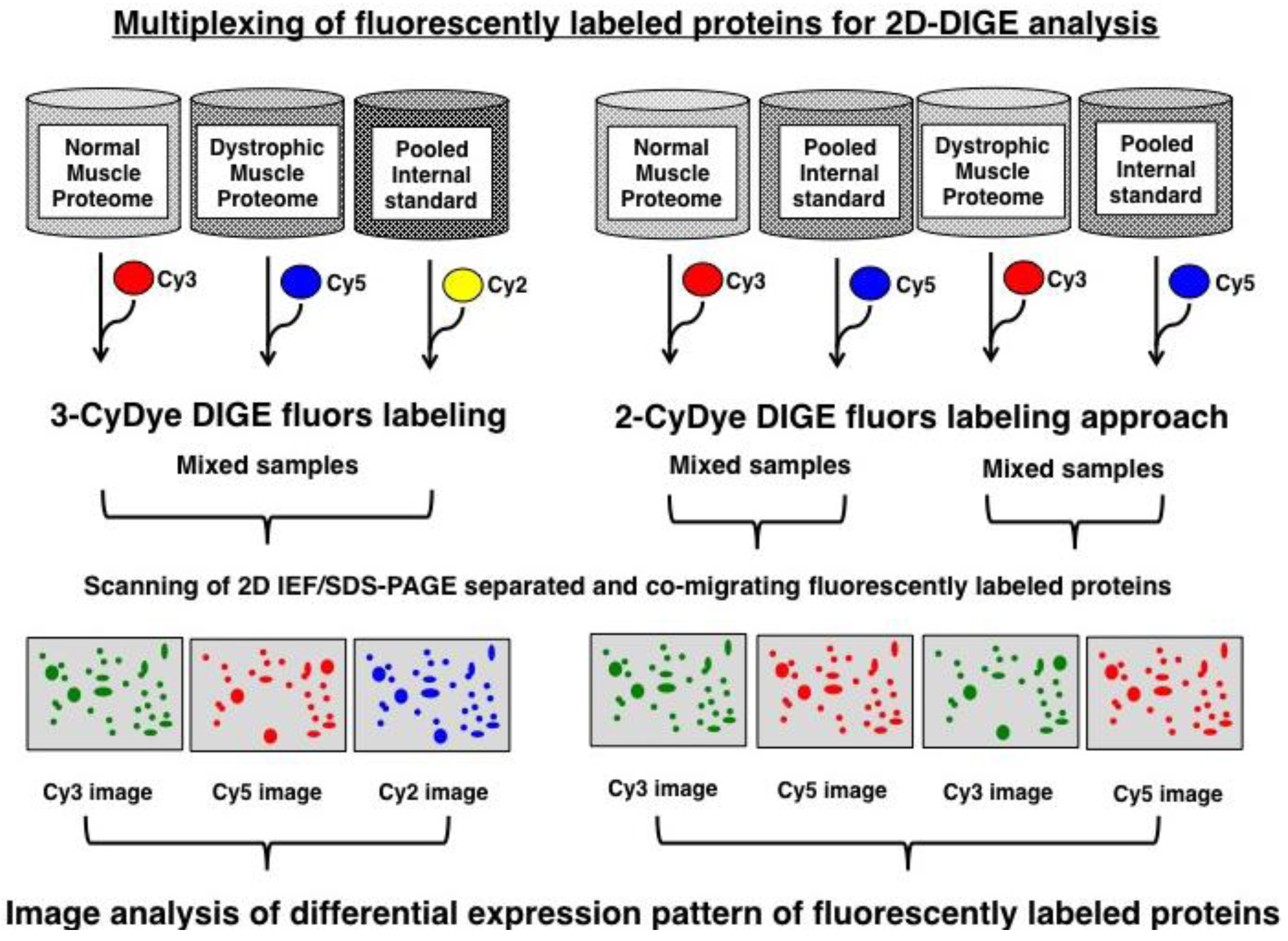
2. Experimental Section
2.1. Chemicals and Materials
2.2. Preparation of Diaphragm Extracts from Aged mdx Mice
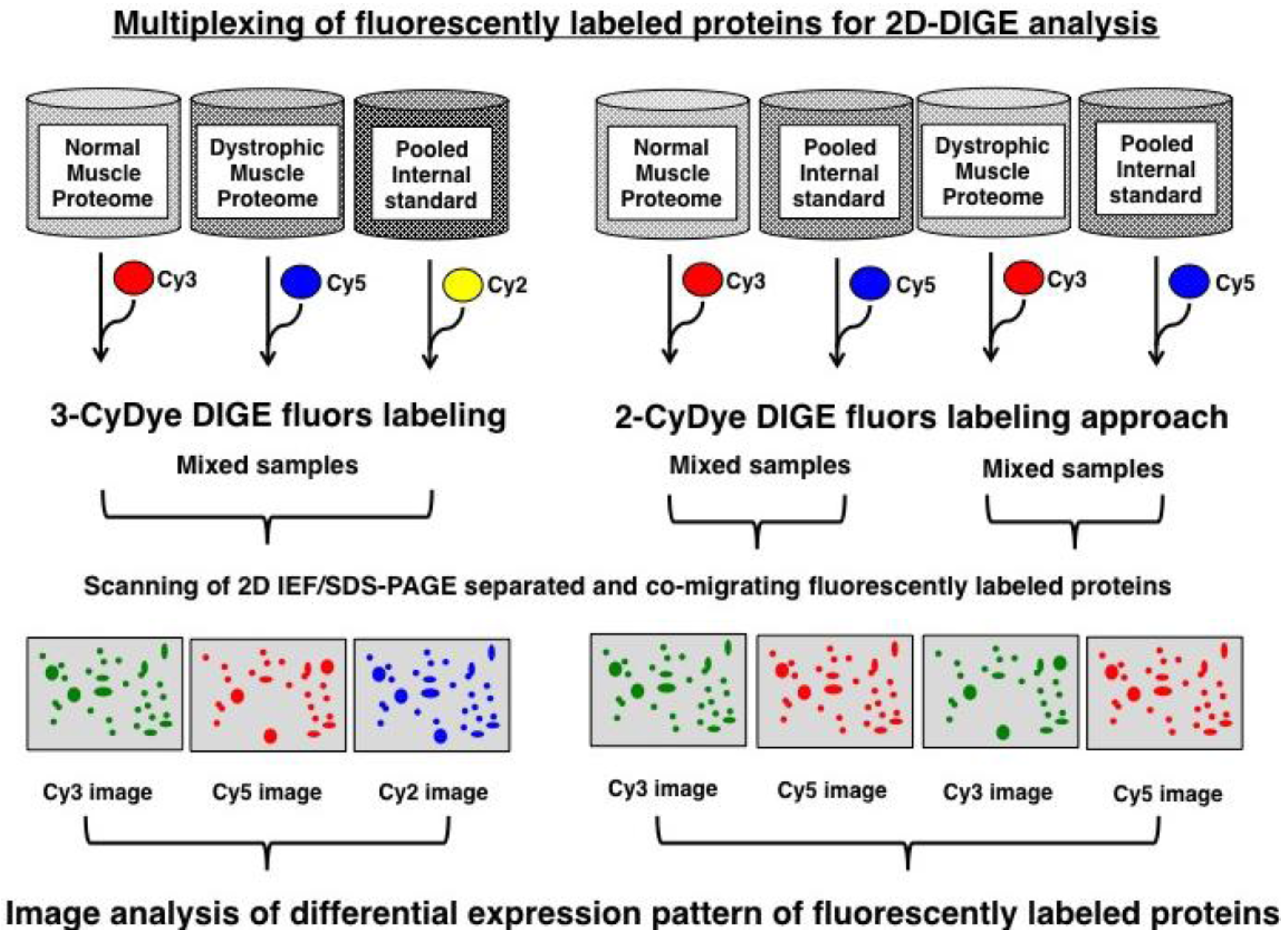
2.3. Fluorescence Two-Dimensional Difference In-Gel Electrophoretic Analysis of the Diaphragm Muscle Proteome
2.4. Mass Spectrometric Identification of Diaphragm Proteins
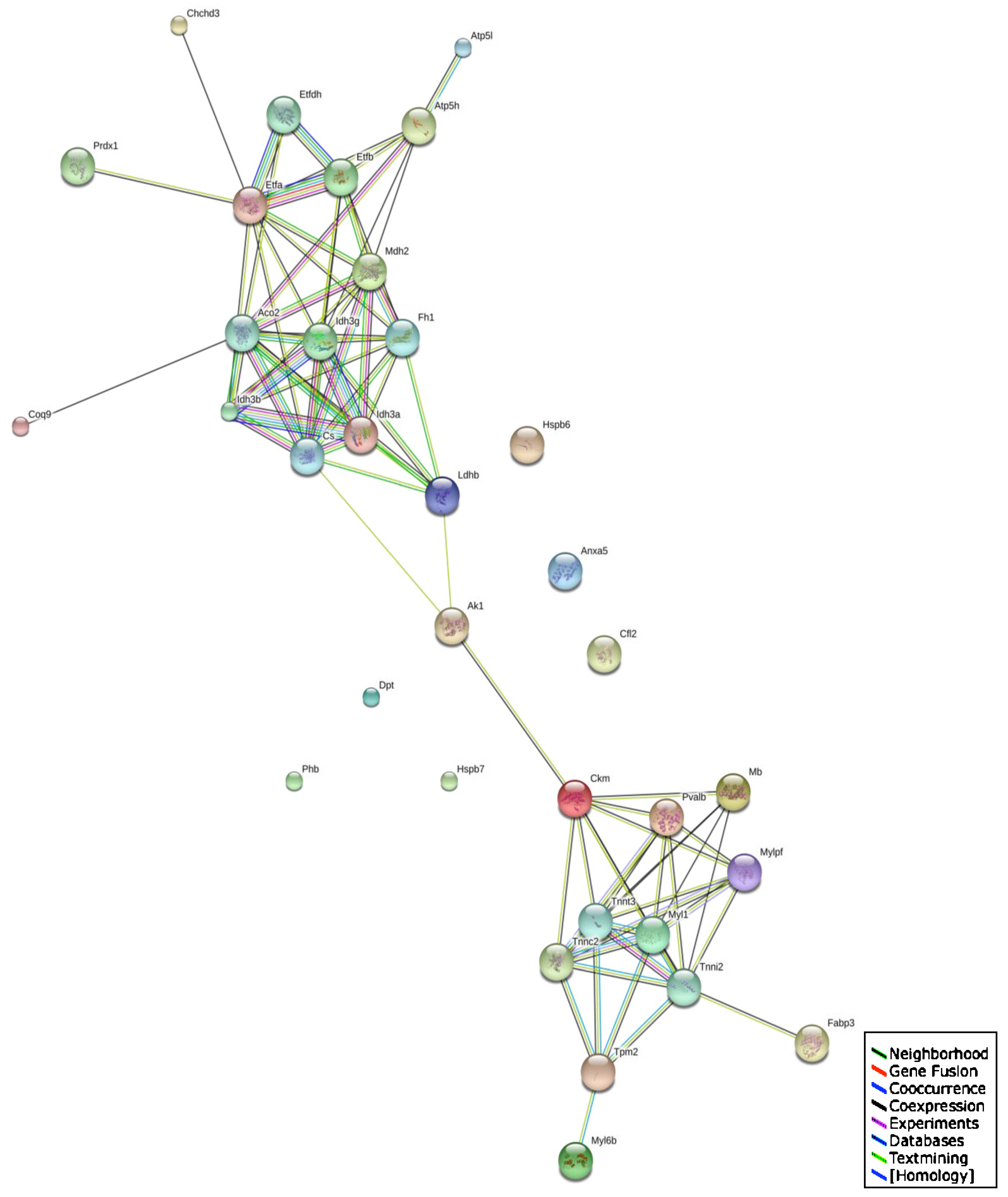
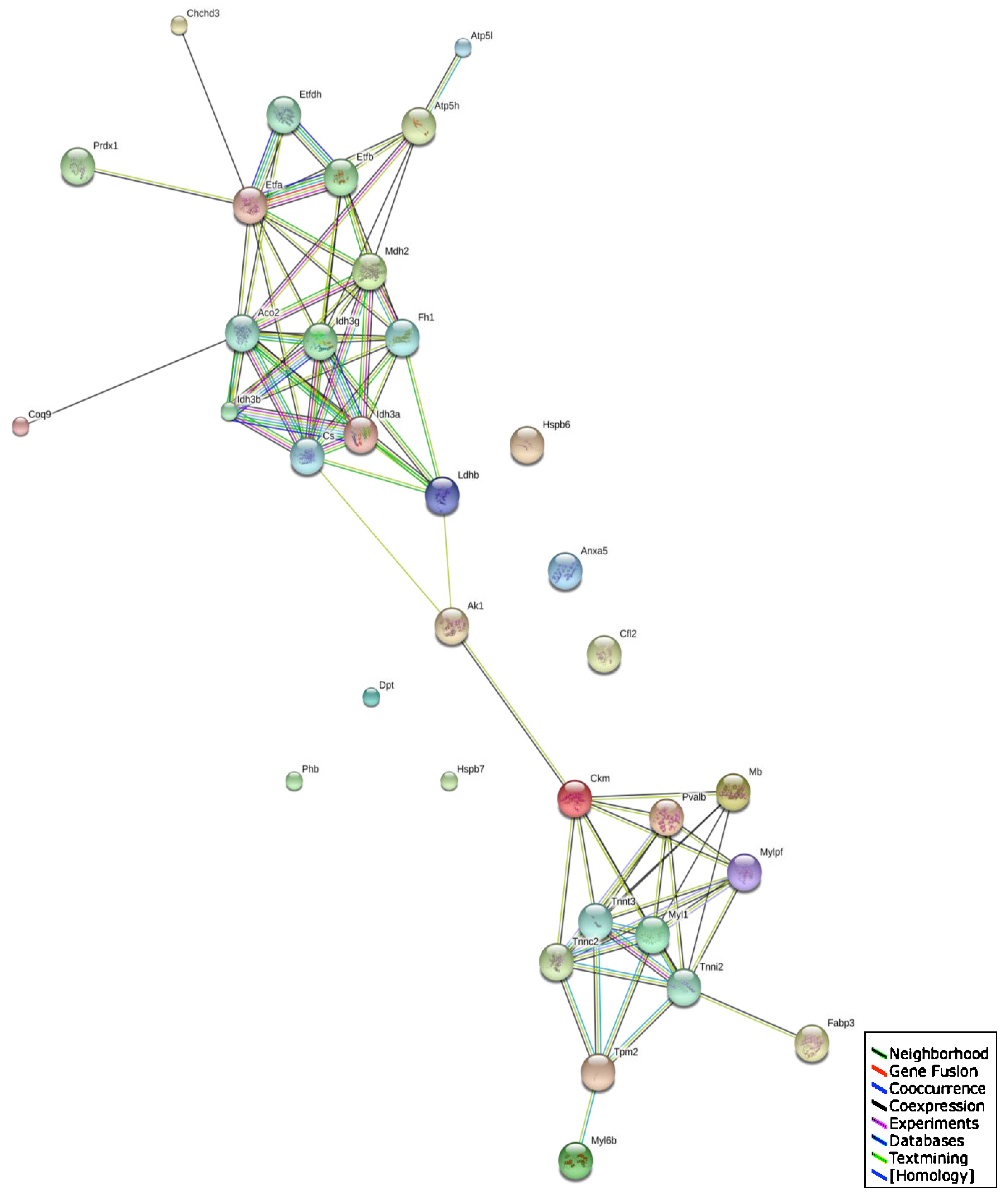
2.5. Bioinformatics Analysis of Protein Classes and Potential Protein Interactions
2.6. Immunoblot Analysis
3. Results
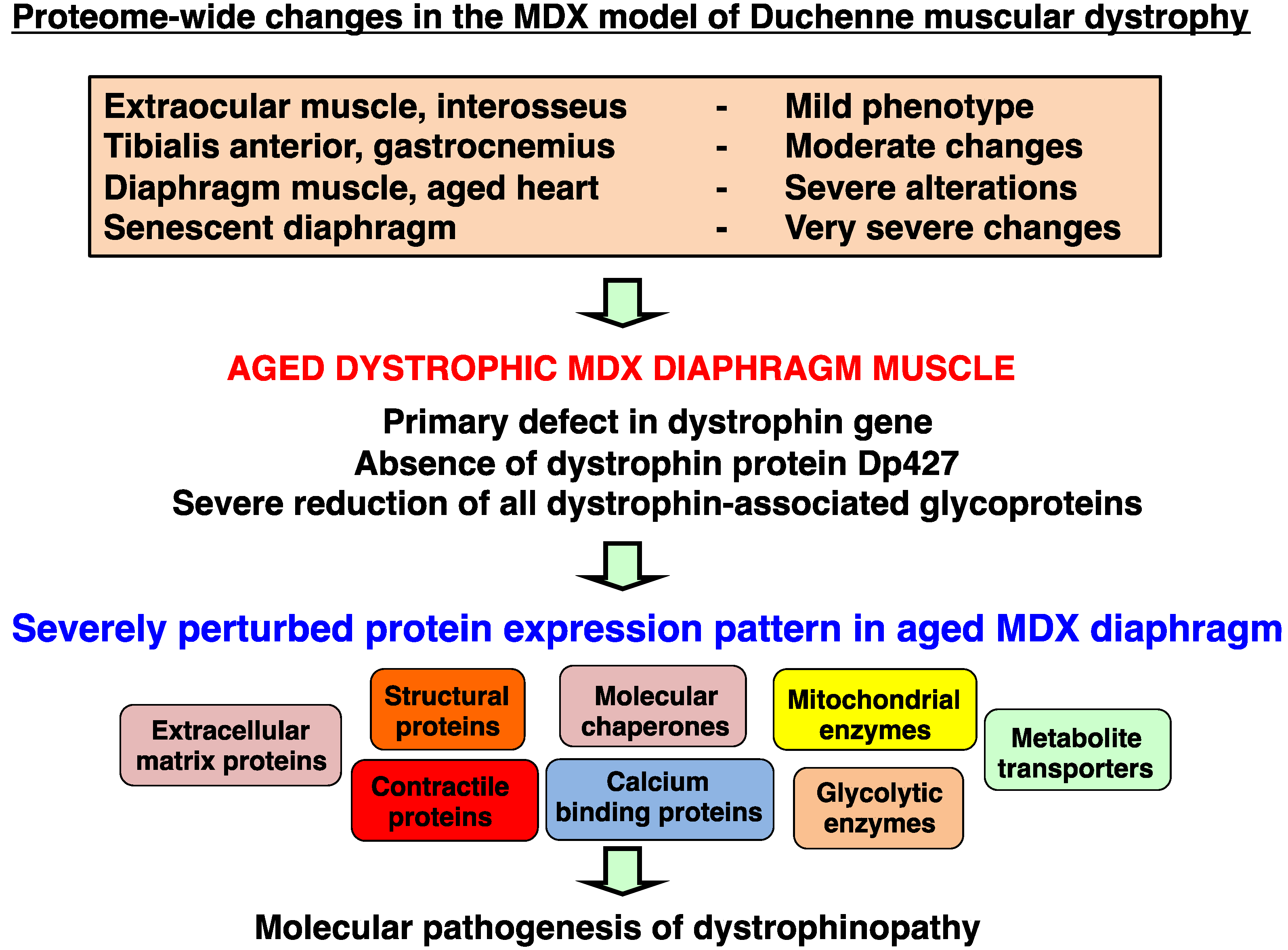
3.1. Overview of Proteomic Surveys of the mdx Animal Model of Duchenne Muscular Dystrophy
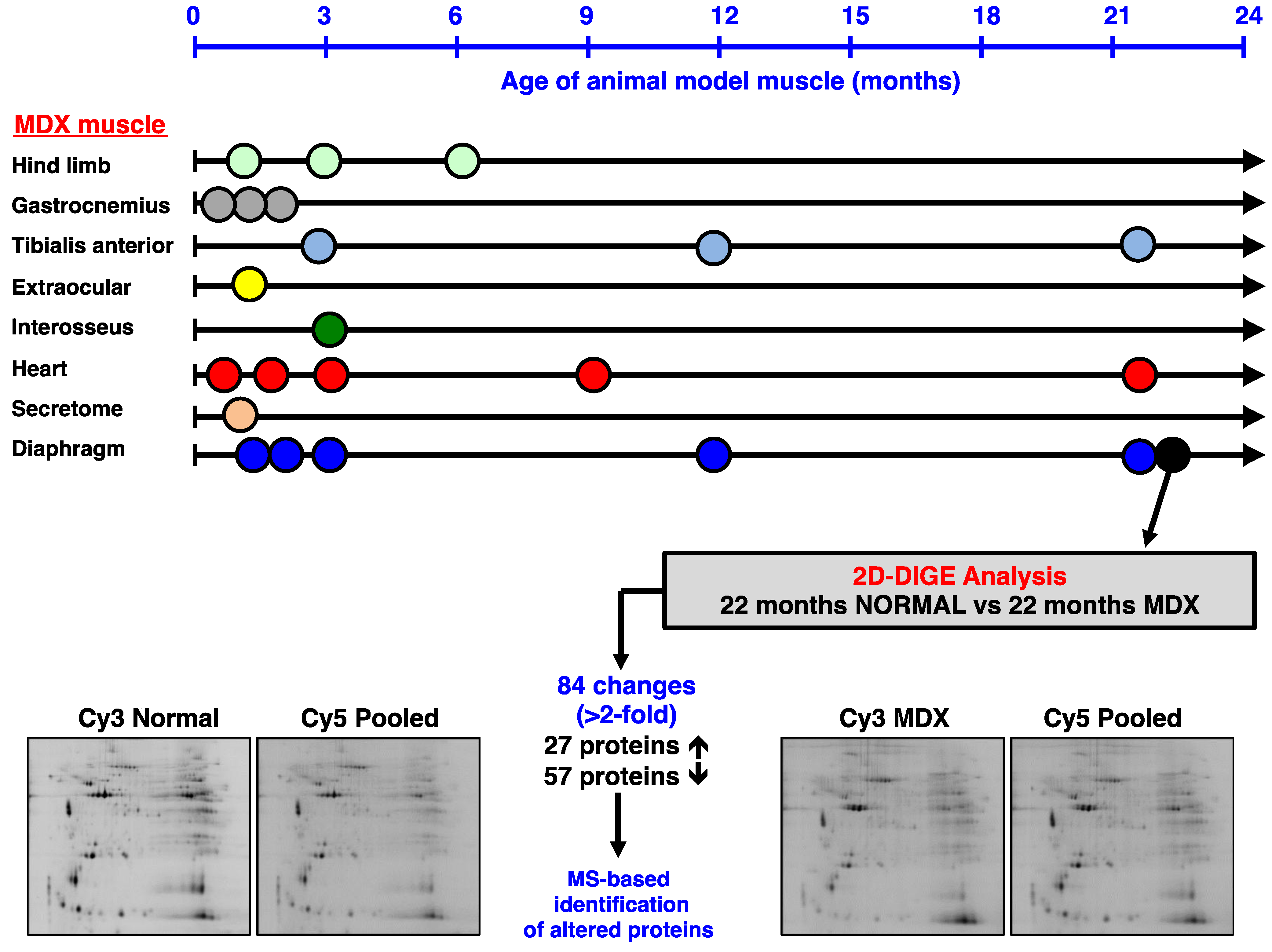
3.2. Fluorescence Two-Dimensional Difference In-Gel Electrophoretic Analysis of the Naturally Aged versus the Aged mdx Diaphragm Muscle
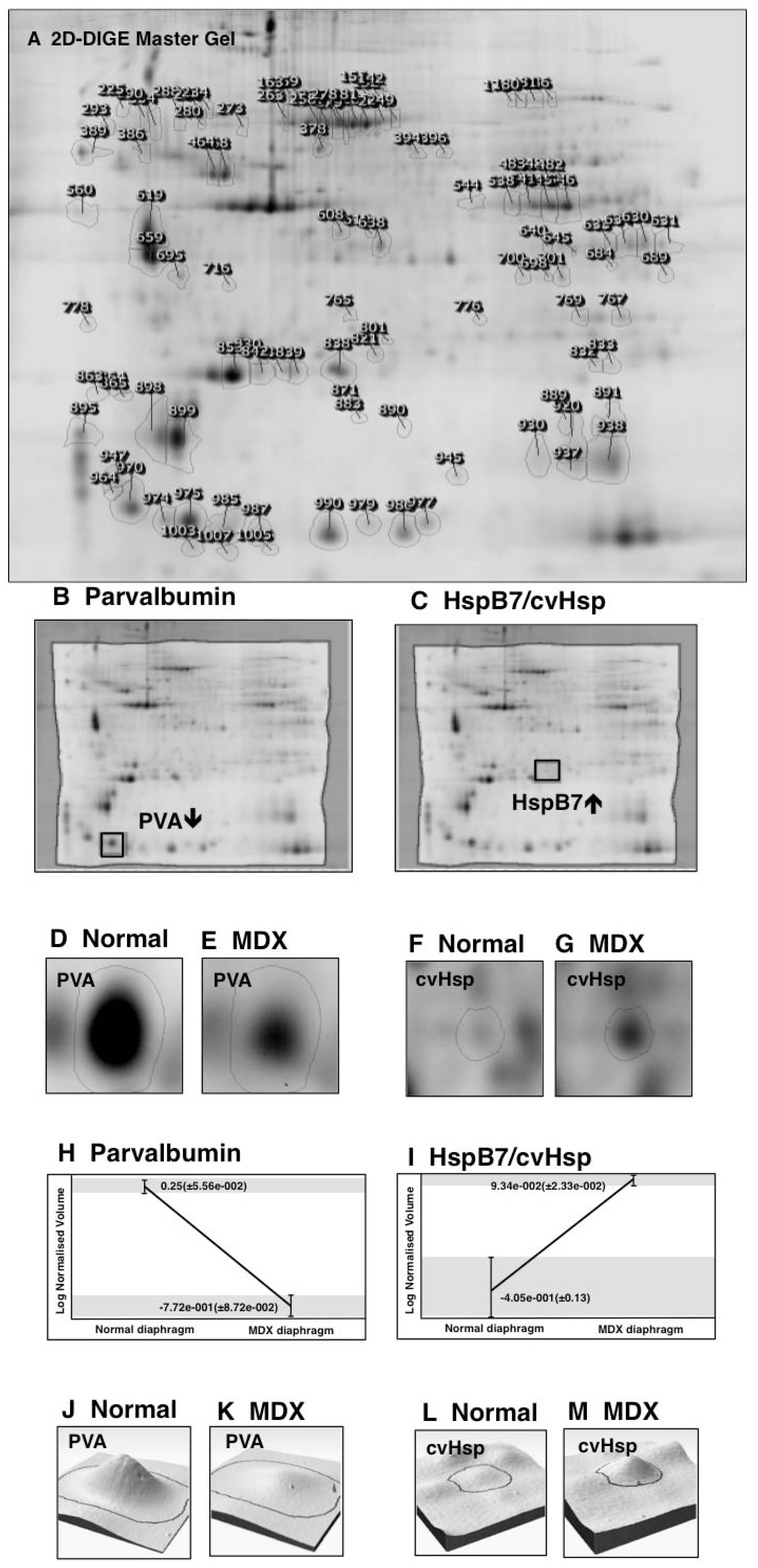
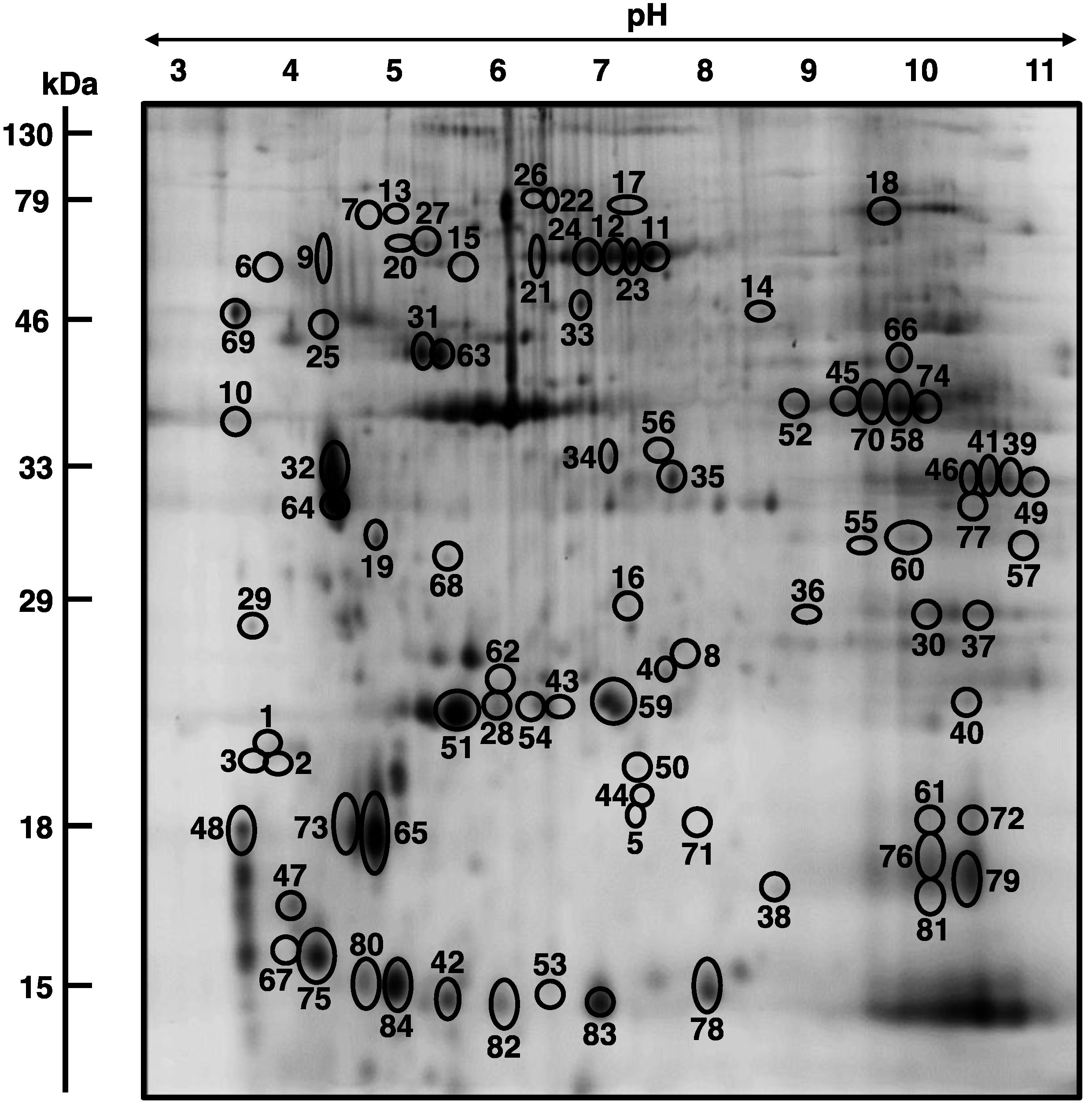
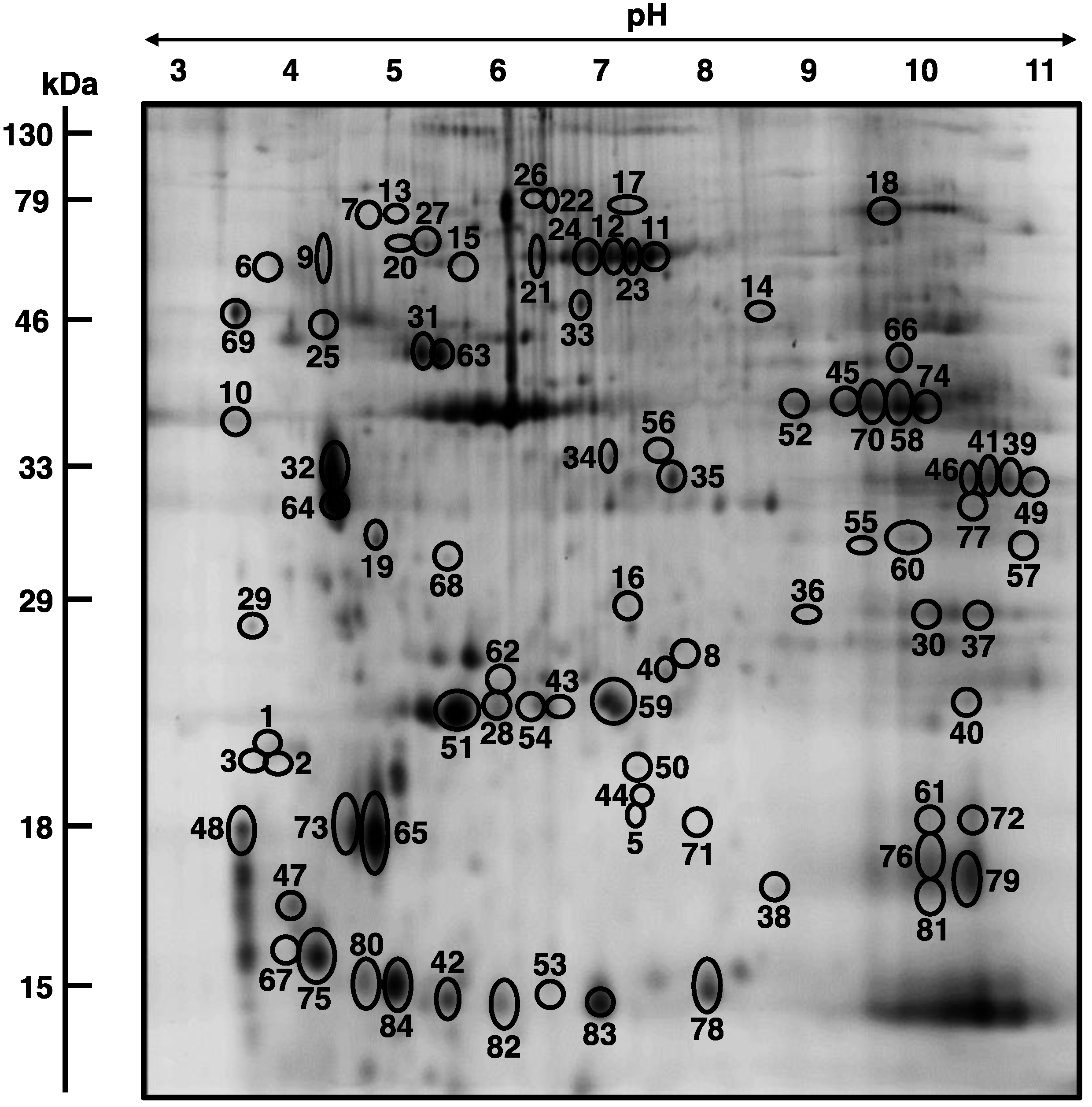
3.3. Mass Spectrometric Identification of Altered Proteins in Aged mdx Diaphragm Muscle

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spot No. | Protein Name | Accession No. | MS/MS Score | pI | Da | Number of peptides | Coverage (%) | Fold change |
|---|---|---|---|---|---|---|---|---|
| 1 | Dermatopontin | NP_062733 | 73 | 4.7 | 24,559 | 5 | 28 | 5.9 |
| 2 | Dermatopontin | NP_062733 | 111 | 4.7 | 24,559 | 7 | 37 | 4.4 |
| 3 | Dermatopontin | NP_062733 | 100 | 4.7 | 24,559 | 6 | 26 | 3.4 |
| 4 | Heat shock protein HspB7 (beta7, cvHsp) | NP_038896 | 87 | 5.95 | 18,681 | 5 | 38 | 3.3 |
| 5 | Myosin light chain 6B | NP_758463 | 181 | 5.41 | 22,851 | 14 | 74 | 3.1 |
| 6 | Tropomyosin, beta chain isoform 1 | NP_033442 | 319 | 4.66 | 32,933 | 22 | 61 | 2.9 |
| 7 | Unnamed protein product | BAE35818 | 286 | 5.78 | 70,776 | 15 | 30 | 2.8 |
| 8 | Myosin light chain 1/3, isoform 1f, muscle | NP_067260 | 97 | 4.98 | 20,697 | 2 | 12 | 2.7 |
| 9 | Tropomyosin, beta chain isoform 1 | NP_033442 | 107 | 4.66 | 32,933 | 11 | 35 | 2.7 |
| 10 | Actin, beta (aa 27–375) | CAA27396 | 124 | 5.78 | 39,451 | 9 | 21 | 2.6 |
| 11 | Heat shock protein Hsp70 cognate | AAA37869 | 110 | 5.37 | 71,025 | 11 | 23 | 2.6 |
| 12 | Mitochondrial stress-70 protein | BAA04493 | 57 | 5.91 | 73,773 | 1 | 1 | 2.6 |
| 13 | Actin, gamma, smooth muscle | AAC52237 | 81 | 5.36 | 43,258 | 5 | 16 | 2.6 |
| 14 | Nebulin (fragment) | AAF59979 | 53 | 9.19 | 87,694 | 4 | 7 | 2.5 |
| 15 | Lumican | AAB35361 | 103 | 5.84 | 38,733 | 2 | 5 | 2.4 |
| 16 | Prohibitin | NP_032857 | 350 | 5.57 | 29,860 | 15 | 69 | 2.4 |
| 17 | Unnamed protein product | BAC34145 | 129 | 5.75 | 70,766 | 8 | 14 | 2.4 |
| 18 | Transferrin | AAL34533 | 168 | 6.92 | 78,832 | 19 | 24 | 2.3 |
| 19 | Annexin A5 | NP_033803 | 249 | 4.83 | 35,788 | 16 | 45 | 2.3 |
| 20 | Alpha-fetoprotein | AAA37190 | 112 | 5.47 | 48,819 | 2 | 6 | 2.2 |
| 21 | Heat shock protein Hsp70 cognate | AAA37869 | 110 | 5.37 | 71,025 | 11 | 23 | 2.2 |
| 22 | Heat shock protein Hsp70 cognate | AAA37869 | 77 | 5.37 | 71,025 | 4 | 6 | 2.2 |
| 23 | Heat shock protein 8 | AAH66191 | 110 | 5.28 | 71,060 | 11 | 23 | 2.2 |
| 24 | Mitochondrial stress-70 protein | BAA04493 | 71 | 5.91 | 73,773 | 5 | 9 | 2.1 |
| 25 | Fmod protein | AAH52673 | 92 | 5.56 | 46,637 | 2 | 4 | 2.1 |
| 26 | Unnamed protein product | BAC34145 | 254 | 5.75 | 70,766 | 8 | 17 | 2.1 |
| 27 | 78 kDa glucose-regulated protein | BAA11462 | 112 | 5.09 | 72,528 | 7 | 15 | 2.0 |
| 28 | Myosin light chain 1/3, isoform 1f, muscle | NP_067260 | 222 | 4.98 | 20,697 | 15 | 63 | −2.0 |
| 29 | Parvalbumin, alpha | NP_038673 | 156 | 5.02 | 11,923 | 6 | 57 | −2.0 |
| 30 | Malate dehydrogenase | AAA39509 | 90 | 8.93 | 36,052 | 3 | 11 | −2.1 |
| 31 | ATP synthase, beta-subunit | AAB86421 | 98 | 5.14 | 56,344 | 2 | 4 | −2.1 |
| 32 | Tropomyosin, beta chain | gi|11875203 | 180 | 4.66 | 32,933 | 11 | 32 | −2.1 |
| 33 | Alpha-fetoprotein | AAA37190 | 95 | 5.47 | 48,819 | 2 | 5 | −2.1 |
| 34 | Isocitrate dehydrogenase subunit alpha | NP_083849 | 242 | 6.27 | 40,077 | 6 | 20 | −2.1 |
| 35 | L-lactate dehydrogenase B chain | NP_032518 | 54 | 5.7 | 36,839 | 5 | 12 | −2.1 |
| 36 | Actin, alpha, cardiac | gi|387090 | 51 | 5.23 | 42,048 | 2 | 7 | −2.1 |
| 37 | Coiled-coil-helix-coiled-coil-helix domain-containing protein 3, mitochondrial | NP_079612 | 80 | 8.56 | 26,550 | 5 | 24 | −2.1 |
| 38 | Cu/Zn superoxide dismutase | 1513495A | 147 | 6.03 | 15,926 | 5 | 41 | −2.1 |
| 39 | Malate dehydrogenase mitochondrial | NP_032643 | 256 | 8.93 | 36,053 | 12 | 45 | −2.1 |
| 40 | Peroxiredoxin Prdx1 | NP_035164 | 97 | 8.26 | 22,394 | 11 | 47 | −2.1 |
| 41 | Malate dehydrogenase mitochondrial | NP_032643 | 343 | 8.93 | 36,053 | 15 | 56 | −2.1 |
| 42 | Cytochrome c oxidase, subunit Va | CAA34085 | 125 | 6.08 | 16,252 | 8 | 48 | −2.2 |
| 43 | ATP synthase, mitochondrial F0 complex | AAH16547 | 148 | 5.52 | 18,810 | 5 | 47 | −2.2 |
| 44 | Heat shock protein Hsp beta-6 | NP_001012401 | 130 | 5.64 | 17,568 | 6 | 51 | −2.2 |
| 45 | Creatine kinase, M-type | NP_031736 | 276 | 6.58 | 43,250 | 9 | 27 | −2.2 |
| 46 | Malate dehydrogenasemitochondrial | NP_032643 | 243 | 8.93 | 36,053 | 9 | 36 | −2.2 |
| 47 | Troponin C, skeletal muscle | NP_033420 | 238 | 4.07 | 18,156 | 3 | 28 | −2.2 |
| 48 | Troponin C, skeletal muscle | NP_033420 | 774 | 4.07 | 18,156 | 8 | 53 | −2.3 |
| 49 | Malate dehydrogenase 2, mitochondrial | ABD77283 | 365 | 7.7 | 32,121 | 7 | 28 | −2.3 |
| 50 | Myoglobin | NP_038621 | 61 | 7.07 | 17,117 | 1 | 9 | −2.3 |
| 51 | Myosin light chain 1/3, isoform 1f, muscle | NP_067260 | 93 | 4.98 | 20,697 | 4 | 17 | −2.3 |
| 52 | Creatine kinase, M-type | NP_031736 | 297 | 6.58 | 43,250 | 10 | 28 | −2.3 |
| 53 | Fatty acid-binding protein FABP3 | NP_034304 | 157 | 6.11 | 14,810 | 6 | 48 | −2.3 |
| 54 | Calcium-binding protein NCS-1-like | XP_003945963 | 48 | 6.49 | 12,957 | 1 | 17 | −2.4 |
| 55 | Electron transferring flavoprotein, alpha | AAH03432 | 449 | 8.62 | 35,366 | 11 | 44 | −2.4 |
| 56 | Isocitrate dehydrogenase subunit alpha | NP_083849 | 167 | 6.27 | 40,077 | 6 | 19 | −2.4 |
| 57 | Malate dehydrogenase 2, mitochondrial | ABD77283 | 85 | 7.7 | 32,121 | 2 | 8 | −2.4 |
| 58 | Creatine kinase, M-type | NP_031736 | 404 | 6.58 | 43,250 | 11 | 33 | −2.4 |
| 59 | ATP synthase, subunit d, mitochondrial | NP_082138 | 168 | 5.52 | 18,796 | 11 | 70 | −2.4 |
| 60 | Electron transfer flavoprotein, subunit alpha | NP_663590 | 195 | 8.62 | 35,336 | 8 | 39 | −2.5 |
| 61 | Cofilin-2 | NP_031714 | 158 | 7.66 | 18,814 | 5 | 36 | −2.5 |
| 62 | Adenylate kinase, isoenzyme AK1 | NP_067490 | 104 | 5.7 | 23,334 | 6 | 36 | −2.5 |
| 63 | ATP synthase, beta-subunit | AAB86421 | 301 | 5.14 | 56,344 | 6 | 16 | −2.6 |
| 64 | Tropomyosin, beta chain | gi|11875203 | 129 | 4.66 | 32,920 | 10 | 25 | −2.6 |
| 65 | Myosin light chain MLC2, skeletal muscle | NP_058034 | 93 | 4.82 | 19,059 | 2 | 8 | −2.7 |
| 66 | Fumarate hydratase 1 | AAH06048 | 173 | 9.12 | 54,568 | 6 | 17 | −2.8 |
| 67 | Myosin light chain 1/3, isoform 1f, muscle | NP_067260 | 201 | 4.98 | 20,697 | 11 | 53 | −2.9 |
| 68 | Ubiquinone biosynthesis protein COQ9 | NP_080728 | 150 | 5.6 | 35,235 | 5 | 21 | −3.0 |
| 69 | Calsequestrin, skeletal muscle | AAC63616 | 143 | 3.93 | 45,619 | 8 | 22 | −3.1 |
| 70 | Creatine kinase, M-type | NP_031736 | 505 | 6.58 | 43,250 | 10 | 31 | −3.1 |
| 71 | Myoglobin | NP_038621 | 124 | 7.07 | 17,117 | 6 | 48 | −3.1 |
| 72 | Myoglobin | NP_038621 | 59 | 7.07 | 17,117 | 1 | 11 | −3.2 |
| 73 | Myosin light chain MLC2, skeletal muscle | NP_058034 | 267 | 4.82 | 19,059 | 15 | 68 | −3.4 |
| 74 | Creatine kinase, M-type | NP_031736 | 266 | 6.58 | 43,250 | 11 | 31 | −3.5 |
| 75 | Myosin light chain 1/3, isoform 1f, muscle | NP_067260 | 87 | 4.98 | 20,697 | 2 | 12 | −3.6 |
| 76 | Myoglobin | NP_038621 | 91 | 7.07 | 17,117 | 2 | 21 | −3.7 |
| 77 | Malate dehydrogenase | AAA39509 | 126 | 8.93 | 36,052 | 5 | 20 | −3.7 |
| 78 | Fatty acid-binding protein FABP3 | NP_034304 | 426 | 6.11 | 14,810 | 11 | 77 | −3.7 |
| 79 | Myoglobin | NP_038621 | 129 | 7.07 | 17,117 | 6 | 46 | −4.0 |
| 80 | Parvalbumin, alpha | NP_038673 | 285 | 5.02 | 11,923 | 6 | 57 | −4.0 |
| 81 | Myoglobin | NP_038621 | 89 | 7.07 | 17,117 | 6 | 41 | −4.3 |
| 82 | Fatty acid-binding protein FABP3 | NP_034304 | 188 | 6.11 | 14,810 | 7 | 63 | −5.3 |
| 83 | Fatty acid-binding protein FABP3 | NP_034304 | 532 | 6.11 | 14,810 | 9 | 71 | −5.3 |
| 84 | Parvalbumin alpha | NP_038673 | 431 | 5.02 | 11,923 | 8 | 57 | −10.5 |
3.4. Changed Protein Classes and Predicted Interaction Patterns of Altered Proteins
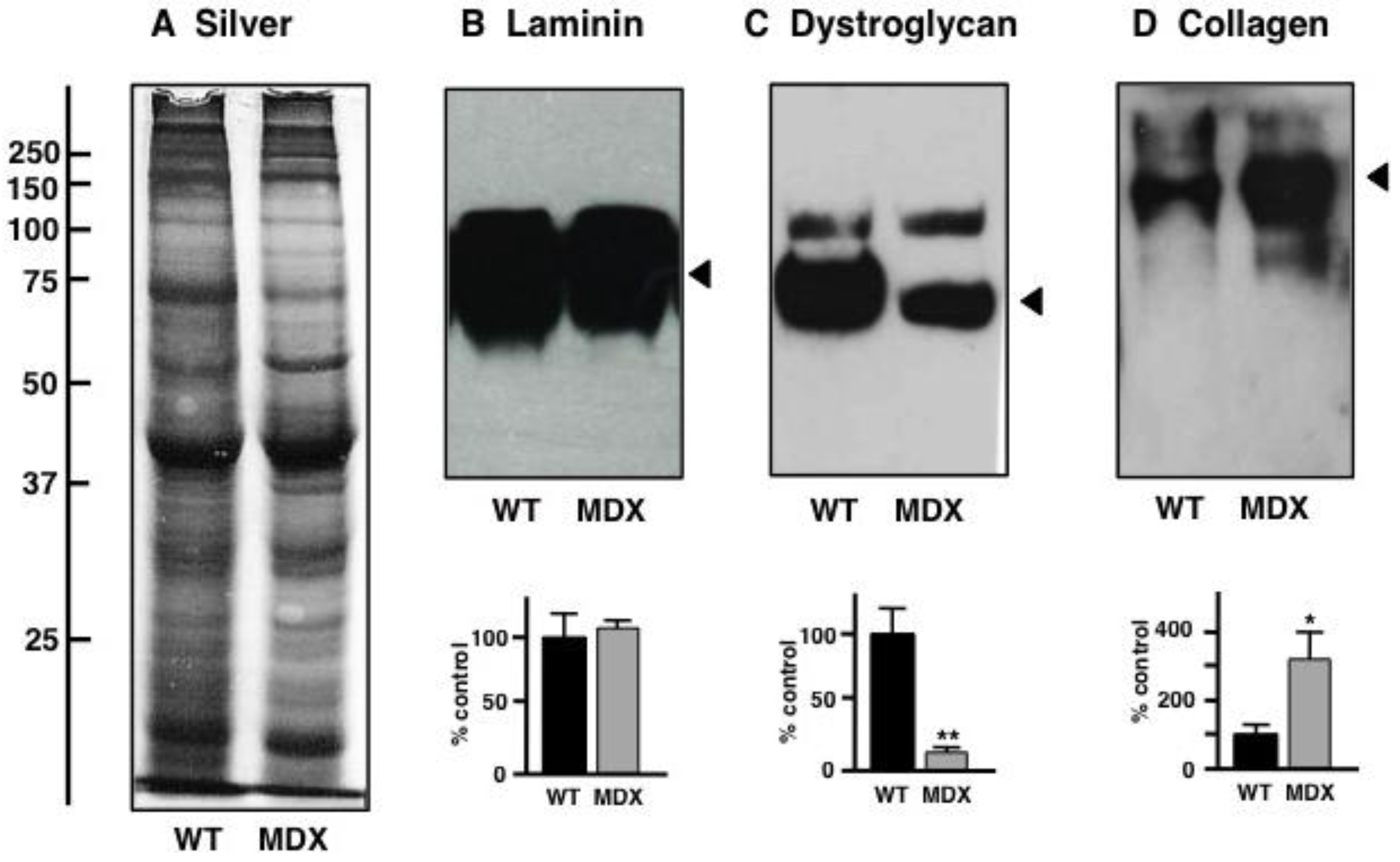
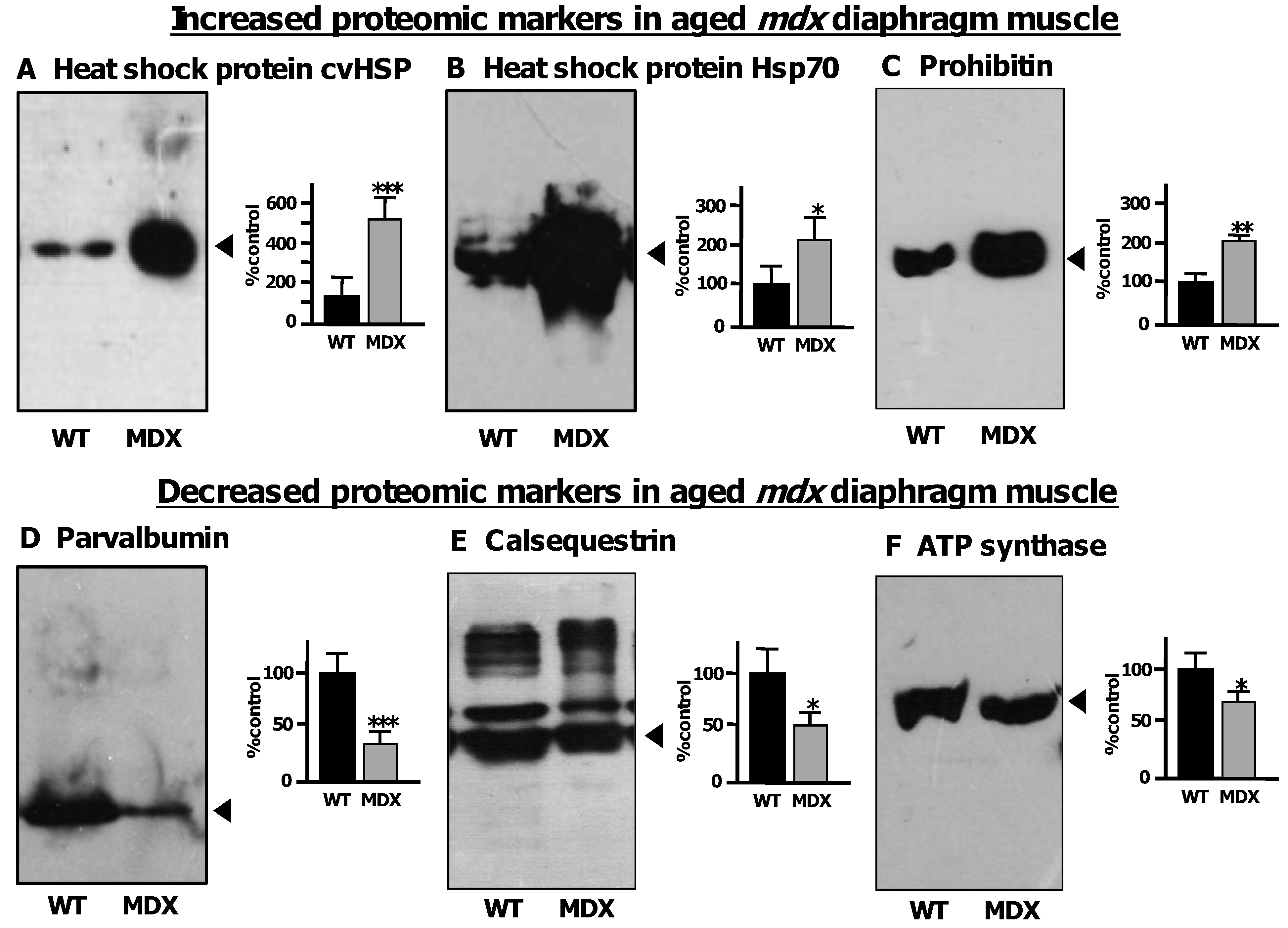
3.5. Immunoblot Analysis of Altered Proteins in Aged mdx Diaphragm Muscle

4. Discussion and Conclusions
Acknowledgments
Conflicts of Interest
References
- Isfort, R.J. Proteomic analysis of striated muscle. J. Chromatogr. B 2002, 771, 155–165. [Google Scholar] [CrossRef]
- Ohlendieck, K. Proteomics of skeletal muscle differentiation, neuromuscular disorders and fiber aging. Expert Rev. Proteomics 2010, 7, 283–296. [Google Scholar] [CrossRef]
- Gelfi, C.; Vasso, M.; Cerretelli, P. Diversity of human skeletal muscle in health and disease: Contribution of proteomics. J. Proteomics 2011, 74, 774–795. [Google Scholar] [CrossRef]
- Ohlendieck, K. Proteomic identification of biomarkers of skeletal muscle disorders. Biomark. Med. 2013, 7, 169–186. [Google Scholar] [CrossRef]
- Chevalier, F. Highlights on the capacities of “Gel-based” proteomics. Proteome Sci. 2010, 8, 23. [Google Scholar] [CrossRef]
- Rabilloud, T.; Chevallet, M.; Luche, S.; Lelong, C. Two-dimensional gel electrophoresis in proteomics: Past, present and future. J. Proteomics 2010, 73, 2064–2077. [Google Scholar] [CrossRef] [Green Version]
- Westermeier, R.; Görg, A. Two-dimensional electrophoresis in proteomics. Methods Biochem. Anal. 2011, 54, 411–439. [Google Scholar] [CrossRef]
- Ohlendieck, K. Skeletal muscle proteomics: Current approaches, technical challenges and emerging techniques. Skelet. Muscle 2011, 1, 6. [Google Scholar] [CrossRef]
- Parker, K.C.; Walsh, R.J.; Salajegheh, M.; Amato, A.A.; Krastins, B.; Sarracino, D.A.; Greenberg, S.A. Characterization of human skeletal muscle biopsy samples using shotgun proteomics. J. Proteome Res. 2009, 8, 3265–3277. [Google Scholar] [CrossRef]
- Holland, A.; Ohlendieck, K. Proteomic profiling of the contractile apparatus from skeletal muscle. Expert Rev. Proteomics 2013, 10, 239–257. [Google Scholar] [CrossRef]
- Maughan, D.W.; Henkin, J.A.; Vigoreaux, J.O. Concentrations of glycolytic enzymes and other cytosolic proteins in the diffusible fraction of a vertebrate muscle proteome. Mol. Cell. Proteomics 2005, 4, 1541–1549. [Google Scholar] [CrossRef]
- Højlund, K.; Yi, Z.; Hwang, H.; Bowen, B.; Lefort, N.; Flynn, C.R.; Langlais, P.; Weintraub, S.T.; Mandarino, L.J. Characterization of the human skeletal muscle proteome by one-dimensional gel electrophoresis and HPLC-ESI-MS/MS. Mol. Cell. Proteomics 2008, 7, 257–267. [Google Scholar]
- Nishimura, R.N.; Sharp, F.R. Heat shock proteins and neuromuscular disease. Muscle Nerve 2005, 32, 693–709. [Google Scholar] [CrossRef]
- Choi, Y.S. Reaching for the deep proteome: Recent nano liquid chromatography coupled with tandem mass spectrometry-based studies on the deep proteome. Arch. Pharm. Res. 2012, 35, 1861–1870. [Google Scholar] [CrossRef]
- Hamano, K.; Takeya, T.; Iwasaki, N.; Okoshi, N.; Fukubayashi, T.; Kirinoki, M.; Yao, Y.; Hirabayashi, T.; Takita, H. Analysis of dystrophin in muscular diseases by two-dimensional gel electrophoresis using agarose gels in the first dimension. Acta Neurol. Belg. 1996, 96, 102–107. [Google Scholar]
- Oh-Ishi, M.; Maeda, T. Disease proteomics of high-molecular-mass proteins by two-dimensional gel electrophoresis with agarose gels in the first dimension (Agarose 2-DE). J. Chromatogr. B 2007, 849, 211–222. [Google Scholar] [CrossRef]
- Reisinger, V.; Eichacker, L.A. Isolation of membrane protein complexes by blue native electrophoresis. Methods Mol. Biol. 2008, 424, 423–431. [Google Scholar] [CrossRef]
- Wittig, I.; Schägger, H. Native electrophoretic techniques to identify protein-protein interactions. Proteomics 2009, 9, 5214–5223. [Google Scholar] [CrossRef]
- Froemming, G.R.; Murray, B.E.; Ohlendieck, K. Self-aggregation of triadin in the sarcoplasmic reticulum of rabbit skeletal muscle. Biochim. Biophys. Acta 1999, 1418, 197–205. [Google Scholar] [CrossRef]
- Brennan, J.P.; Wait, R.; Begum, S.; Bell, J.R.; Dunn, M.J.; Eaton, P. Detection and mapping of widespread intermolecular protein disulfide formation during cardiac oxidative stress using proteomics with diagonal electrophoresis. J. Biol. Chem. 2004, 279, 41352–41360. [Google Scholar]
- Fraterman, S.; Zeiger, U.; Khurana, T.S.; Rubinstein, N.A.; Wilm, M. Combination of peptide OFFGEL fractionation and label-free quantitation facilitated proteomics profiling of extraocular muscle. Proteomics 2007, 7, 3404–3416. [Google Scholar] [CrossRef]
- Gannon, J.; Ohlendieck, K. Subproteomic analysis of basic proteins in aged skeletal muscle following offgel pre-fractionation. Mol. Med. Rep. 2012, 5, 993–1000. [Google Scholar]
- Staunton, L.; Ohlendieck, K. Mass spectrometric characterization of the sarcoplasmic reticulum from rabbit skeletal muscle by on-membrane digestion. Protein Pept. Lett. 2012, 19, 252–263. [Google Scholar] [CrossRef]
- Ohlendieck, K. On-membrane digestion technology for muscle proteomics. J. Membr. Sep. Technol. 2013, 2, 1–12. [Google Scholar]
- Guevel, L.; Lavoie, J.R.; Perez-Iratxeta, C.; Rouger, K.; Dubreil, L.; Feron, M.; Talon, S.; Brand, M.; Megeney, L.A. Quantitative proteomic analysis of dystrophic dog muscle. J. Proteome Res. 2011, 10, 2465–2478. [Google Scholar] [CrossRef]
- Rayavarapu, S.; Coley, W.; Cakir, E.; Jahnke, V.; Takeda, S.; Aoki, Y.; Grodish-Dressman, H.; Jaiswal, J.K.; Hoffman, E.P.; Brown, K.J.; et al. Identification of disease specific pathways using in vivo SILAC proteomics in dystrophin deficient mdx mouse. Mol. Cell. Proteomics 2013, 12, 1061–1073. [Google Scholar] [CrossRef]
- Duguez, S.; Duddy, W.; Johnston, H.; Lainé, J.; Le Bihan, M.C.; Brown, K.J.; Bigot, A.; Hathout, Y.; Butler-Browne, G.; Partridge, T. Dystrophin deficiency leads to disturbance of LAMP1-vesicle-associated protein secretion. Cell. Mol. Life Sci. 2013, 70, 2159–2174. [Google Scholar] [CrossRef]
- Malm, C.; Hadrevi, J.; Bergström, S.A.; Pedrosa-Domellöf, F.; Antti, H.; Svensson, M.; Frängsmyr, L. Evaluation of 2-D DIGE for skeletal muscle: Protocol and repeatability. Scand. J. Clin. Lab. Invest. 2008, 68, 793–800. [Google Scholar] [CrossRef]
- Unlü, M.; Morgan, M.E.; Minden, J.S. Difference gel electrophoresis: A single gel method for detecting changes in protein extracts. Electrophoresis 1997, 18, 2071–2077. [Google Scholar] [CrossRef]
- Alban, A.; David, S.O.; Bjorkesten, L.; Andersson, C.; Sloge, E.; Lewis, S.; Currie, I. A novel experimental design for comparative two-dimensional gel analysis: Two-dimensional difference gel electrophoresis incorporating a pooled internal standard. Proteomics 2003, 3, 36–44. [Google Scholar] [CrossRef]
- Viswanathan, S.; Unlü, M.; Minden, J.S. Two-dimensional difference gel electrophoresis. Nat. Protoc. 2006, 1, 1351–1358. [Google Scholar] [CrossRef]
- Minden, J.S. Two-dimensional difference gel electrophoresis. Methods Mol. Biol. 2012, 869, 287–304. [Google Scholar]
- Tonge, R.; Shaw, J.; Middleton, B.; Rowlinson, R.; Rayner, S.; Young, J.; Pognan, F.; Hawkins, E.; Currie, I.; Davison, M. Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics 2001, 1, 377–396. [Google Scholar] [CrossRef]
- Marouga, R.; David, S.; Hawkins, E. The development of the DIGE system: 2D fluorescence difference gel analysis technology. Anal. Bioanal. Chem. 2005, 382, 669–678. [Google Scholar] [CrossRef]
- Karp, N.A.; Kreil, D.P.; Lilley, K.S. Determining a significant change in protein expression with DeCyder during a pair-wise comparison using two-dimensional difference gel electrophoresis. Proteomics 2004, 4, 1421–1432. [Google Scholar] [CrossRef]
- Karp, N.A.; Lilley, K.S. Maximising sensitivity for detecting changes in protein expression: Experimental design using minimal CyDyes. Proteomics 2005, 5, 3105–3115. [Google Scholar] [CrossRef]
- Doran, P.; Martin, G.; Dowling, P.; Jockusch, H.; Ohlendieck, K. Proteome analysis of the dystrophin-deficient MDX diaphragm reveals a drastic increase in the heat shock protein cvHSP. Proteomics 2006, 6, 4610–4621. [Google Scholar] [CrossRef]
- Corzett, T.H.; Fodor, I.K.; Choi, M.W.; Walsworth, V.L.; Chromy, B.A.; Turteltaub, K.W.; McCutchen-Maloney, S.L. Statistical analysis of the experimental variation in the proteomic characterization of human plasma by two-dimensional difference gel electrophoresis. J. Proteome Res. 2006, 5, 2611–2619. [Google Scholar] [CrossRef]
- Guenet, J.L. Animal models of human genetic diseases: Do they need to be faithful to be useful? Mol. Genet. Genomics 2011, 286, 1–20. [Google Scholar] [CrossRef]
- Vainzof, M.; Ayub-Guerrieri, D.; Onofre, P.C.; Martins, P.C.; Lopes, V.F.; Zilberztajn, D.; Maia, L.S.; Sell, K.; Yamamoto, L.U. Animal models for genetic neuromuscular diseases. J. Mol. Neurosci. 2008, 34, 241–248. [Google Scholar]
- Doran, P.; O’Connell, K.; Gannon, J.; Ohlendieck, K. Proteomic profiling of animal models mimicking skeletal muscle disorders. Proteomics Clin. Appl. 2007, 1, 1169–1184. [Google Scholar] [CrossRef]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar]
- Nakamura, A.; Takeda, S. Mammalian models of Duchenne Muscular Dystrophy: Pathological characteristics and therapeutic applications. J. Biomed. Biotechnol. 2011, 2011. Article ID 184393. [Google Scholar]
- Ng, R.; Banks, G.B.; Hall, J.K.; Muir, L.A.; Ramos, J.N.; Wicki, J.; Odom, G.L.; Konieczny, P.; Seto, J.; Chamberlain, J.R.; et al. Animal models of muscular dystrophy. Prog. Mol. Biol. Transl. Sci. 2012, 105, 83–111. [Google Scholar] [CrossRef]
- Banks, G.B.; Chamberlain, J.S. The value of mammalian models for Duchenne muscular dystrophy in developing therapeutic strategies. Curr. Top. Dev. Biol. 2008, 84, 431–453. [Google Scholar] [CrossRef]
- Kornegay, J.N.; Bogan, J.R.; Bogan, D.J.; Childers, M.K.; Li, J.; Nghiem, P.; Detwiler, D.A.; Larsen, C.A.; Grange, R.W.; Bhavaraju-Sanka, R.K.; et al. Canine models of Duchenne muscular dystrophy and their use in therapeutic strategies. Mamm. Genome 2012, 23, 85–108. [Google Scholar] [CrossRef]
- Ge, Y.; Molloy, M.P.; Chamberlain, J.S.; Andrews, P.C. Proteomic analysis of mdx skeletal muscle: Great reduction of adenylate kinase 1 expression and enzymatic activity. Proteomics 2003, 3, 1895–1903. [Google Scholar] [CrossRef]
- Holland, A.; Dowling, P.; Zweyer, M.; Swandulla, D.; Henry, M.; Clynes, M.; Ohlendieck, K. Proteomic profiling of cardiomyopathic tissue from the aged mdx model of Duchenne muscular dystrophy reveals a drastic decrease in laminin, nidogen and annexin. Proteomics 2013, 13, 2312–2323. [Google Scholar] [CrossRef]
- Doran, P.; Wilton, S.D.; Fletcher, S.; Ohlendieck, K. Proteomic profiling of antisense-induced exon skipping reveals reversal of pathobiochemical abnormalities in dystrophic mdx diaphragm. Proteomics 2009, 9, 671–685. [Google Scholar] [CrossRef]
- Lewis, C.; Carberry, S.; Ohlendieck, K. Proteomic profiling of x-linked muscular dystrophy. J. Muscle Res. Cell Motil. 2009, 30, 267–269. [Google Scholar] [CrossRef]
- Lefaucheur, J.P.; Pastoret, C.; Sebille, A. Phenotype of dystrophinopathy in old mdx mice. Anat. Rec. 1995, 242, 70–76. [Google Scholar] [CrossRef]
- Head, S.I. Branched fibres in old dystrophic mdx muscle are associated with mechanical weakening of the sarcolemma, abnormal Ca2+ transients and a breakdown of Ca2+ homeostasis during fatigue. Exp. Physiol. 2010, 95, 641–656. [Google Scholar] [CrossRef]
- Chamberlain, J.S.; Metzger, J.; Reyes, M.; Townsend, D.; Faulkner, J.A. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. 2007, 21, 2195–2204. [Google Scholar] [CrossRef]
- Mouisel, E.; Vignaud, A.; Hourde, C.; Butler-Browne, G.; Ferry, A. Muscle weakness and atrophy are associated with decreased regenerative capacity and changes in mTOR signaling in skeletal muscles of venerable (18-24-month-old) dystrophic mdx mice. Muscle Nerve 2010, 41, 809–818. [Google Scholar] [CrossRef]
- Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Proteomics reveals drastic increase of extracellular matrix proteins collagen and dermatopontin in aged mdx diaphragm muscle. Int. J. Mol. Med. 2012, 30, 229–234. [Google Scholar]
- Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Profiling of age-related changes in the tibialis anterior muscle proteome of the mdx mouse model of dystrophinopathy. J. Biomed. Biotechnol. 2012, 2012, 691641. [Google Scholar]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science 1989, 244, 1578–1580. [Google Scholar]
- Stedman, H.H.; Sweeney, H.L.; Shrager, J.B.; Maguire, H.C.; Panettieri, R.A.; Petrof, B.; Narusawa, M.; Leferovich, J.M.; Sladky, J.T.; Kell, A.M. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 1991, 352, 536–539. [Google Scholar] [CrossRef]
- Staunton, L.; Jockusch, H.; Ohlendieck, K. Proteomic analysis of muscle affected by motor neuron degeneration: The wobbler mouse model of amyotrophic lateral sclerosis. Biochem. Biophys.Res. Commun. 2011, 406, 595–600. [Google Scholar] [CrossRef]
- O’Connell, K.; Ohlendieck, K. Proteomic DIGE analysis of the mitochondria-enriched fraction from aged rat skeletal muscle. Proteomics 2009, 9, 5509–5524. [Google Scholar] [CrossRef]
- Kang, Y.; Techanukul, T.; Mantalaris, A.; Nagy, J.M. Comparison of three commercially available DIGE analysis software packages: Minimal user intervention in gel-based proteomics. J. Proteome Res. 2009, 8, 1077–1084. [Google Scholar]
- Carberry, S.; Ohlendieck, K. Gel electrophoresis-based proteomics of senescent tissues. Methods Mol. Biol. 1048, 229–246. [Google Scholar]
- Staunton, L.; Jockusch, H.; Wiegand, C.; Albrecht, T.; Ohlendieck, K. Identification of secondary effects of hyperexcitability by proteomic profiling of myotonic mouse muscle. Mol. Biosyst. 2011, 7, 2480–2489. [Google Scholar] [CrossRef]
- PANTHER Gene List Analysis. Available online: http://pantherdb.org/ (accessed on 15 November 2013).
- Mi, H.; Muruganujan, A.; Thomas, P.D. PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013, 41, D377–D386. [Google Scholar] [CrossRef]
- STRING Functional Protein Association Network. Available online: http://string-db.org/ (accessed on 17 November 2013).
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef]
- Staunton, L.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Mass spectrometry-based proteomic analysis of middle-aged vs. aged vastus lateralis reveals increased levels of carbonic anhydrase isoform 3 in senescent human skeletal muscle. Int. J. Mol. Med. 2012, 30, 723–733. [Google Scholar]
- Holland, A.; Carberry, S.; Ohlendieck, K. Proteomics of the dystrophin-glycoprotein complex and dystrophinopathy. Curr. Protein Pep. Sci. 2013, in press. [Google Scholar]
- Culligan, K.; Banville, N.; Dowling, P.; Ohlendieck, K. Drastic reduction of calsequestrin-like proteins and impaired calcium binding in dystrophic mdx muscle. J. Appl. Physiol. 2002, 92, 435–445. [Google Scholar]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar]
- Pontén, F.; Schwenk, J.M.; Asplund, A.; Edqvist, P.H. The Human Protein Atlas as a proteomic resource for biomarker discovery. J. Intern. Med. 2011, 270, 428–446. [Google Scholar] [CrossRef]
- Theodoridis, G.A.; Gika, H.G.; Want, E.J.; Wilson, I.D. Liquid chromatography-mass spectrometry based global metabolite profiling: A review. Anal. Chim. Acta 2012, 711, 7–16. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. Quantitative, high-resolution proteomics for data-driven systems biology. Annu. Rev. Biochem. 2011, 80, 273–299. [Google Scholar]
- Stastna, M.; van Eyk, J.E. Secreted proteins as a fundamental source for biomarker discovery. Proteomics 2012, 12, 722–735. [Google Scholar] [CrossRef]
- Mallick, P.; Kuster, B. Proteomics: A pragmatic perspective. Nat. Biotechnol. 2010, 28, 695–709. [Google Scholar] [CrossRef]
- Graham, K.M.; Singh, R.; Millman, G.; Malnassy, G.; Gatti, F.; Bruemmer, K.; Stefanski, C.; Curtis, H.; Sesti, J.; Carlson, C.G. Excessive collagen accumulation in dystrophic (mdx) respiratory musculature is independent of enhanced activation of the NF-kappaB pathway. J. Neurol. Sci. 2010, 294, 43–50. [Google Scholar]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Winkler, K.; Wiedemann, F.R.; von Bossanyi, P.; Dietzmann, K.; Kunz, W.S. Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Mol. Cell. Biochem. 1998, 183, 87–96. [Google Scholar] [CrossRef]
- Percival, J.M.; Siegel, M.P.; Knowels, G.; Marcinek, D.J. Defects in mitochondrial localization and ATP synthesis in the mdx mouse model of Duchenne muscular dystrophy are not alleviated by PDE5 inhibition. Hum. Mol. Genet. 2013, 22, 153–167. [Google Scholar]
- Doran, P.; Dowling, P.; Donoghue, P.; Buffini, M.; Ohlendieck, K. Reduced expression of regucalcin in young and aged mdx diaphragm indicates abnormal cytosolic calcium handling in dystrophin-deficient muscle. Biochim. Biophys. Acta 2006, 1764, 773–785. [Google Scholar]
- Doran, P.; Dowling, P.; Lohan, J.; McDonnell, K.; Poetsch, S.; Ohlendieck, K. Subproteomics analysis of Ca2+-binding proteins demonstrates decreased calsequestrin expression in dystrophic mouse skeletal muscle. Eur. J. Biochem. 2004, 271, 3943–3952. [Google Scholar]
- Carberry, S.; Brinkmeier, H.; Zhang, Y.; Winkler, C.K.; Ohlendieck, K. Comparative proteomic profiling of soleus, extensor digitorum longus, flexor digitorum brevis and interosseus muscle from the mdx mouse model of Duchenne muscular dystrophy. Int. J. Mol. Med. 2013, 32, 544–556. [Google Scholar]
- Gardan-Salmon, D.; Dixon, J.M.; Lonergan, S.M.; Selsby, J.T. Proteomic assessment of the acute phase of dystrophin deficiency in mdx mice. Eur. J. Appl. Physiol. 2011, 111, 2763–2773. [Google Scholar] [CrossRef]
- Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Comparative proteomic analysis of the contractile protein-depleted fraction from normal versus dystrophic skeletal muscle. Anal. Biochem. 2013. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Application of Fluorescence Two-Dimensional Difference In-Gel Electrophoresis as a Proteomic Biomarker Discovery Tool in Muscular Dystrophy Research. Biology 2013, 2, 1438-1464. https://doi.org/10.3390/biology2041438
Carberry S, Zweyer M, Swandulla D, Ohlendieck K. Application of Fluorescence Two-Dimensional Difference In-Gel Electrophoresis as a Proteomic Biomarker Discovery Tool in Muscular Dystrophy Research. Biology. 2013; 2(4):1438-1464. https://doi.org/10.3390/biology2041438
Chicago/Turabian StyleCarberry, Steven, Margit Zweyer, Dieter Swandulla, and Kay Ohlendieck. 2013. "Application of Fluorescence Two-Dimensional Difference In-Gel Electrophoresis as a Proteomic Biomarker Discovery Tool in Muscular Dystrophy Research" Biology 2, no. 4: 1438-1464. https://doi.org/10.3390/biology2041438