The Preparation and Properties of Fluoroacrylate-Modified Polysiloxane as a Fabric Coating Agent

1
Department of Fashion Design, Wenzhou Vocational and Technical College, Wenzhou 325035, China
2
Zhejiang Wenzhou Research Institute of Light Industry, Wenzhou 325003, China
3
College of Bioresources Chemical and Materials Engineering, Shaanxi University of Science and Technology, Xi’an 710021, China
*
Author to whom correspondence should be addressed.
Coatings 2018, 8(1), 31; https://doi.org/10.3390/coatings8010031
Submission received: 11 December 2017
/
Revised: 6 January 2018
/
Accepted: 9 January 2018
/
Published: 11 January 2018
Abstract
:Polysiloxanes, which can add high softness and lubricity to treated textiles, have been proposed as softening post-finishing agents for fabric. However, the hydrophobicity of the finished fabric is not satisfactory. In this work, long-chain fluoroalkyl acrylate was used to modify the polysiloxane, aimed at improving hydrophobicity of the finished fabric and retaining its softness simultaneously. Fourier transform infrared spectroscopy (FT-IR), nuclear magnetic resonance spectra (1H/13C NMR), and thermogravimetric analysis (TGA) were used to characterize the chemical structure and thermal stability of the as-prepared polymer. The modified polysiloxane was tested as a finishing agent. Its film morphologies on the fabric surface and on a silicon wafer were determined. Chemical compositions and performance properties of the finished fabric were investigated. By bonding long-chain fluoroalkyl, the modified polysiloxane presented good thermal stability. Due to the combined effect of the low surface free energy of the perfluorinated side chains and the relatively high surface roughness of the cotton fibers, the treated fabric had favorable hydrophobicity with a WCA of 144.7° on its surface. In addition, their softness was increased, but the color remained unchanged.
1. Introduction
With flexible segments, prominent film-forming capacity and low glass transition temperature, polysiloxanes have often been chosen as softening post-finishing agents for fabric [1,2]. However, because the surface free energies of the silicone copolymer films are not low enough, the waterproofing property of the treated fabrics is suboptimal. Fluorinated polyacrylate is another “triple-proofing” textile finish and endows the treated fabric with excellent water- and oil-repellency due to its very low surface free energy [3,4,5]. However, the handfeel of the finished fabric was too stiff to obtain comfortable wearability. Fluorosilicon copolymer, prepared by modifying polysiloxane with fluorinated acrylate, presents the characteristics of these two polymers, providing polysiloxane not only with the high elasticity and flexibility of organosilicon, but with the low surface energy and strong mechanical and adhesive properties of fluorinated polyacrylate. This has recently been a hot topic in the field of textile post-finishing agents.
Currently, concerning fluoropolymer-modified polysiloxane materials, preparation methods mainly include hydrosilylation, emulsion polymerization, atom transfer radical polymerization (ATRP), photopolymerization, and grafting methods [6,7,8,9,10,11,12,13,14]. Furukawa et al. [6,7] utilized the hydrosilylation of fluorinated olefins with polyhydromethylsiloxane to synthesize a series of fluorosilicones and used them as finishing agents for polyester fabrics. Miniemulsion polymerization was adopted by Guo to prepare a fluorinated siloxane-acrylate polymer latex, which was then used as a new kind of waterborne textile finishing agent [8]. Finally, the treated fabric could be rendered with both desirable softness and excellent water resistance. Luo et al. [9,10] used the ATRP technique to prepare a poly(dimethylsiloxane)-block-poly(methyl methacrylate)-block-poly(2,2,3,3,4,4,4-heptafluorobutyl methacrylate) triblock copolymer and poly(methacryloxypropyltrimethoxysilane-b-2,2,3,3,4,4,4-heptafluorobutyl methacrylate -grafted silica hybrid nanoparticles. Martinelli et al. [11,12] prepared a series of amphiphilic diblock copolymers containing a poly(dimethylsiloxane) block and a PEGylated-fluoroalkyl-modified polystyrene block via ATRP and tested them as biofouling coatings. Novel photopolymerized network films on the basis of a polysiloxane matrix containing varied amounts of polyoxyethylene or perfluorohexylethyl dangling side chains were studied by Martinelli et al. [13]. Among the above methods, hydrosilylation is most frequently employed by scientists and technicians. However, due to the difference in chemical structure between long-chain fluorine-containing monomer and polyhydromethylsiloxane, their intersolubility is too poor to make the hydrosilylation reaction proceed well under the condition of bulk polymerization. Thus, it becomes an obstacle in the preparation of fluorosilicon copolymer via the hydrosilylation method. In some cases, if some of the fluoroalkyl segments were previously imported into the polysiloxane chains by the equilibration reaction, the compatibility between the long-chain fluorine monomer and the modified polysiloxane was enhanced, which probably benefited the following Si–H addition reaction. Therefore, the above obstacle is likely to be overcome.
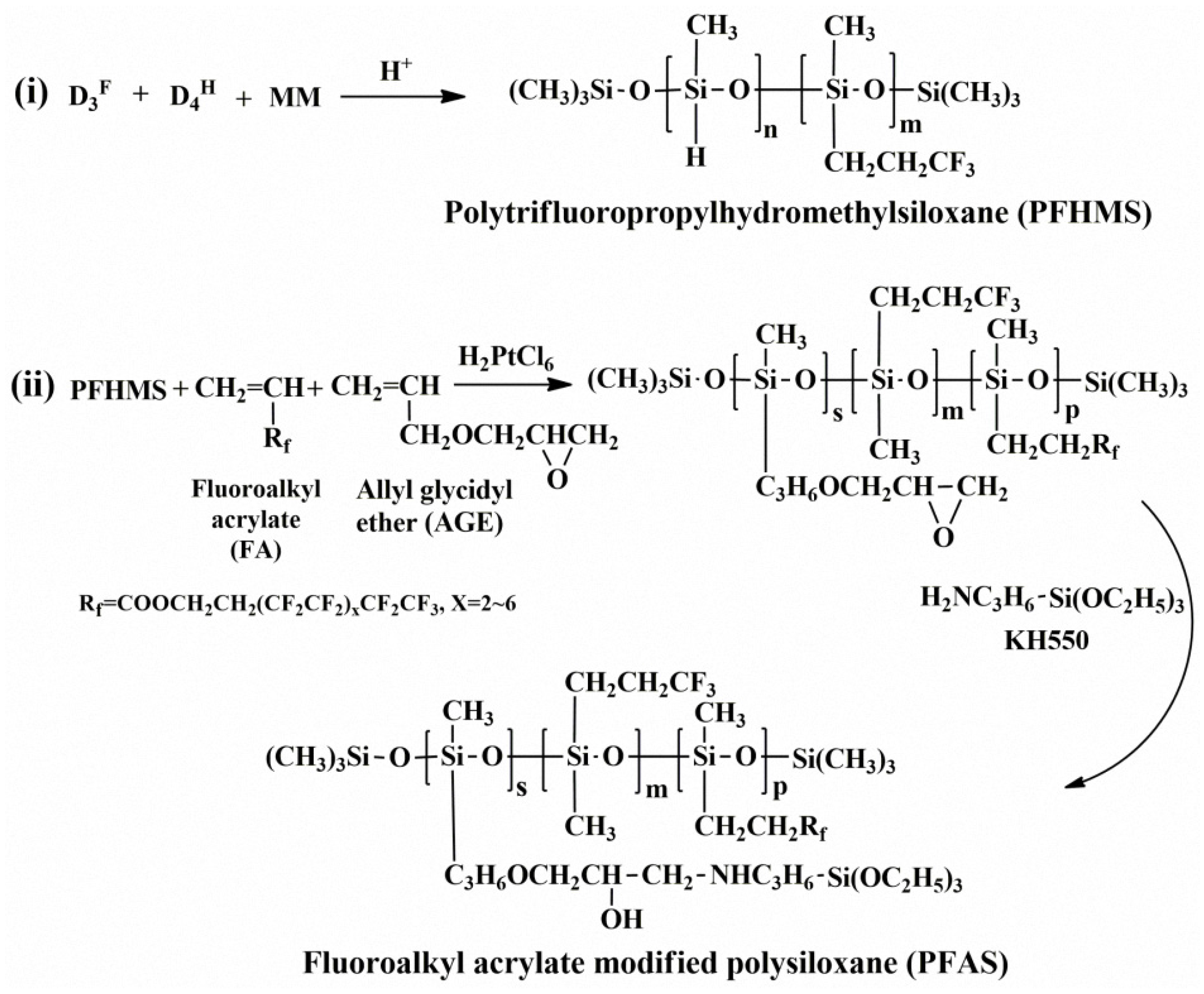
Therefore, we firstly prepared a polytrifluoropropylhydromethylsiloxane (PFHMS) via the equilibration reaction of 1,3,5-trimethyltris(3,3,3-trifluoropropyl) cyclotrisiloxane (D3F), 1,3,5,7-tetramethylcyclotetrasiloxane (D4H), and hexamethyldisiloxane (MM) under the acidic catalyst condition. Hydrosilylation was performed with fluoroalkyl acrylate (FA), allyl glycidyl ether (AGE), and PFHMS to synthesize a sort of comb-like long-chain-fluoroalkyl- and epoxy-group-modified polysiloxane, which was then reacted with aminopropyltriethoxysilane (KH550) to create a novel comb-like long-chain fluoroalkyl- and siloxy-modified polysiloxane (PFAS). Here, the dangling side siloxy groups will help to form a more compact film on the fabric surface during the subsequent curing, which is beneficial for the hydrophobicity of the treated fabric, and will provide some reactive sites for later modifications, such as the in situ condensation reaction with silica sols to prepare nanocomposite. The whole preparation process can be viewed in Scheme 1. The aim of our present study is to examine the application properties of our as-prepared PFAS as the fabric coating material and to shed light on the molecular design and the development of new fluorosilicon copolymers as functional coating finishes.
2. Materials and Methods
2.1. Materials
Polytrifluoropropylhydromethylsiloxane (PFHMS) was prepared in our lab with a viscosity of 580 mPa·s and an Si–H content of 0.23% (expressed by the moles of Si–H groups contained in 1 g of silicone). Fluoroalkyl acrylate (FA) and allyl glycidyl ether (AGE) were industrial products and purchased from Shandong Zhongfu Chemical Co. Ltd. (Jinan, China) and Jiangshu Jintan Chemical Co. Ltd. (Changzhou, China). Chloroplatinic acid hexahydrate (H2PtCl6·6H2O), ethyl acetate, and aminopropyltriethoxysilane (KH550) were of analytic grade and supplied by Shanghai Tuosi Chemical Co. Ltd. (Shanghai, China), Xi’an Chemical reagent factory (Xi’an, China) and Hangzhou Dadi Chemical Co. Ltd. (Hangzhou, China), all in China. Silicon wafers, kindly presented by Shanghai Songjiang Silicon Material Co. (Shanghai, China), were cleaned and dried according to our previous work [15,16,17]. A white pure cotton fabric was obtained from the local store and used as FESEM investigation and application experiments. The specifications of the used fabric are given in Table 1.
2.2. Preparation of Fluoroalkyl Acrylate Modified Polysiloxane (PFAS)
Thirty grams of PFHMS, 34.43 g of FA (68.3 mmol), and 0.87 g of AGE (7.6 mmol) were successively added into a 100 mL three-necked flask equipped with a mechanical stirrer, a reflux condenser, and a thermometer and then dry nitrogen gas was bubbled into the flask for half an hour. After the admixture was heated to 80–85 °C, several drops of H2PtCl6 was added and the reaction was retained for 3–4 h at this temperature. After the temperature of the system was lowered to 65–70 °C, 1.68 g of KH550 (7.6 mmol) was poured into the mixture to take further action for another 3–4 h. After this, the temperature was increased to 125–130 °C to remove the low-boiling residues by vacuum distillation. Finally, a viscous fluoroalkyl-acrylate-modified polysiloxane (PFAS) was obtained with a clear appearance.
2.3. Coating of Cotton Fabric with PFAS
At first, all cotton samples were ultrasonicated in an orderly manner using deionized water and acetone at 25 °C for 20 min and then dried at 100 °C for 5 min. This procedure was to ensure that the slurry and contaminants were removed from the fiber/fabric substrates.
First, different doses of PFAS were severally dissolved with redistilled ethyl acetate to form different PFAS solutions with around 0–0.5% mass concentrations. The above cotton samples were then immersed into the PFAS solution for several seconds and padded to wet pick-up at about 70% on the weight of the dry fabric. Next, the cotton samples were dried at 100 °C for 10 min and cured at 160 °C for 1 min. Finally, treated samples were kept in a desiccator to balance at room temperature for 24 h.
2.4. Characterization
The structure of the as-prepared resultant was confirmed by FTIR and NMR. FTIR spectra of the samples were recorded on a Bruker VECTOR-22 spectrophotometer (Bruker, Billerica, MA, USA) between 4000 and 500 cm−1 using a KBr pellet technique. NMR analysis was performed with an INOVA-400 spectrometer (Varian, San Francisco, CA, USA) using CDCl3 as a solvent and tetramethylsilane (TMS) as an internal standard.
Thermal stability was interpreted with thermogravimetry analysis (TGA), which was conducted on a Q500 thermogravimeter (TA Instruments Company, New Castle, DE, USA) at a heating rate of 10 °C/min from ambient temperature to 600 °C in an inert atmosphere.
Film morphologies of the PFAS-coated fabric or silicon wafer were observed by SEM and AFM, respectively. After the fabrics/fibers were coated with gold in a vacuum, SEM observations were carried out on an S-4800 scanning electron microscope (Hitachi Company, Tokyo, Japan) at a magnification of 5000 times. The method concerning deposition of the PFAS on the silicon wafer is described in [18]. After the PFAS film was formed on the silicon wafer, it was detected by a Nanoscope IIIA atomic force microscope (AFM, Digital Instruments, Tonawanda, NY, USA) in tapping mode. All determinations were performed at 22 °C and in air with a relative humidity of 48%.
Surface chemical compositions of the PFAS-coated fabric were detected by a Physical Electronics Model 5700 XPS instruments (PHI, Chanhassen, MN, USA). This apparatus was equipped with a monochromatic Al Kα source (150 W, 15 KV, KE = 1486.6 eV). Parameters 30° and 6.7 × 10−8 Pa were set as the nominal photoelectron take-off angle and vacuum degree, respectively, in the analysis chamber. During the XPS detecting process, since hydrocarbon contamination existed on the sample surface, the C1s photoelectron emission signal was often utilized to correct the deviation of the binding energy at 284.8 eV.
Static water contact angles (WCAs) on the treated fabrics surface were detected by a JC2000A contact angle goniometer (Shanghai Zhong Chen Powereach Co., Shanghai, China) using the sessile drop method at 20 °C. The injection volume of liquid was 5 μL and the average of at least five readings was used as the final value of each sample.
Whiteness and bending rigidity (BR) of the PFAS-treated fabrics were severally measured with a YQ-Z-48B fluorescent whiteness tester (Wenzhou Instrument Co., Wenzhou, China) and a Kawabata Evaluation System (KES) instrument (KATO TECH Co. Ltd., Kyoto, Japan).
3. Results and Discussion
3.1. Structure Characterization
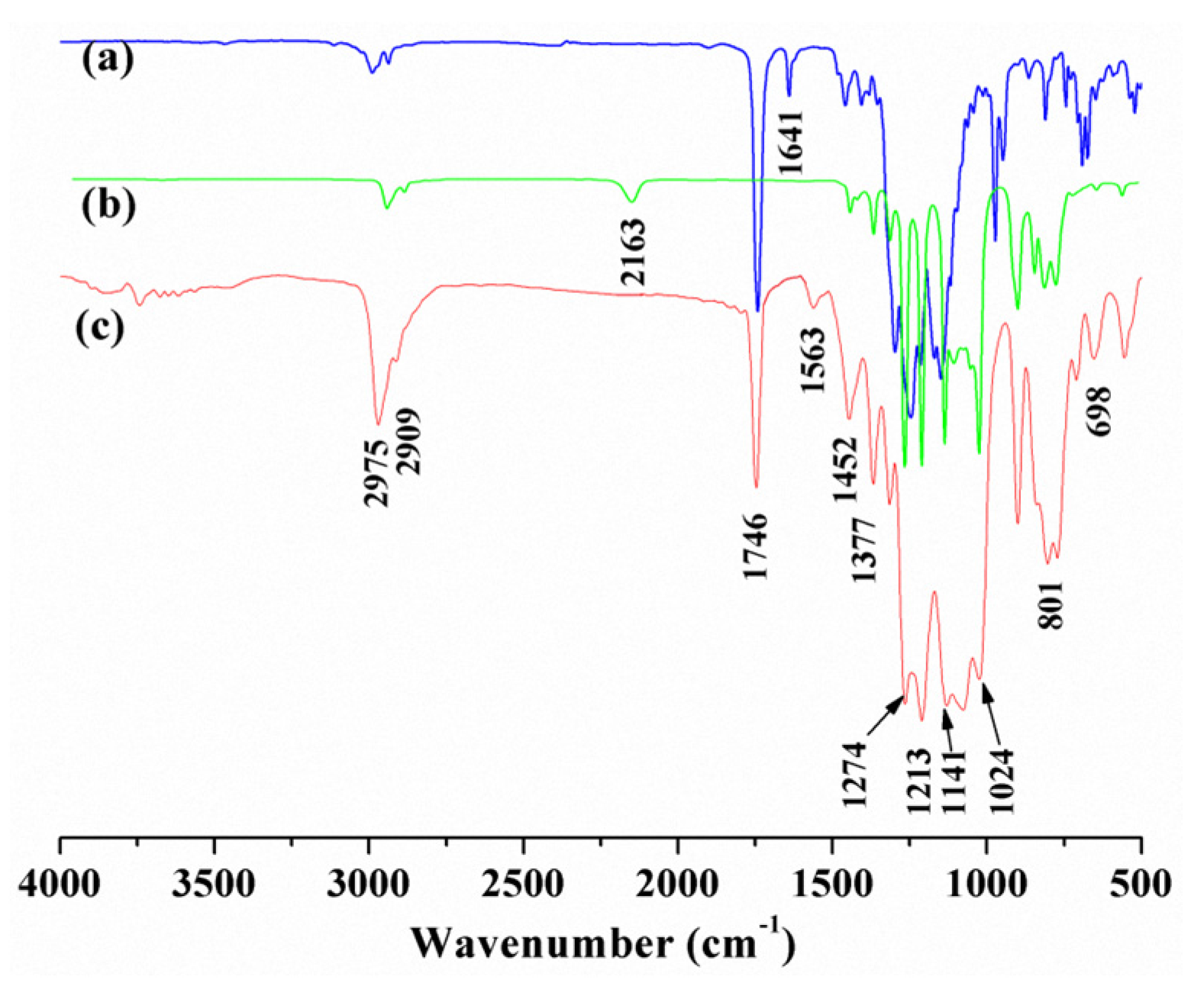
FTIR spectra of the reactants, FA and PFHMS, and the outcome, PFAS, were compared and are shown in Figure 1. Figure 1a denotes the spectrum of the reactant FA. The absorption peaks at about 2975 cm−1, 2909 cm−1, 1452 cm−1, 1377 cm−1, and 1641 cm−1 resulted from the stretching and bending vibrations of alkyl C–H and the stretching vibration of the C=C bond in FA, respectively. The absorption peaks at 1746 cm−1, 1213 cm−1, and 698 cm−1 were associated with the stretching vibration of C=O, the stretching and wagging vibrations of the C–F bond, respectively [15,16,19]. Figure 1b represents the spectrum of the reactant PFHMS. There were typical absorption peaks at around 2163 cm−1, 2975 cm−1, 2909 cm−1, 1452 cm−1, 1377 cm−1, 1274 cm−1, and 801 cm−1, which denote the stretching vibration of the Si–H bond, the stretching and bending vibrations of alkyl C–H, the bending vibration of the C–H bond in Si–CH3, and the stretching vibration of the Si–C bond in PFHMS, respectively. The signals between 1141 cm−1 and 1024 cm−1 are supposed to originate from the stretching vibrations of Si–O–Si bond. Figure 1c is the spectrum of the resultant PFAS. Except for the above signals, the absorption peaks of the C=C bond and the Si–H bond completely disappeared at 1641 cm−1 and 2163 cm−1, indicating that the hydrosilylation reaction proceeded well. Besides, a new absorption peak emerged at 1563 cm−1, resulting from the bending vibration of the secondary amine. The above FTIR results demonstrate that PFAS was successfully acquired.
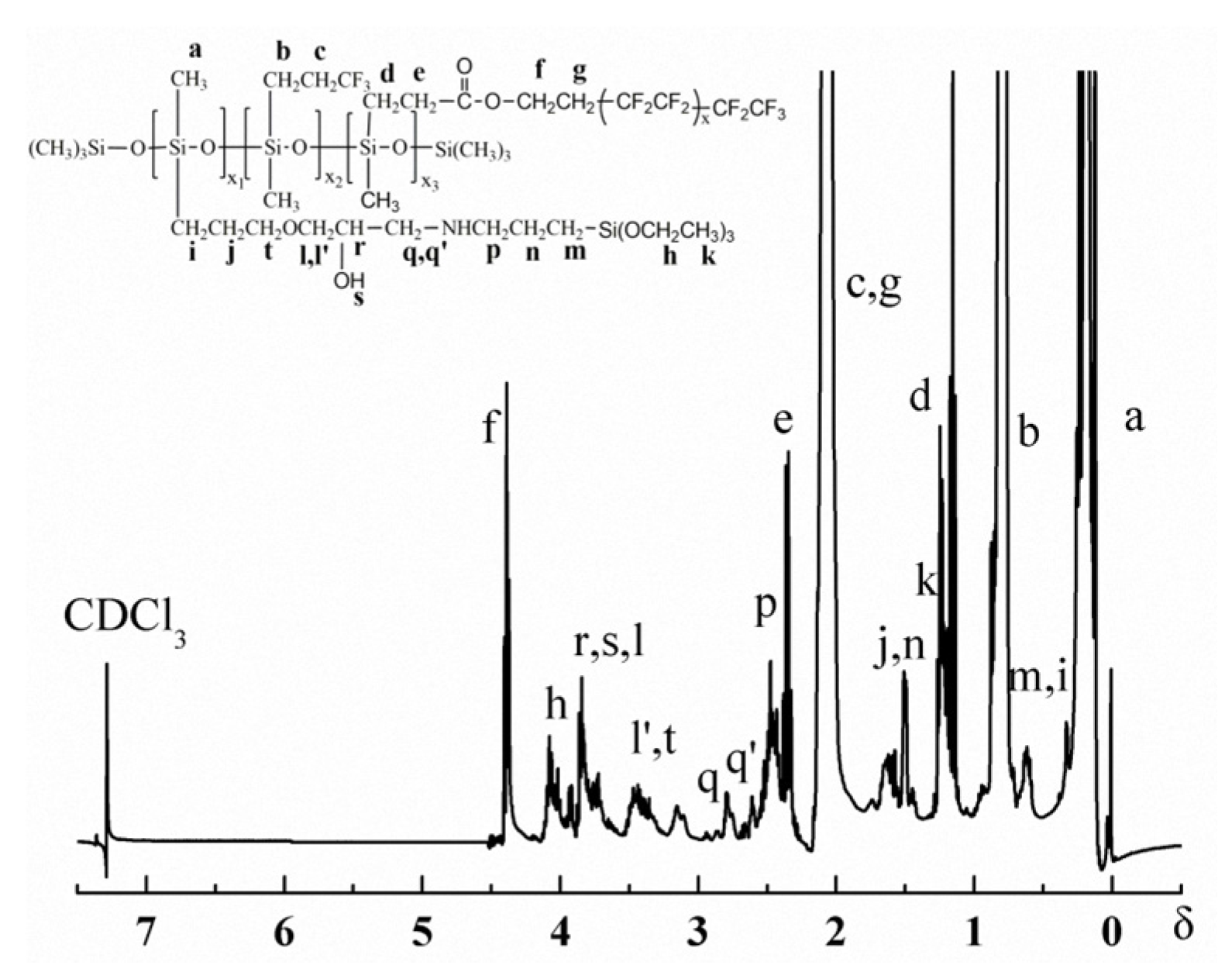
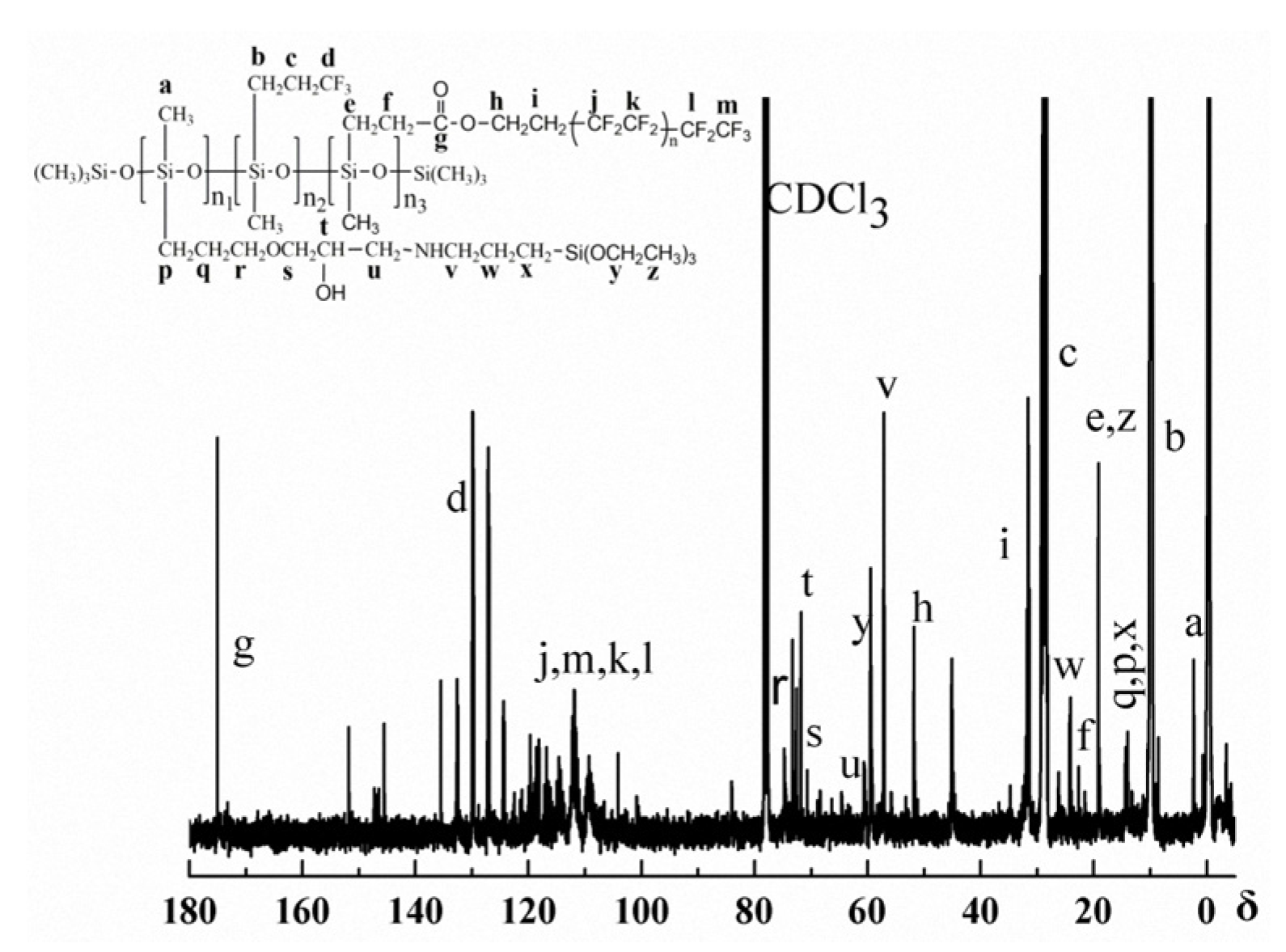
Figure 2 and Figure 3 are the 1H-NMR spectrum and the 13C-NMR spectrum of the PFAS, respectively. Obviously, the relevant chemical shift values of the protons from the PFAS were observed at δ 0.1 (aH), 0.6 (mH and iH), 0.9 (bH), 1.2 (kH), 1.4 (dH), 1.5 (jH and nH), 1.9 (cH and gH), 2.3 (eH), 2.4 (pH), 2.6–2.8 (q’H and qH), 3.4 (l’H and tH), 3.7 (lH, rH and sH), and 4.3 (fH), respectively. In addition, the corresponding 13C chemical shift values of the PFAS could be identified at δ 2.1 (aC), 10.1 (bC), 14.1 (qC, pC and xC), 19.1 (eC and zC), 22.4 (fC), 24.4 (wC), 28.4 (cC), 31.4 (iC), 52.0 (hC), 57.0 (vC), 59.7 (yC), 61.0 (uC), 72.2 (tC), 73.0 (sC), 75.3 (rC), 108–120 (jC, mC, kC and lC), 130.1 (dC), and 175.3 (gC), respectively. Thus, on the basis of the FTIR and NMR results, the conclusion can be drawn that the as-prepared PFAS molecule has the due structure.
3.2. Thermal Stability
As is known, the Si–O bond possesses strong strength of about 108 kcal·mol−1 [20], which endows the polysiloxane materials with high thermal stability. In general, data concerning decomposition temperatures (Td) at different weight losses, such as 5% and 50% (noted as Td5% and Td50%), are often given to indicate the thermal stability of a polymer.
To obtain thermal stability of our as-synthesized polysiloxane-based polymer, TGA analysis was used in this work. Figure 4 is a TGA graph of the PFAS polymer. It can be seen that Td5% and Td50% were 331 °C and 426 °C, respectively. The entire decomposition temperature reached 570 °C. By comparison with the TGA results of other polysiloxane polymers, such as the poly(methylchloromethyl)siloxane (PCMS) [21], the polysiloxane-ferroferric oxide composite [22], and the photosensitive polysiloxane [23], our as-prepared polysiloxane has a higher thermal stability than that of the above polymers. Two cases may account for this phenomenon. One is the effect of the short- and long-chain fluoroalkyl in the side chain of the Si–O backbone, which can shield and protect the inner polysiloxane skeleton and enhance its thermal stability [15,16,17]; the other is the reactive siloxy, which can react with the Si–O and form crosslinking structures among polysiloxane chains during the heating process. As a result, the thermal stability of the polysiloxane polymer is improved.
3.3. Film Morphology of the PFAS
As a sort of coating material with low surface tension, polysiloxane is easily spread on various substrates. To acquire its film morphology, we used SEM. Figure 5 shows the SEM images of the blank cotton fabric and the fabric treated with PFAS. The average diameter of one fiber was about 10 μm, and there were many intrinsic stripes and some white granules on the fiber surface, which are likely uncleaned slurries, whereas the PFAS-treated cotton fabric became relatively smooth, and a flat polysiloxane film was distinctly coated on its surface. A finer film morphology of the PFAS on a nanoscale cannot be obtained from SEM observation.
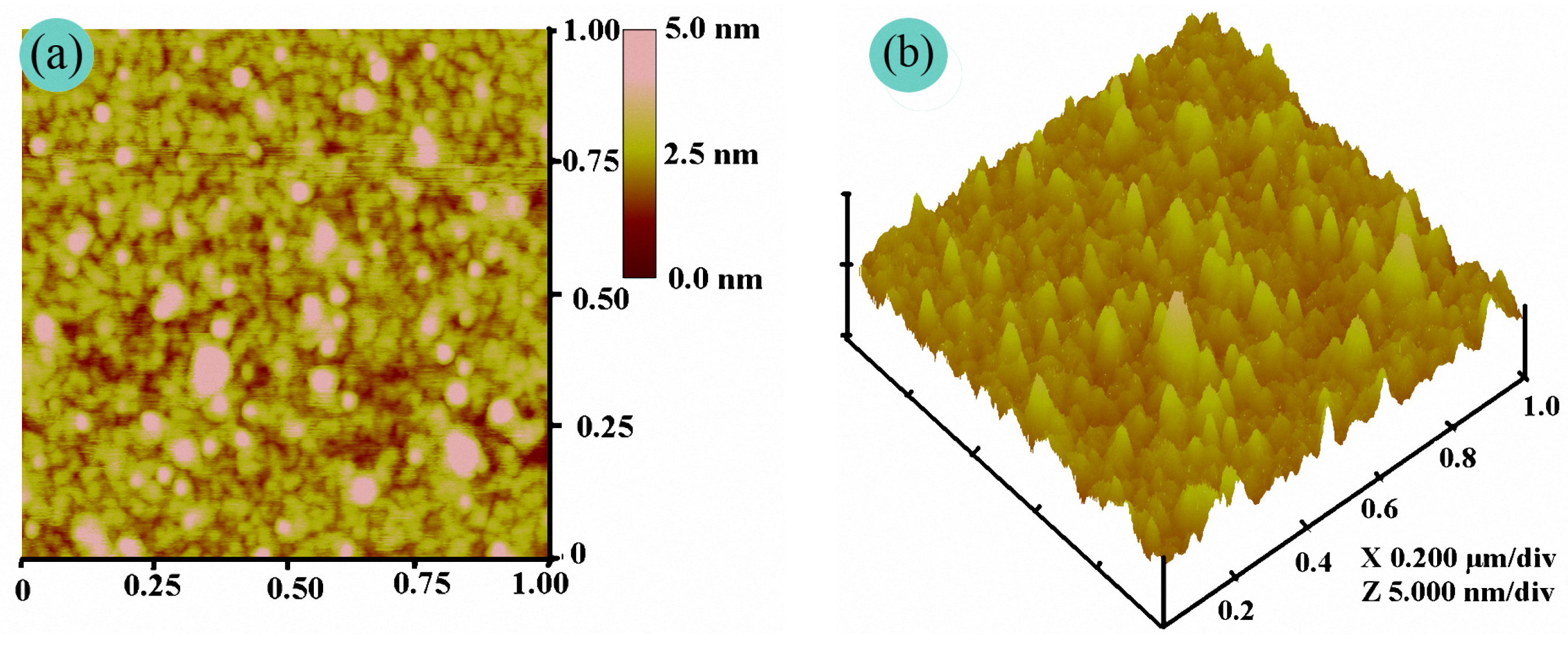
To date, AFM has been widely used as an instrument to investigate the fine morphology of polymer films on a nanometer scale [24]. Since unwanted deformation takes place on the surface of natural fabrics or fibers during AFM observation, a rigid silicon wafer is often selected as a substituted matrix to examine the fine film morphology of a polymer [15,16,17,18]. Therefore, the precise morphology of the PFAS film was carried out by AFM. Figure 6 shows the AFM images. As shown in Figure 5, an inhomogeneous film was deposited on the silicon wafer surface with many strong or weak bright peaks in 3-D topography. The root-mean-square roughness (Rq), average roughness (Ra), and maximum roughness (Rmax) were 0.650 nm, 0.465 nm, and 6.911 nm in the 1 × 1 μm2 scanning field, respectively. By contrast, the Rq of the uniform PDMS film is only 0.081 nm according to [19]. In fact, PFAS is a typical polysiloxane with hydrophobic short-chain trifluoropropyl and long-chain fluoroalkyl pendant groups and hydrophilic side groups containing ether, hydroxyl, and siloxy as shown in Scheme 1. Thus, after deposition of the PFAS on the silicon wafer, the PFAS molecules encountered the hydrophilic matrix and aggregated on its surface, and the hydrophilic side groups and dipolar Si–O bonds pointed to the silicon surface. The hydrophobic ones should be expelled out of surface and oriented outward from the silicon surface. In addition, the hydrogen-bond interactions or intermolecular forces made these hydrophobic parts curl and spiral, and an inhomogeneous morphology was formed.
3.4. Chemical Compositions of the Fabric Coatings
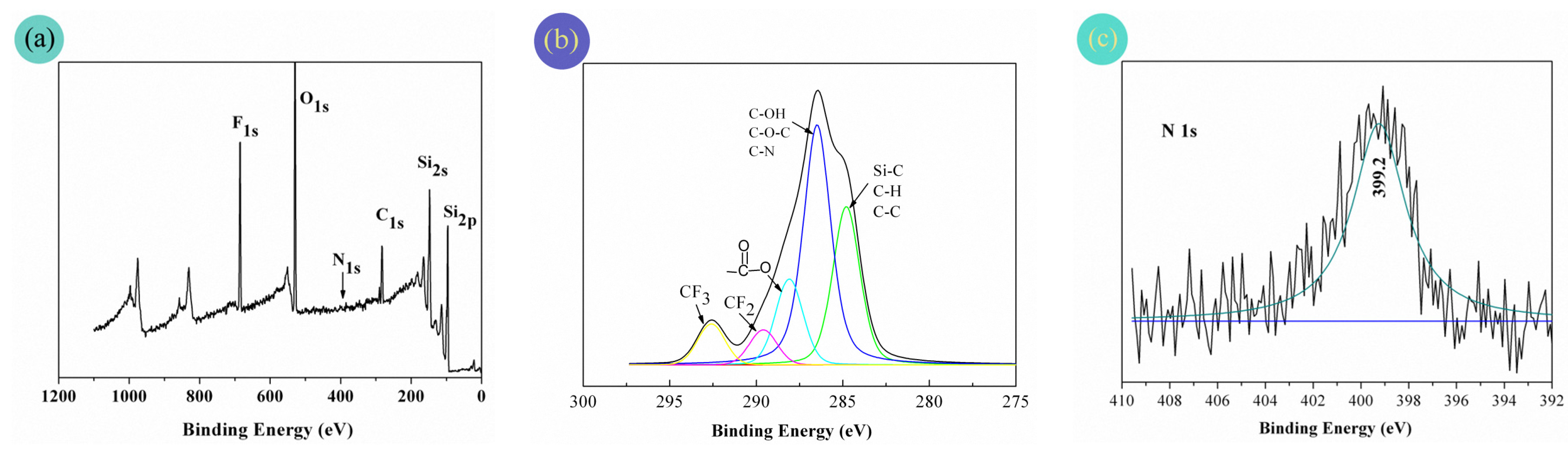
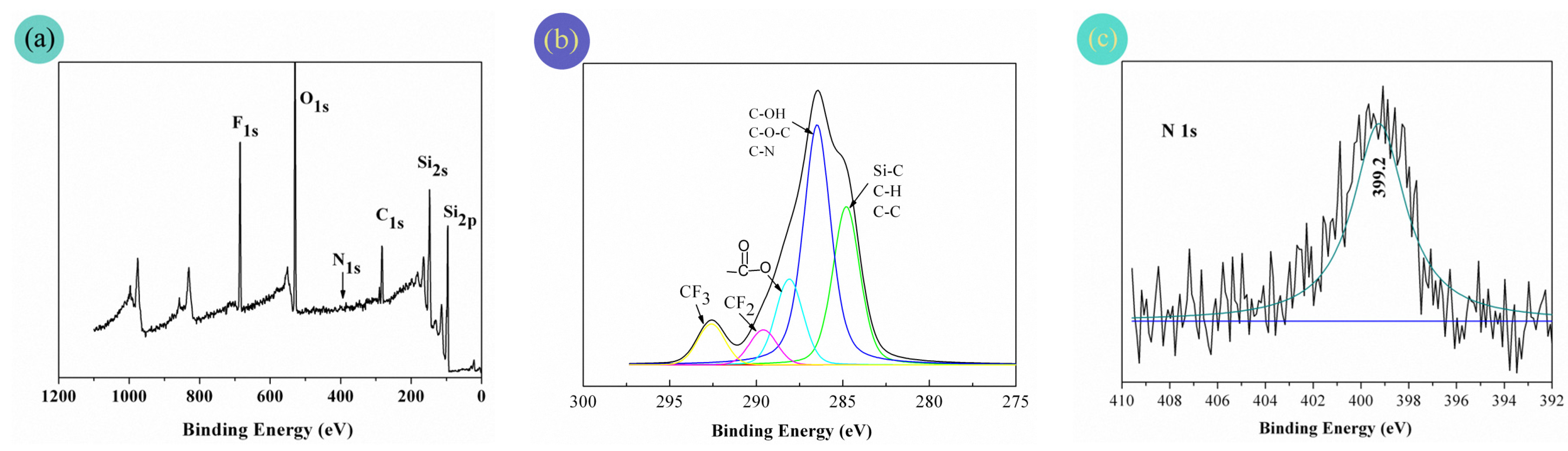
The XPS wide spectrum of the PFAS-treated fabrics is shown in Figure 7a. Meanwhile, to make clear the detailed chemical environment of certain elements, the high resolution C1s and N1s spectra are also respectively presented in Figure 7b,c.
Strong O1s (O1s at 532.0 and 533.5 eV) and F1s (F1s at 688.4 and 691.0 eV), moderate Si2s and Si2p (Si2s at 153.0 eV and Si2p at 102.0 eV), and C1s spectra appeared for the PFAS-coated fabric surface. Owing to the very low level of nitrogen content, almost no N1s signal was found at 399.2 eV in the wide spectrum, but a slender N1s peak indeed existed in Figure 7c of the high-resolution N1s spectrum. Furthermore, in Figure 7b of the high resolution C1s spectrum, a complex pattern of fivefold peaks was presented. More narrowly, these fivefold peaks could be assigned to the following carbon bonds (–CF3 at 292.6 eV, –CF2– at 289.6 eV, –O–C=O at 288.1 eV, C–N, C–O–C, and C–OH at 286.5 eV, and C–C, C–H, and Si–C at 284.8 eV). Thus, the XPS results demonstrated that the fabric surface was indeed covered by the PFAS film.
3.5. Performance Properties of the Fabrics Finished with PFAS
In addition to hydrophobicity and softness of the fabrics treated with different amount of PFAS, attention was also paid to the color change of the treated fabrics in the experiment. The fabrics showed a static water contact angle (WCA), had a bending rigidity, and were white. Performance properties of the fabrics treated with different amounts of PFAS are presented in Table 2.
As is shown in Table 2, the WCAs on the PFAS-treated fabric surface slowly increased with the increase in PFAS content. It did not change until the PFAS content was more than 0.4 g in 100 g ethyl acetate solutions. The final WCA reached 144.7°. Compared to WCA values on the fabrics treated with fluorine-free polysiloxane finishing agents (137.5° for CLPS, a crosslinked polysiloxane with end-capped epoxy groups, and 130.5° for EPVMS, an epoxy group-terminated polyvinylmethylsiloxane) [20], waterproofing property of the fabrics treated with PFAS was preferably improved. Therefore, in terms of improving the waterproofing property of the polysiloxane, the purpose of this experiment seems to be acquired. However, these high CA values appear to be too high to be totally ascribed to the effect of surface enrichment in the fluorinated components. In fact, even longer perfluoroalkyl homopolymer is generally reported to display a static water contact angle (CA) of 120° [25]. Thus, such high CA values might be due to the combination of two effects: the low surface free energy of the perfluorinated side chains and the relatively high surface roughness of the cotton fibers. In addition, the whiteness of the PFAS-treated fabrics showed almost no change compared with the untreated one. This consequence may result from the fact that the polysiloxane film is transparent and its color did not change during the baking process. As for the bending rigidity, both warp and fill values of the PFAS-treated fabrics, compared to those of the untreated fabric, continued to reduce as the PFAS dose was augmented. This indicates that the softness of the PFAS-treated fabrics increased compared to the blank. The embedding of long-chain fluoroalkyl does not affect the hand feel of the treated fabric, which is exactly caused by the inherent properties of silicone softeners, which can endow the treated fabrics with very high softness, lubricity, and special tear strength, among other things, partly as a result of a reduction of interfibers and interyarns friction [26,27].
4. Conclusions
In summary, a long-chain fluoroacrylate was used to modify a polysiloxane so as to improve the hydrophobic properties of the polysiloxane and simultaneously retaining the softness of a fabric. The structure of the PFAS was confirmed by FT-IR and NMR. As a result of the bonding of long-chain fluoroalkyl, the PFAS presented good thermal stability. Due to the combined effect of the low surface free energy of the perfluorinated side chains and the relatively high surface roughness of the cotton fibers, the treated fabric had favorable hydrophobicity with a WCA of 144.7° on its surface. Its softness was increased, but the color remained unchanged due to the inherent property of silicone softeners.
Acknowledgements
This project was financially supported by the Public Welfare Technology Program of Wenzhou Science and Technology Bureau (No. G20170017).
Author Contributions
All the research work for this study was designed and conducted by Wei Xu. The article draft was written by Hua Jin. Wei Xu supervised and finalized the draft for publication.
Conflicts of Interest
The authors declare no conflict of interest.
References
- An, Q.-F.; Li, L.-S.; Huang, L.-X.; Chen, K.-C. Film morphology and characterization of functional polysiloxane softeners. AATCC Rev. 2006, 6, 39–43. [Google Scholar]
- An, Q.-F.; Wang, K.-F.; Jia, Y. Film morphology, orientation and performance of dodecyl/carboxyl functional polysiloxane on cotton substrates. Appl. Surf. Sci. 2011, 257, 4569–4574. [Google Scholar] [CrossRef]
- Huang, J.-Q.; Meng, W.-D.; Qing, F.-L. Synthesis and repellent properties of vinylidene fluoride-containing polyacrylates. J. Fluorine Chem. 2007, 128, 1469–1477. [Google Scholar] [CrossRef]
- Chang, K.-C.; Chen, H.; Huang, C.-K.; Huang, S.-I. Preparation of super-hydrophobic film with fluorinated-copolymer. J. Appl. Polym. Sci. 2007, 104, 1646–1653. [Google Scholar] [CrossRef]
- Li, J.; Wang, Q.-J.; Su, C.-H.; Chen, Q.-M. Preparation and characterization of fluorine-containing acrylate copolymers by 60Co γ-ray radiation co-polymerization. Eur. Polym. J. 2007, 43, 2928–2934. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kotera, M. Synthesis of fluorosilicone having highly fluorinated alkyl side chains based on the hydrosilylation of fluorinated olefins with polyhydromethylsiloxane. J. Polym. Sci. Polym. Chem. 2002, 40, 3120–3128. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kotera, M. Water and oil repellency of polysiloxanes with highly fluorinated alkyl side chains. J. Appl. Polym. Sci. 2002, 87, 1085–1091. [Google Scholar] [CrossRef]
- Guo, L.-H.; Jiang, S.-J.; Qiu, T.; Zhang, S.-W.; He, L.-F.; Tan, J.; Li, X.-Y. Miniemulsion polymerization of fluorinated siloxane-acrylate latex and the application as waterborne textile finishing agent. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Luo, Z.-H.; He, T.-Y.; Yu, H.-J.; Dai, L.-Z. A novel ABC triblock copolymer with very low surface energy: Poly(dimethylsiloxane)-block-poly(methyl methacrylate)-block-poly(2,2,3,3,4,4,4-heptafluorobutyl methacrylate). Macromol. React. Eng. 2008, 2, 398–406. [Google Scholar] [CrossRef]
- Yu, H.-J.; Luo, Z.-H. Novel superhydrophobic silica/poly(siloxane-fluoro-acrylate) hybrid nanoparticles prepared via two-step surface-initiated ATRP: Synthesis, characterization, and wettability. J. Polym. Sci. Polym. Chem. 2010, 48, 5570–5580. [Google Scholar] [CrossRef]
- Martinelli, E.; Suffredini, M.; Galli, G.; Glisenti, A.; Pettitt, M.-E.; Callow, M.-E.; Callow, J.-A.; Williams, D.; Lyall, G. Amphiphilic block copolymer/poly(dimethylsiloxane) (PDMS) blends and nanocomposites for improved fouling-release. Biofouling 2011, 27, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Galli, G.; Martinelli, E. Amphiphilic polymer platforms: Surface engineering of films for marine antibiofouling. Macromol. Rapid Commun. 2017, 38. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, E.; Moro, I.-D.; Galli, G.; Barbaglia, M.; Bibbiani, C.; Mennillo, E.; Oliva, M.; Pretti, C.; Antonioli, D.; Laus, M. Photopolymerized network polysiloxane films with dangling hydrophilic/hydrophobic chains for the biofouling release of invasive marine serpulid ficopomatus enigmaticus. ACS Appl. Mater. Interfaces 2015, 7, 8293–8301. [Google Scholar] [CrossRef] [PubMed]
- Baradie, B.; Lai, P.H.M.; Shoichet, M.-S. Synthesis and characterization of novel polysiloxane-grafted fluoropolymers. Can. J. Chem. 2005, 83, 553–558. [Google Scholar] [CrossRef]
- Xu, W.; An, Q.-F.; Hao, L.-F.; Zhang, D.; Zhang, M. Synthesis and characterization of self-crosslinking fluorinated polyacrylate soap-free latices with core-shell structure. Appl. Surf. Sci. 2013, 268, 373–380. [Google Scholar] [CrossRef]
- Xu, W.; An, Q.-F.; Hao, L.-F.; Huang, L.-X. Synthesis, film morphology and performance of cationic fluorinated polyacrylate emulsion with core-shell structure. J. Appl. Polym. Sci. 2012, 125, 2376–2383. [Google Scholar] [CrossRef]
- Hao, L.-F.; An, Q.-F.; Xu, W.; Huang, L.-X. Synthesis, film morphology and hydrophobicity of novel fluorinated polyacrylate emulsion and solution on silicon wafer. Colloids Surf. A 2012, 396, 83–89. [Google Scholar] [CrossRef]
- An, Q.-F.; Cheng, G.-W.; Li, L.-S. Synthesis, characterization, and film morphology of dodecylpolysiloxane. J. Appl. Polym. Sci. 2006, 101, 4480–4486. [Google Scholar] [CrossRef]
- Chen, L.-J.; Wu, F.-Q. BA-MMA-POMA copolymer latexes prepared by using HMPS polymerizable emulsifier. J. Appl. Polym. Sci. 2011, 122, 819–826. [Google Scholar] [CrossRef]
- Hao, L.-F.; Xu, W.; An, Q.-F. Synthesis, film morphology and performance of novel crosslinked polysiloxane with end-capped epoxy groups on cotton substrates. Fibers Polym. 2014, 15, 1567–1574. [Google Scholar] [CrossRef]
- Dong, F.-Y.; Sun, X.-R.; Feng, S.-Y. Thermal degradation kinetics of functional polysiloxanes containing chloromethyl groups. Thermochim. Acta 2016, 639, 14–19. [Google Scholar] [CrossRef]
- Xu, Y.-S.; Liu, M.-Y. Corrosion behaviors of polysiloxane-ferroferric oxide coating coated on carbon steel in NaCl solution and geothermal water. Geothermics 2017, 70, 339–350. [Google Scholar] [CrossRef]
- Jiang, B.; Zhang, T.; Zhao, L.-W.; Xu, Z.-M.; Huang, Y.-D. Effect of polymerizable photoinitiators on the UV-polymerization behaviors of photosensitive polysiloxane. J. Polym. Sci. Pol. Chem. 2017, 55, 1696–1705. [Google Scholar] [CrossRef]
- Song, X.-Y.; Zhai, J.; Wang, Y.-L.; Jiang, L. Fabrication of superhydrophobic surfaces by self-assembly and their water-adhesion properties. J. Phys. Chem. B 2005, 109, 4048–4052. [Google Scholar] [CrossRef] [PubMed]
- Corpart, J.-M.; Girault, S.; Juhué, D. Structure and surface properties of liquid crystalline fluoroalkyl polyacrylates: Role of the spacer. Langmuir 2001, 17, 7237–7244. [Google Scholar] [CrossRef]
- Montazer, M.; Hashemikia, S. Application of polyurethane/citric acid/silicone softener composite on cotton/polyester knitted fabric producing durable soft and smooth surface. J. Appl. Polym. Sci. 2012, 124, 4141–4148. [Google Scholar] [CrossRef]
- Fouda, M.M.G.; Fahmy, H.M. Multifunctional finish and cotton cellulose fabric. Carbohydr. Polym. 2011, 86, 625–629. [Google Scholar] [CrossRef]
Scheme 1.
Synthetic process of the fluoroalkyl-acrylate-modified polysiloxane (PFAS).
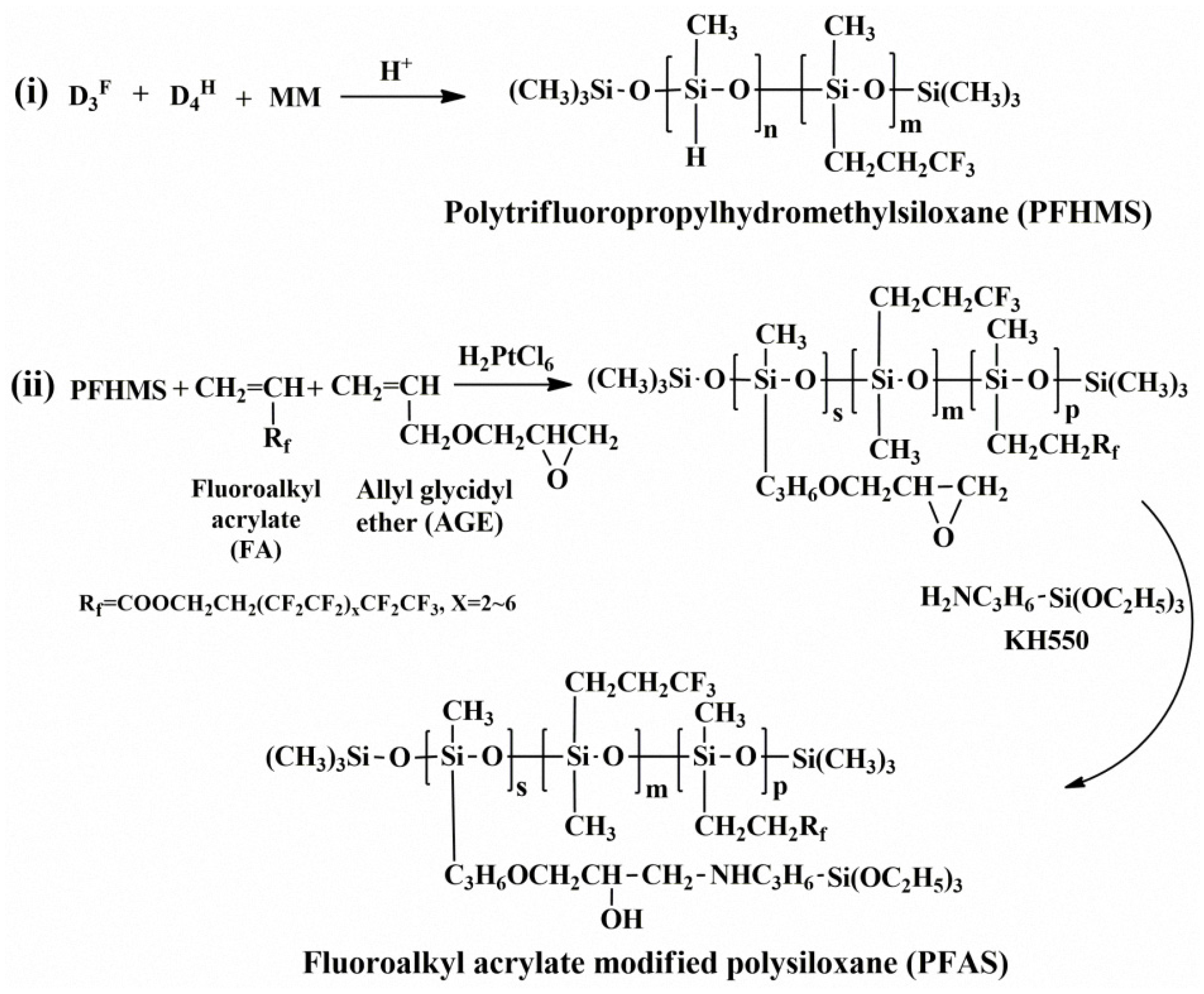
Figure 1.
IR spectra of (a) fluoroalkyl acrylate (FA), (b) polytrifluoropropylhydromethylsiloxane (PFHMS), and (c) PFAS.
Figure 1.
IR spectra of (a) fluoroalkyl acrylate (FA), (b) polytrifluoropropylhydromethylsiloxane (PFHMS), and (c) PFAS.
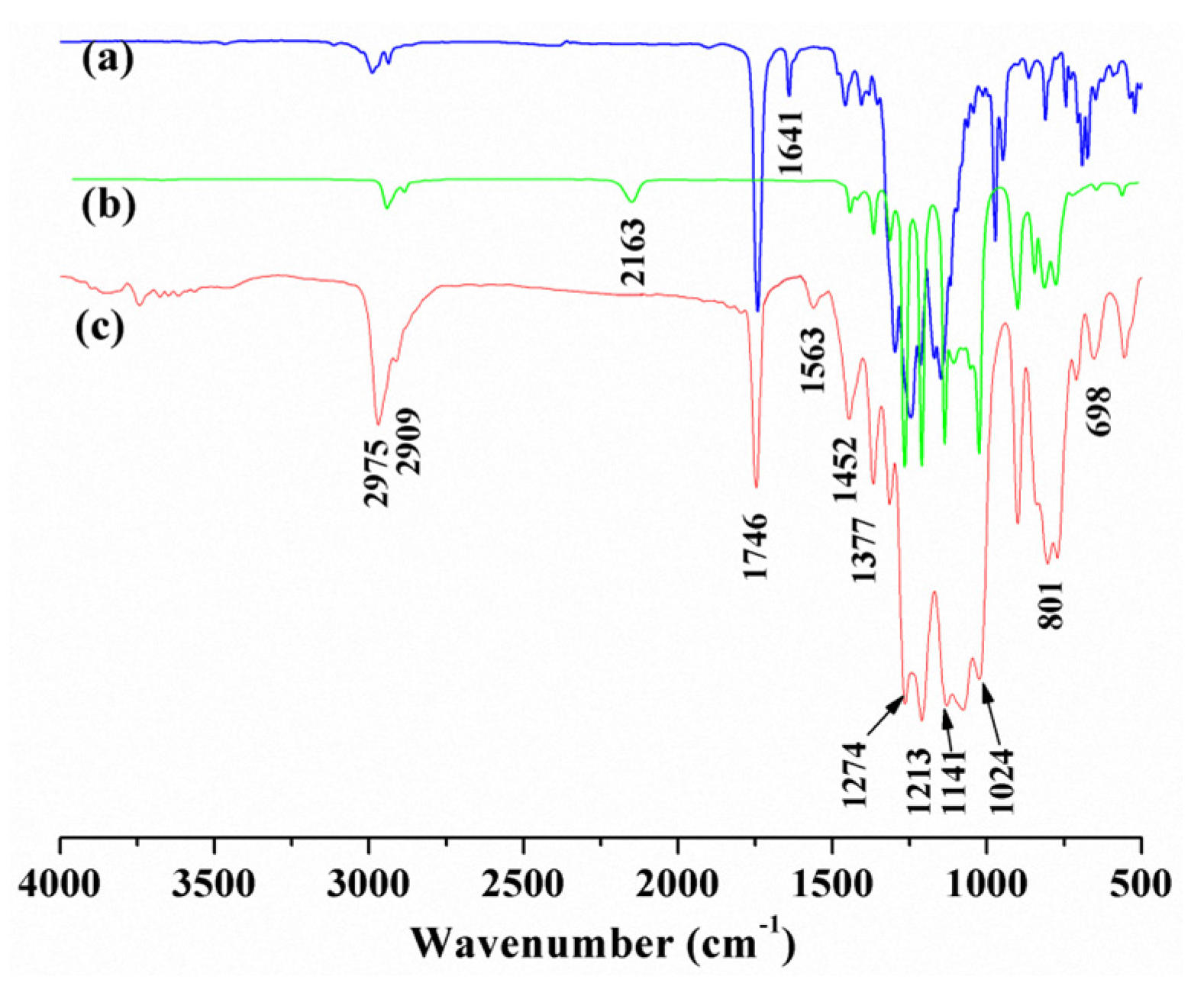
Figure 2.
1H-NMR of the PFAS.
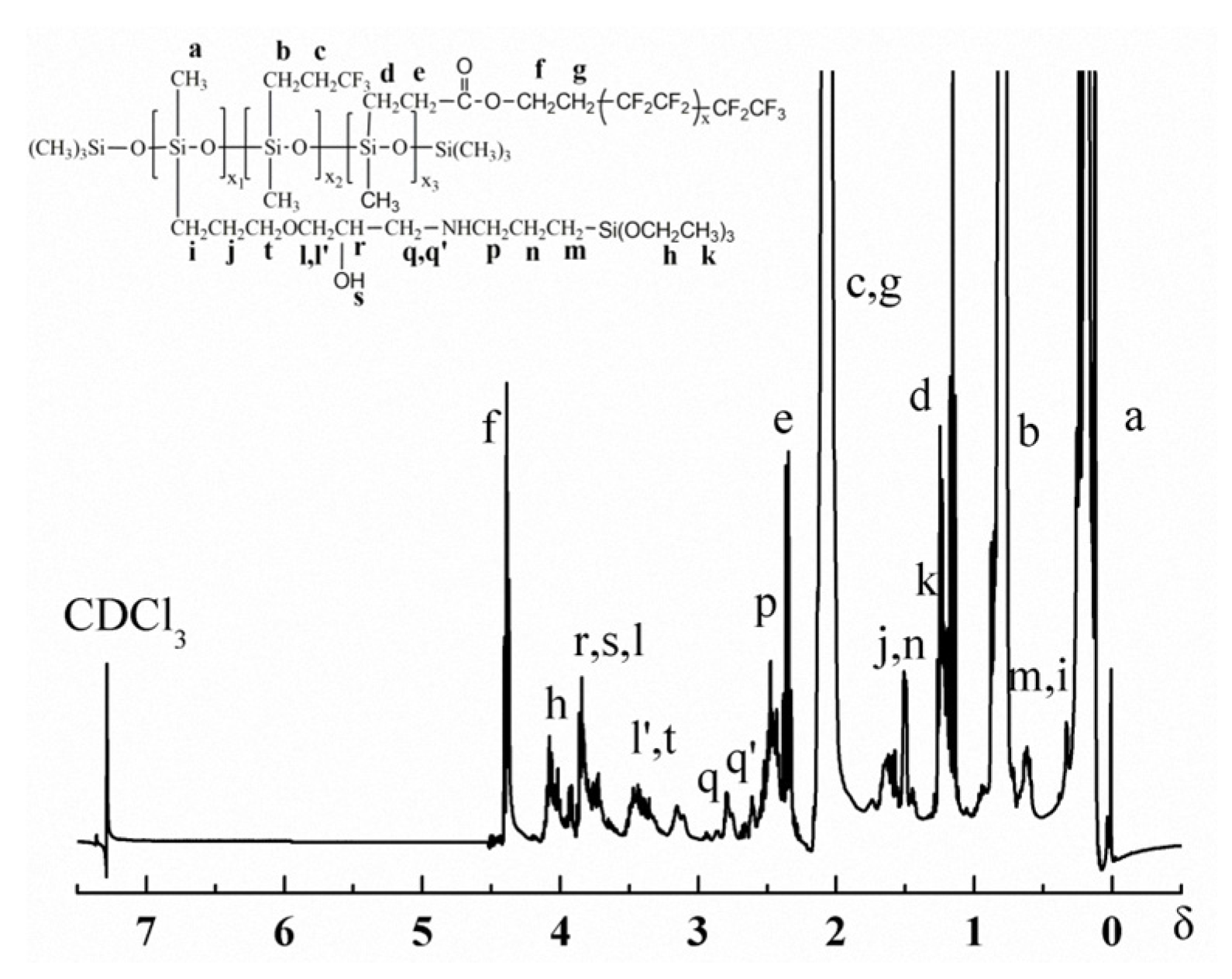
Figure 3.
13C-NMR of the PFAS.
Figure 4.
TGA graph of the PFAS polymer.
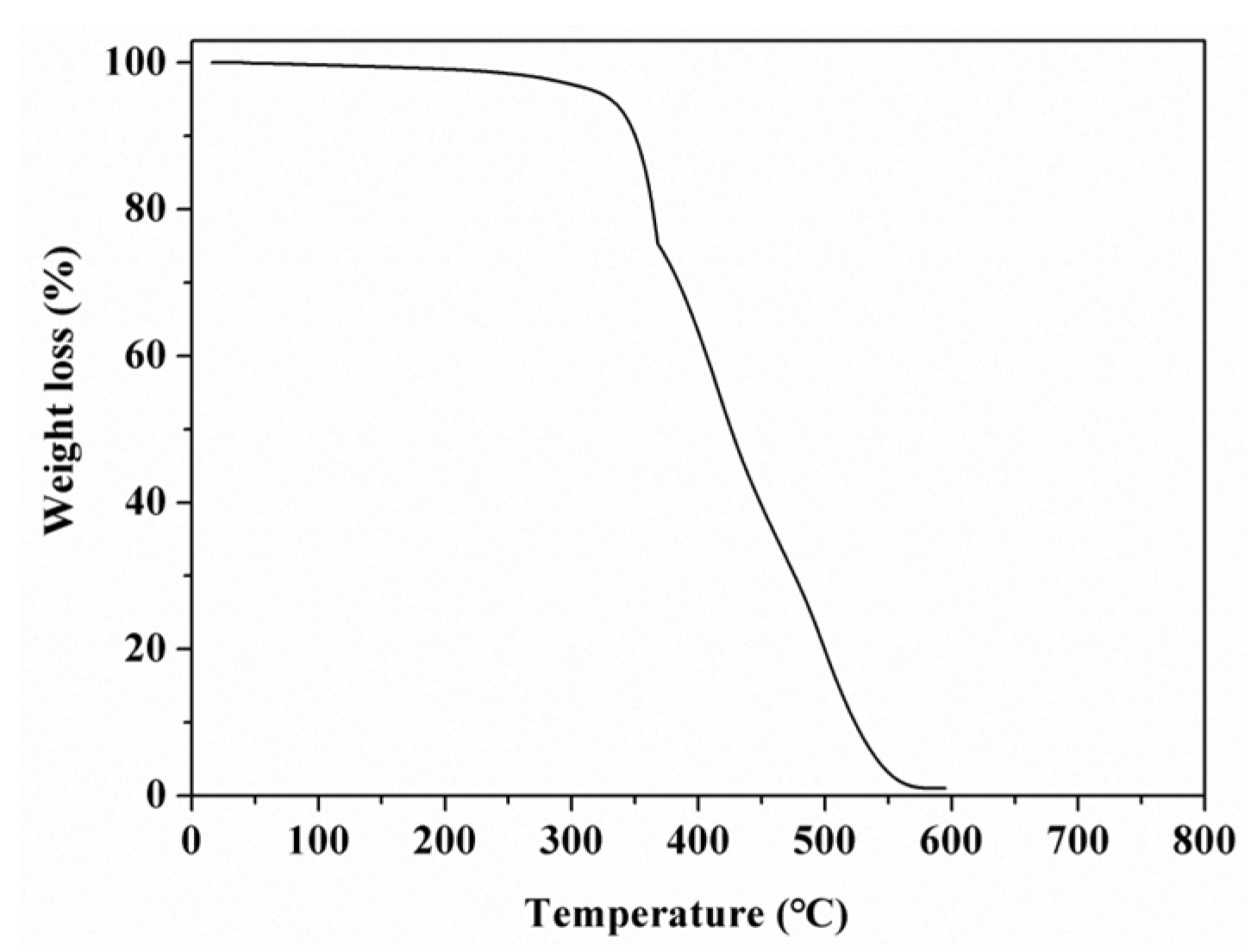
Figure 5.
SEM images of (a) the blank cotton fabric and (b) the PFAS-treated one.

Figure 6.
AFM images: (a) the PFAS height image and (b) the PFAS topography.
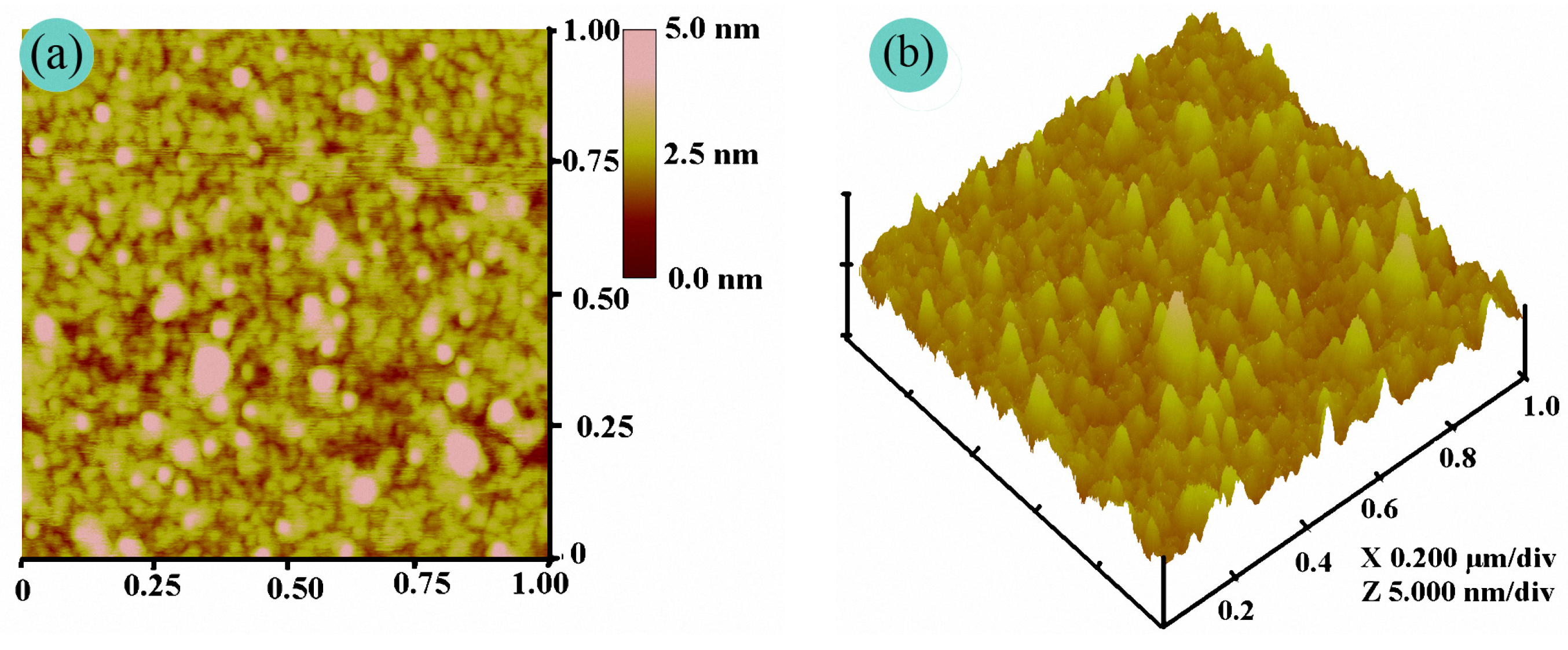
Figure 7.
XPS spectra: (a) wide spectrum; (b) C12S high resolution spectrum and (c) N1S high resolution spectrum.
Figure 7.
XPS spectra: (a) wide spectrum; (b) C12S high resolution spectrum and (c) N1S high resolution spectrum.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Cotton fabric specifications.
| Fabric Weight | Yarn Count | Fabric Density | ||
|---|---|---|---|---|
| (g/m2) | Warp | Weft | End/cm | Picks/cm |
| 119 | 40 | 40 | 133 | 72 |
Table 2.
Performance properties of the cotton fabrics treated with PFAS.
| Dose of the Finishing Agent | WCA (°) | Whiteness | BR (mN) | |
|---|---|---|---|---|
| (g/100 g Ethyl Acetate) | (°) | w | f | |
| 0 | 0 | 76.89 | 239 | 175 |
| 0.1 | 139.3 ± 1.2 | 76.80 | 234 | 153 |
| 0.2 | 141.4 ± 1.8 | 76.31 | 230 | 152 |
| 0.3 | 143.9 ± 1.3 | 76.82 | 224 | 149 |
| 0.4 | 144.7 ± 1.1 | 76.75 | 218 | 141 |
| 0.5 | 144.9 ± 1.9 | 76.88 | 215 | 137 |
Notes: WCA: Water contact angles, BR: bending rigidity, w: warp, f: fill.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jin, H.; Xu, W. The Preparation and Properties of Fluoroacrylate-Modified Polysiloxane as a Fabric Coating Agent. Coatings 2018, 8, 31. https://doi.org/10.3390/coatings8010031
AMA Style
Jin H, Xu W. The Preparation and Properties of Fluoroacrylate-Modified Polysiloxane as a Fabric Coating Agent. Coatings. 2018; 8(1):31. https://doi.org/10.3390/coatings8010031
Chicago/Turabian StyleJin, Hua, and Wei Xu. 2018. "The Preparation and Properties of Fluoroacrylate-Modified Polysiloxane as a Fabric Coating Agent" Coatings 8, no. 1: 31. https://doi.org/10.3390/coatings8010031
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.