1. Introduction
The development of bio-based materials as an alternative to traditional polymers and composites from non-renewable resources has gained significant attention in the past couple of decades due to economical and environmental factors. As a result, numerous vegetable oil-based thermosetting resin systems have been recently studied [
1,
2,
3,
4,
5,
6]. Vegetable oil-based resins recently developed include polyester amides [
7], and cyanate esters [
8]. Due to their abundance, low cost, readily availability, and renewable nature, vegetable oils have been used for the preparation of films, coatings, and thermosetting resins with interesting thermo-mechanical properties [
9,
10].
With over 80% of its fatty acid chains bearing a conjugated triene, tung oil can react with vinyl co-monomers via cationic, thermal, or free radical polymerizations without the need of any structural modification of the triglyceride [
9,
10,
11]. The resulting materials from the polymerization of tung oil consist of highly crosslinked thermosets. Whenever vegetable oil-based thermosetting resins are reinforced with materials exhibiting a polar surface, there is an intrinsic incompatibility between the hydrophobic resin and the hydrophilic reinforcement, which can be overcome by the addition of compatibilizers to the resin formulation [
3,
12]. Very recently, the successful use of asolectin as a natural compatibilizer for cellulose-reinforced tung oil-based composites has been reported [
13].
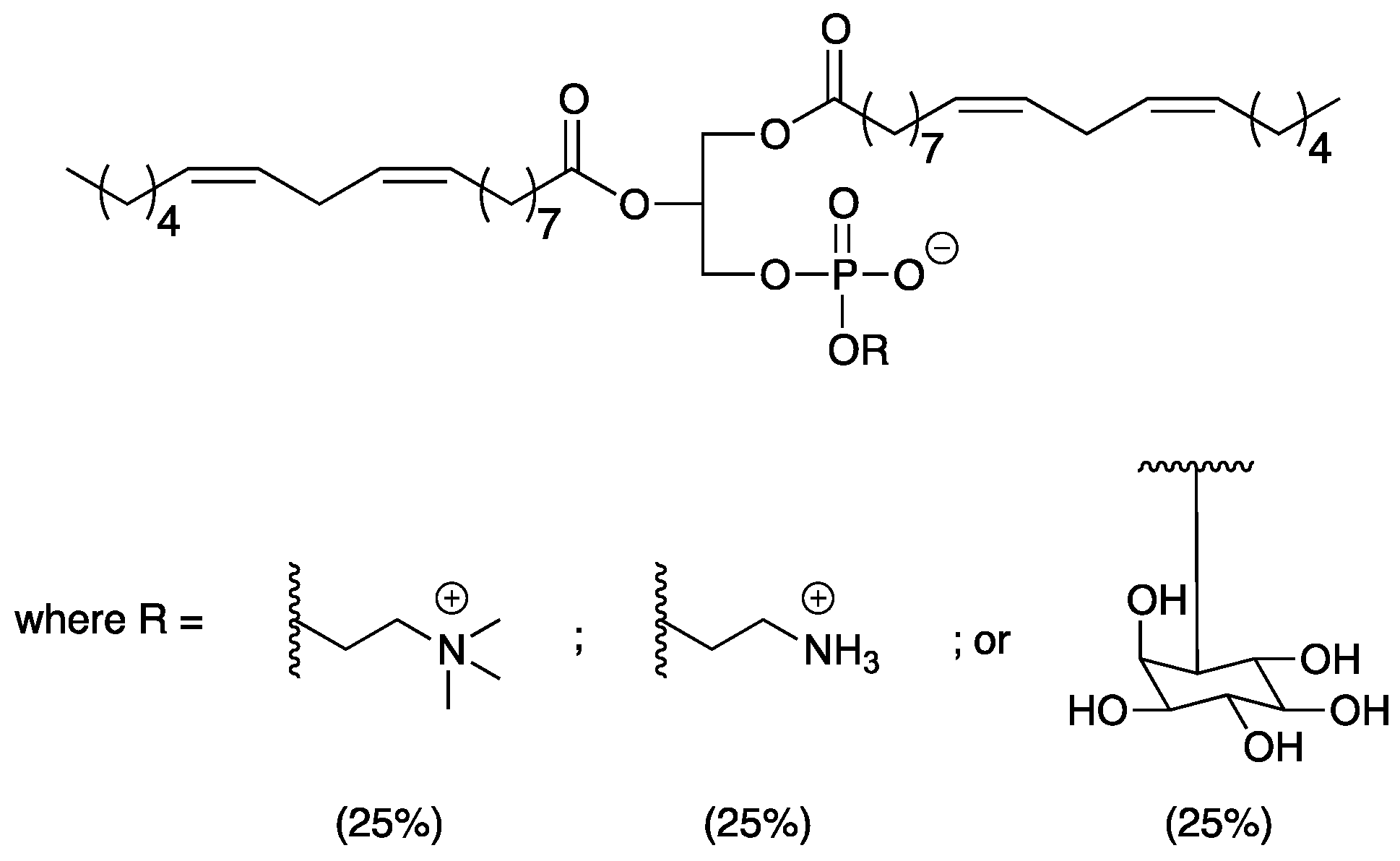
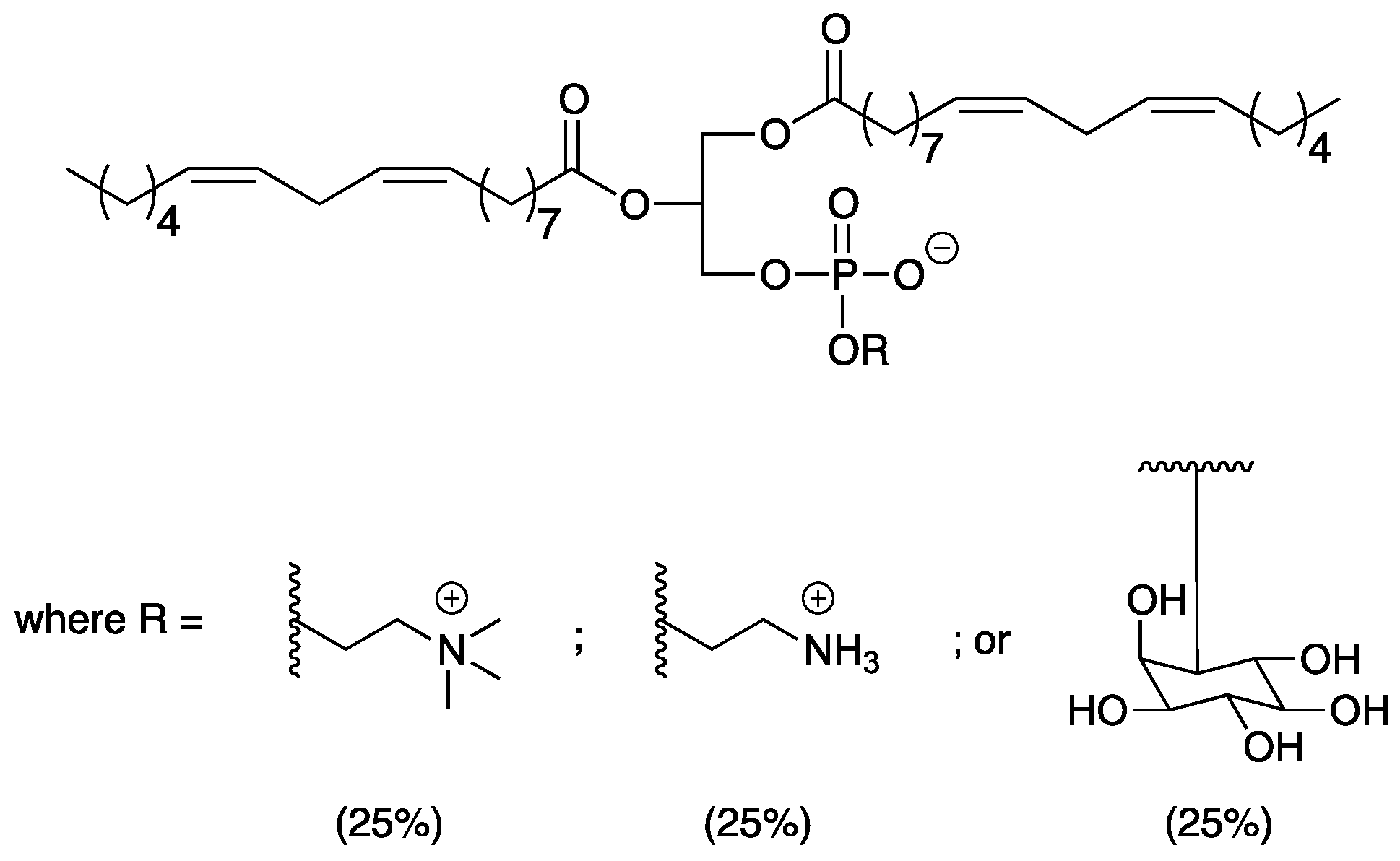
Asolectin consists of a mixture of phospholipids from soybeans containing 76% of polyunsaturated fatty acids [
14]. Indeed, the mixture contains approximately 25% of phosphatidylcholine, 25% of cephalin, and 25% of phosphatidylinositol (
Figure 1), with the remainder consisting of small amounts of other phospholipids from soybeans [
15]. Due to its amphiphilic nature, asolectin interacts with the non-polar resin components through its fatty acid chains and with the hydrophilic reinforcement through the phosphate and ammonium groups [
13]. The polyunsaturated fatty acids in asolectin enable the phospholipid to be free radically crosslinked to other reactive and compatible olefins upon cure [
13]. Due to obvious similarities in the structures of phospholipids and triglycerides, a copolymer of tung oil and asolectin is readily formed in the presence of a free radical initiator [
13]. When cellulose/tung oil-based composites are prepared with asolectin, improved thermo-mechanical properties are obtained in comparison to composites prepared without asolectin [
13].
The preparation of latexes and emulsions containing bio-based polymers allow for smooth film depositions leading to bio-based coatings [
15,
16]. Vegetable oil-based anionic [
15], and cationic [
16] polyurethane latexes with uniform particle sizes have been previously prepared. In these instances, the final polyurethane latexes were obtained by the co-polycondensation of vegetable oil-based polyols and diisocyanates in a water medium [
15,
16]. Polyurethane-acrylic hybrid systems have been developed with a similar concept [
17]. In such systems, unreacted carbon-carbon double bonds from the vegetable oil-based polyols were crosslinked with acrylic co-monomers through free radical emulsion polymerization, resulting in hybrid latexes [
17,
18]. In such hybrid systems, the polyurethane forms a polar shell that serves as a high molecular weight emulsifier, while the vinyl polymers form a non-polar core [
19].
Typically, emulsifiers are required in order to achieve successful free radical emulsion polymerization in solvent mixtures containing water [
20]. In this work, asolectin is used for the first time as an effective bio-based emulsifier for the emulsion free radical co-polymerization of tung oil, divinylbenzene, and
n-butyl methacrylate in a xylene/water mixture. The incorporation of asolectin in the crosslinked polymer results in stable bio-based polymeric vesicles with potential application in drug delivery and/or nano-catalyst encapsulation/stabilization. The successful free radical polymerization of xylene/water emulsions containing tung oil and asolectin was verified through Matrix-assisted Laser Desorption/Ionization-Time of Flight (Maldi-TOF). The cure of the monomer mixture was monitored by Dielectric Analysis (DEA), while changes in the Raman spectrum of all co-monomers before and after the reaction, along with differential scanning calorimetry (DSC) analysis, indicate the need of a post-cure step in order to attain completion of the polymerization reaction. Products obtained with different amounts of tung oil were further characterized by Thermogravimetric Analysis (TGA). The formation of vesicles was confirmed by Scanning Transmission Electron Microscopy (STEM) imaging, and vesicle size distribution was evaluated using a Zetasizer.
2. Experimental
2.1. Materials
Tung oil, asolectin from soybeans, di-tert-butyl peroxide (DTBP), and n-butyl methacrylate (BMA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Divinylbenzene (DVB) was acquired from TCI America (Portland, OR, USA), and xylenes and 3,5-dimethoxy-4-hydroxycinnamic acid were procured from Acros Organics (Pittsburg, PA, USA).
2.2. Preparation of Asolectin-Containing Latexes
Designated amounts of a resin containing 40 wt.% of tung oil, 30 wt.% of n-butyl methacrylate (BMA), 20 wt.% of DVB, and 10 wt.% of asolectin was initially prepared and added to a 20 mL scintillation vial. Whenever the percentage of tung oil in the mixture was changed as described in the text, a constant DVB:BMA ratio of 3:2 (by weight) was maintained. The percentage of asolectin was kept constant at 10 wt.% for all emulsions. The mixture was vortexed for 15 s, leading to a seemingly homogenous liquid. After addition of an extra 5 wt.% of DTBP with respect to the total monomer mixture weight, the components were vortexed for another 15 s. 2.75 g of Xylene were then dispensed into the vial, followed by another 15 s of vortexing. Finally, 11.0 g of deionized water was added to the vial to create a 1:4 (xylene:water) by weight mixture. The vial was then capped, sealed with parafilm, and placed in a sonicator for 20 min to create an emulsion. After sonication, the vial was placed in a convection oven for 24 h at 100 °C for initial cure. After the initial curing process, the vial containing the emulsion was uncapped and dried for approximately 24 h in a vacuum oven at 60 °C. The dried contents were subjected to a post-cure step at 140 °C for 2 h in a convection oven to ensure complete polymerization, unless otherwise noted in the text.
2.3. Characterization
Maldi-TOF spectra were collected with a Microflex Maldi-TOF (Bruker Daltonics, Karlsruhe, Germany) using a linear method. Using 3,5-dimethoxy-4-hydroxycinnamic acid as the matrix, 1:50 (sample:matrix) by weight mixtures were prepared by mixing appropriate amounts of the ground post-cured sample and the matrix with a mortar and pestle. The dry sample/matrix mixture was sandwiched between two MSP 96 polished steel target plates and compressed at 1000 lbs in a hydraulic press. The laser was set to 100 shots per run at 66% power and a frequency of 60 Hz.
DSC experiments were conducted on a Q20 DSC instrument (TA Instruments, New Castle, DE, USA). Tests were run in a N2 atmosphere from −20 °C to 200 °C at a heating rate of 10 °C/min with sample sizes of 10 mg.
DEA experiments were conducted with an Epsilon DEA 230/1 cure monitor (Netzsch Instruments North America LLC, Burlington, MA, USA) to analyze the rate of cure. The instrument’s probe was immersed in 10 g of the resin. The resin was cured in a convection oven held constantly at 100 °C. The DEA test was conducted for 24 h with frequencies ranging from 0.1 Hz to 10,000.0 Hz. The permittivity and loss factor were measured as a function of time, and the results displayed in the text convey a plot of ion viscosity (Ohm·cm) versus time. The use of dielectric analysis for monitoring the polymerization of resins relies on the fact that in non-conductive polymers/substances, charge transfer occurs through the mobility of ions within the sample and is measured by the material’s overall permittivity. As the polymerization progresses, longer chains are formed and ion mobility is gradually compromised, which is reflected in the materials permittivity. Once the polymerization is completed and polymer chain growth has ceased, no further change in ion mobility occurs and the permittivity remains constant. The ion viscosity discussed in in the text corresponds to a measurement of the resistance offered by a material to the transfer of a charge in a linear path. This property is measured in Ohm·cm and is referred to as “ion viscosity” due to the analogy with the flow of a substance through a medium.
Raman spectra were acquired with a DXR Raman Microscope (Thermo Fisher Scientific, Waltham, MA, USA). A total of 32 scans were collected per sample at an exposure time of 1 s for each scan. Laser power was set to 7.0 mW, and a 50 µm slit aperture was used.
A Q50 TGA instrument (TA Instruments, New Castle, DE, USA) measured weight loss of samples (10 mg) under an air atmosphere as a function of temperature. Samples were heated from room temperature to 600 °C at a heating rate of 20 °C/min.
The morphology of the vesicles prepared was observed under Scanning Transmission Electron Microscopy mode (STEM) using a JEOL JSM-7600F field emission scanning electron microscope (Peabody, MA, USA) equipped with a transmission detector. The microscope was operated at 30.0 kV accelerating voltage during STEM imaging. The suspension was deposited directly on a TEM copper grid after the heating treatment, and allowed to dry at ambient conditions for 24 h before imaging.
A nano series Zetasizer (Malvern Instruments, Worcestershire, UK) was used in order to obtain particle size and polydispersity index values. An aliquot of each emulsion was placed in a cuvette and diluted 10 times with deionized water. Measurements were made at 25 °C with a 90° angle of detection. Emulsion samples underwent three measurements with 11 runs per measurement for a duration of 10 s per run. Automatic attenuation selection was performed for each sample. The results reported in this manuscript correspond to the average of the three measurements carried out on each sample.
3. Results and Discussion
In order to verify that tung oil/asolectin-based emulsions can be polymerized under free radical conditions in the presence of DTBP, 1:4 (xylene:water) by weight emulsions of various ratios of tung oil and asolectin were cured in a convection oven at 100 °C for 24 h. The 1:4 (xylene:water) by weight solvent ratio was determined based on related literature [
20] and after a major screening of solvent combinations. As described previously, the polymerized emulsions were dried in a vacuum oven at 60 °C for 24 h and the dried materials recovered were subjected to Maldi-TOF analysis.
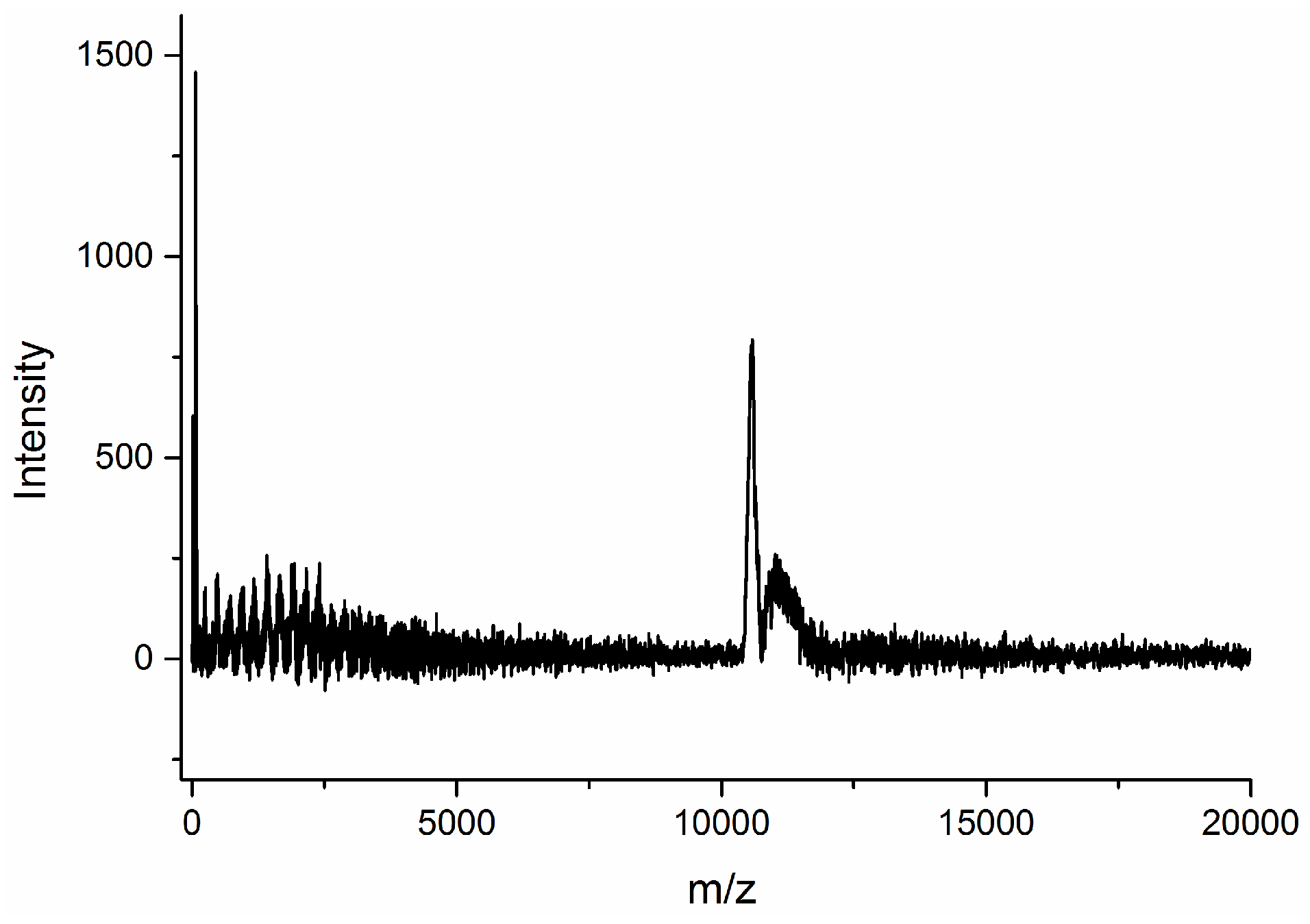
Figure 2 shows the Maldi-TOF spectrum of the dry products from the emulsion containing a 1:1 weight ratio of tung oil and asolectin.
Two major signals can be clearly seen in the 11,000–12,000 m/z range in
Figure 2, confirming the formation of high molecular weight products upon cure of the emulsion. Despite the promising results shown in
Figure 2, the dry products obtained from tung oil and asolectin were exceedingly soft, rubbery, and tacky. In order to enhance the mechanical characteristics of the dry film, as demonstrated previously with related bio-based polyolefin systems [
1], it became necessary to incorporate other reactive olefins, such as DVB and BMA. Upon addition of more reactive components, a natural increase of the dry products’ crosslink density made it impossible to obtain clear Maldi-TOF spectra due to the inherent difficulty of analyzing thermoset materials through this technique.
Previously, the bulk free radical polymerization of 40 wt.% of tung oil, 30 wt.% of BMA, 20 wt.% of DVB, and 10 wt.% of asolectin had been successfully achieved after heating the crude resin in the presence of DTBP at 140 °C for 2 h and 50 min [
13]. The same resin composition was therefore prepared into a xylene/water emulsion as described in the
Experimental section. Xylene was chosen as the most appropriate solvent due to its compatibility with the monomers and also due to its relatively high boiling point, allowing for higher polymerization temperatures. Water has a lower boiling point than xylene, and therefore, the highest temperature at which the system can be cured is 100 °C. A weight ratio of 1:4 (xylene:water) was found to create a stable emulsion after 20 min of sonication. In order to select the optimum resin/solvent ratio, emulsions containing overall monomer weights of 200 mg, 400 mg, and 600 mg were prepared, as shown in
Figure 3.
It is obvious from
Figure 3 that an immediate phase separation occurs for samples containing 400 mg (
Figure 3B) and 600 mg (
Figure 3C) of total monomer weight, while a total monomer weight of 200 mg (
Figure 3A) produced no phase separation and therefore appeared to create a stable emulsion. The latter composition was observed over a period of 30 min in order to assess the stability of the emulsion formed. The results can be seen in
Figure 4.
From the pictures in
Figure 4, the emulsion containing 200 mg of total monomer weight doesn’t change its appearance for at least 30 min after preparation. It is therefore hypothesized that, if the free radical polymerization reaction starts within the first 30 min following the preparation of the emulsion, the monomers present in the non-polar micelles will start crosslinking before disruption of the emulsion, leading to spherical particles. The emulsions prepared held their stability for an extended period of time when kept in a closed container. No phase separation was observed since the emulsion preparation.
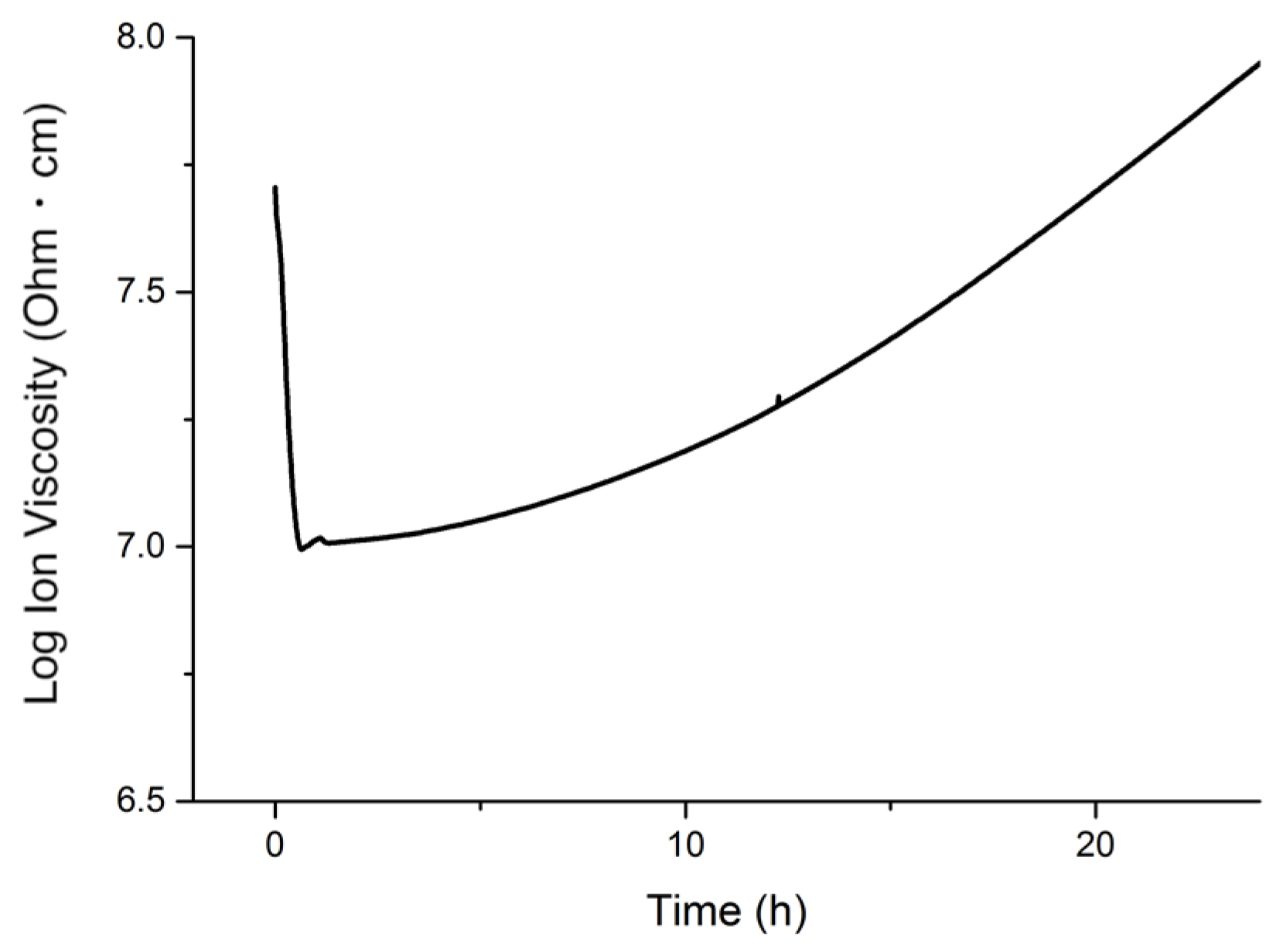
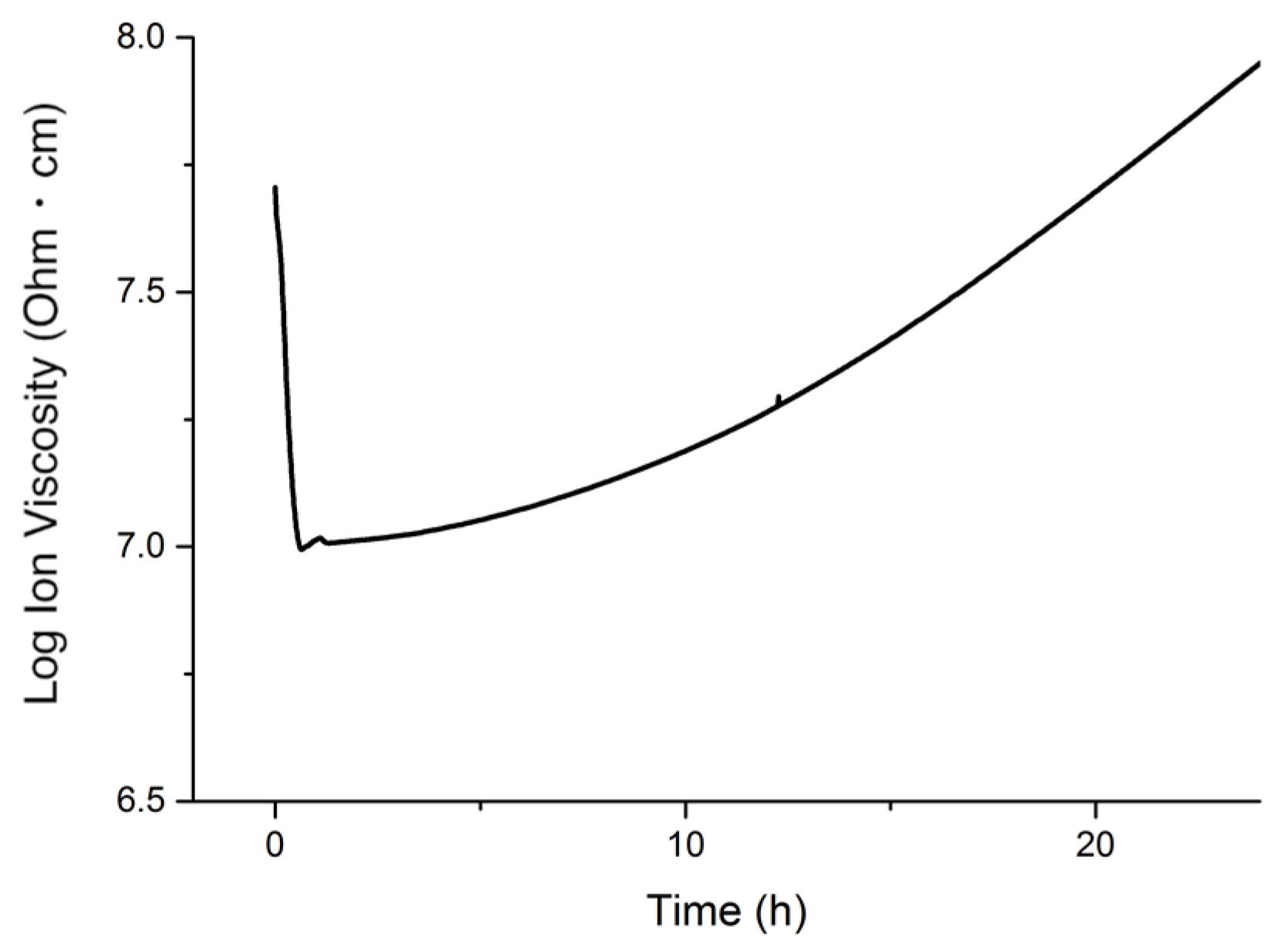
The progress of the polymerization of the monomers at the cure temperature employed was monitored through dielectric analysis (DEA). The pure resin, containing 40 wt.% of tung oil, 30 wt.% of BMA, 20 wt.% of DVB, 10 wt.% of asolectin and an extra 5 wt.% of DTBP, was heated at 100 °C in a convection oven. The resulting DEA curve is displayed in
Figure 5.
In
Figure 5, the initial decrease in ion viscosity is due to an increase in ion mobility as the monomers start heating. After 20 min of heating, ion viscosity begins to increase as the polymerization process begins. Based on the stability experiment results (
Figure 4), the onset polymerization time of 20 min is ideal for the cure of emulsions since within that time frame, the emulsion will still be intact. The continuous increase in the slope of the DEA curve for the remainder of the experiment indicates a continuous progress in the polymerization process. Over the course of 24 h, the DEA curve doesn’t plateau, indicating that the resin isn’t fully polymerized within the duration of the experiment. These results suggest that a post-cure step might be needed in order to complete the polymerization reaction.
In order to investigate the effect of tung oil concentration in the cure of tung oil/asolectin-based emulsions, samples with varying concentrations of tung oil were prepared and cured at 100 °C for 24 h. Maintaining the concentration of asolectin constant at 10 wt.% and the BMA/DVB weight ratio set at 1.5, four samples containing 40 wt.%, 55 wt.%, 70 wt.%, and 80 wt.% of tung oil were prepared and cured at 100 °C for 24 h. The dried products from these samples were analyzed by DSC as shown in
Figure 6.
From the DSC curves in
Figure 6, it can be seen that all samples exhibit an exothermic peak in the 150–160 °C regardless of the sample composition. Those peaks indicate an exothermic reaction occurring at that temperature, suggesting further crosslink of unreacted carbon-carbon double bonds. The presence of such peak in the DSC curves of all four compositions demonstrates that none of the dried products are fully polymerized when subjected to the cure conditions (100 °C, 24 h), revealing a need for a post-cure step after drying.
Table 1 lists the enthalpy and the temperature for the maximum of the exothermic peaks observed for the samples in
Figure 6. The enthalpy, calculated from the integration of the corresponding peaks is highest in the products containing the greatest amount of tung oil. This information suggests that samples with lower amounts of tung oil are polymerized to a higher extent during cure when compared to samples having a higher concentration of tung oil. This is most likely due to the relative reactivity of the oil and the other monomers. Due to its chemical structure, tung oil is expected to be less reactive towards free radicals than DVB or BMA. Therefore, increasing the amount of tung oil in the resin makes it less reactive and a higher portion of the monomers remains unreacted after the cure, generating higher enthalpies upon heating during the DSC experiments.
When comparing the peak max temperatures in
Table 1, there is an apparent trend of decrease in the temperature with increases in the percentage of tung oil in the sample for samples containing up to 70 wt.% of oil. This behavior could be related to the overall number of carbon-carbon double bonds per monomer molecules. With increasing amounts of tung oil, there is a natural increase in the number of carbon-carbon double bonds that remain unreacted in the resin after cure. With a larger number of carbon-carbon double bonds, it is expected that the residual polymerization during the DSC experiments will start more easily, requiring a lower temperature. This trend, however, doesn’t hold true for the sample containing 80 wt.% of tung oil, which exhibits a slightly higher peak max than the sample with 70 wt.% of tung oil. At this stage, after repeating these experiments three times and preparing new samples with consistent results, it is unclear why the sample containing 80 wt.% of tung oil exhibits a different behavior than the other ones. The answer could be in micro-phase separation phenomena happening at a very small scale within the sample, but further investigation would be needed in order to fully understand this behavior. Overall, the DSC results suggest that the sample containing 40 wt.% of tung oil is the one with the highest extent of polymerization after the 24 h curing process, but the implementation of a post-cure step is needed for full polymerization of the sample.
In a previous work [
13], it had been demonstrated that the bulk polymerization of a resin with the same monomer composition as the sample containing 40 wt.% of tung oil had been completed after heating at 140 °C for 2 h and 50 min. Therefore, in order to obtain a completely cured system, a post-cure step at 140 °C was implemented here after drying the emulsions. DSC was used to assess any residual cure in the dried emulsions after post-curing at 140 °C for 1.5 h and 2 h (
Figure 7).
When comparing the DSC curves of samples post-cured for 0 h, 1.5 h, and 2 h (
Figure 7), it becomes evident that the exothermic event at approximately 160 °C decreases with post-cure time. Indeed, the peak is obvious and prominent in the sample that wasn’t post-cured (
Figure 7A). After a post-cure time of 1.5 h (
Figure 7B), the peak is very subtle. When the sample is subjected to a post-cure of 2 h (
Figure 7C), no peaks can be detected at 160 °C. The appearance of a change in the baseline at approximately 70 °C for the post-cured samples (
Figure 7B,C) may be indicative of a glass transition temperature. The absence of considerable exothermic peaks after a post-cure of 2 h suggests that no residual cure takes place during the DSC experiment, indicating that the polymerization process is complete. Therefore, a post-cure step of 2 h at 140 °C is crucial in order to complete the polymerization of the system.
Raman spectroscopy was also used to verify the completion of the reaction after a 2 h post-cure at 140 °C (
Figure 8). The spectra corresponding to asolectin, tung oil, and BMA components (
Figure 8A–C) exhibit a peak in the 1630–1650 cm
−1 range corresponding to C=C streches. The absence of a significant peak in the same region of the spectrum of the post-cured sample (
Figure 8D) suggests that the carbon-carbon double bonds in the monomers were completely reacted during the polymerization and post-cure processes. The additional peak observed in the spectrum of BMA at approximately 1725 cm
−1 can be attributed to the carbonyl group.
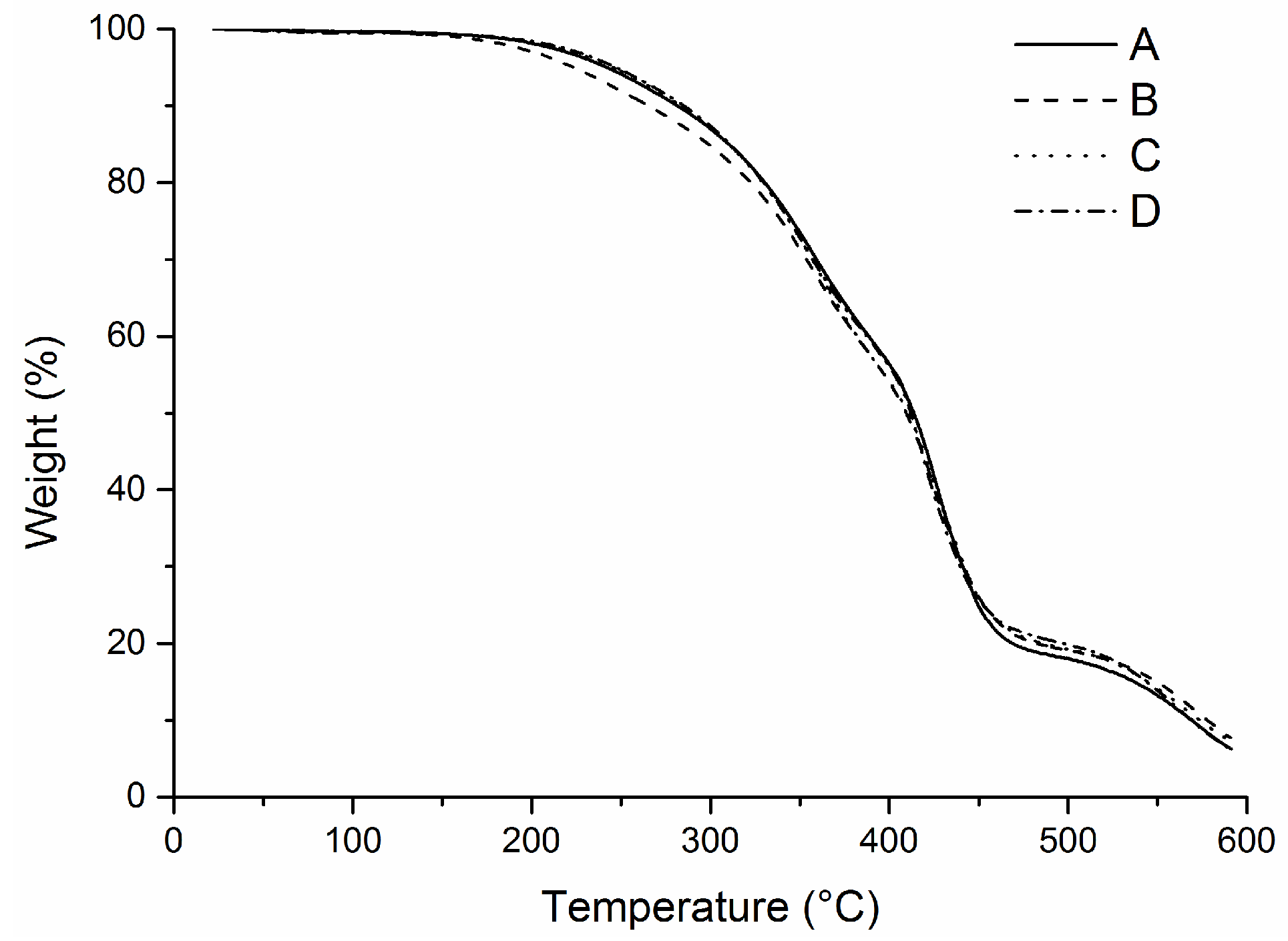
Since fully polymerized samples were obtained from the emulsion containing 40 wt.% tung oil after a post cure step of 2 h at 140 °C, the post cure step was applied to all monomer compositions. After the post cure step, emulsions containing 40–80 wt.% of tung oil were analyzed by DSC (
Figure 9) and TGA (
Figure 10).
The similarity among the DSC curves from samples containing 40–80 wt.% of tung oil (
Figure 9) suggests that a fully cured system can be obtained from any of these compositions after a 2 h post cure step at 140 °C. Indeed, no obvious exothermic peaks can be observed at approximately 160 °C after the post-cure (
Figure 9). It can also be seen that the change in the baseline at approximately 70 °C, possibly indicating a glass transition temperature, is present in all curves in
Figure 9. TGA results (
Figure 10) also illustrate similar thermal properties between post-cured samples prepared with 200 mg of total monomer weight and with 40–80 wt.% of tung oil. Very little variability with no clear trend can be observed among the samples when heated to 600 °C under air.
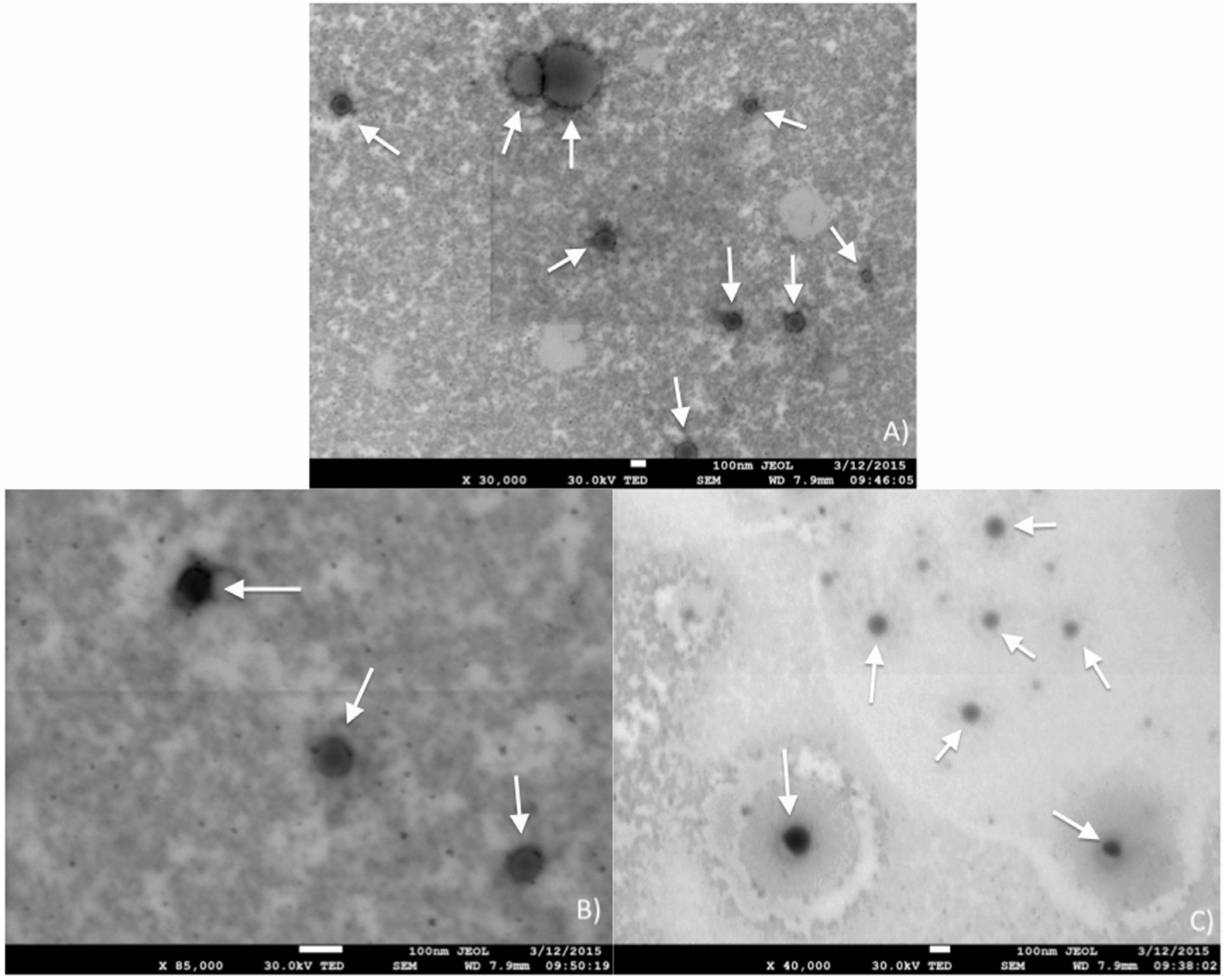
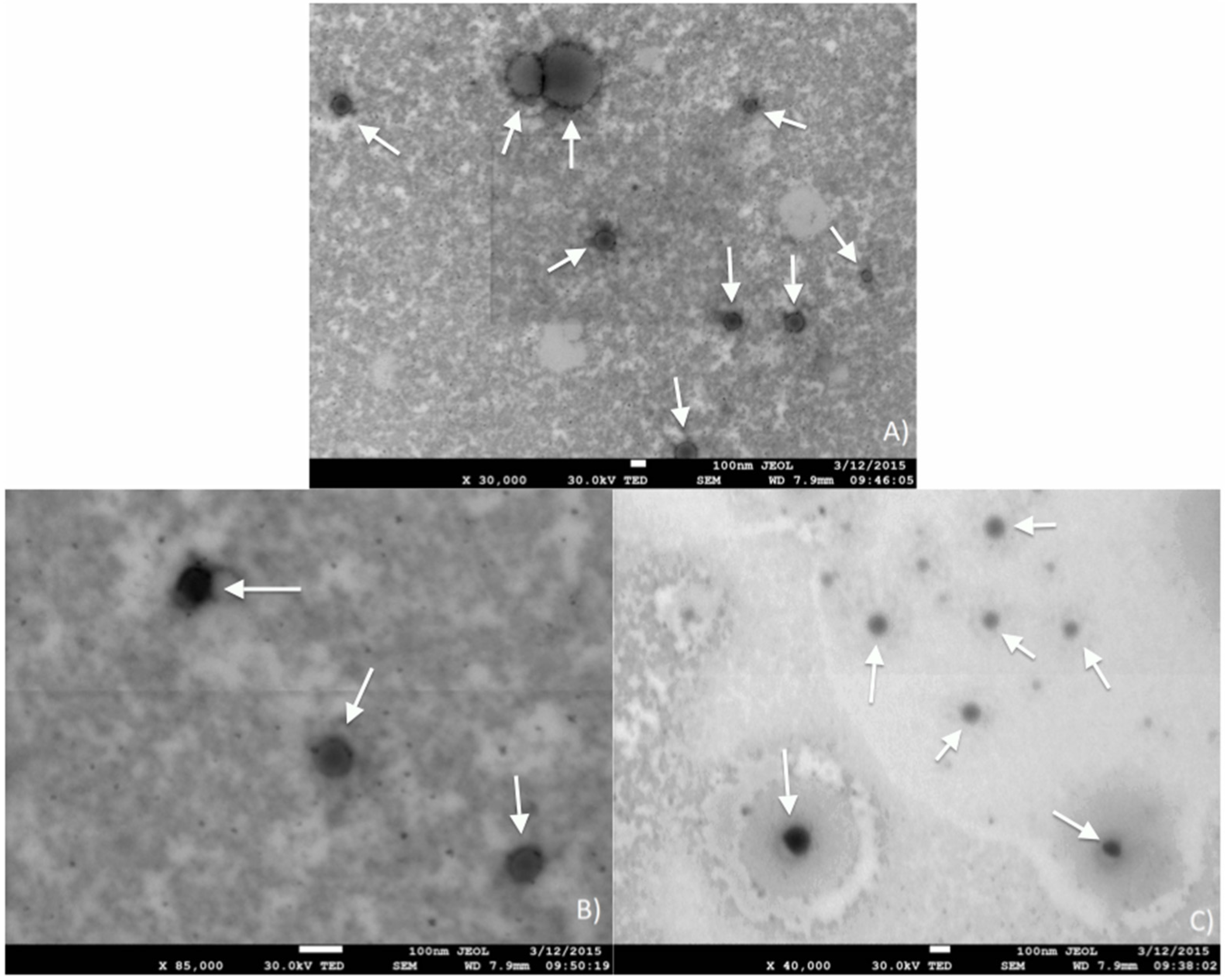
Transmission microscopy was performed on a cured emulsion (
Figure 11), revealing the morphology of the system upon cure. A darker outer layer of the structures seen and a lighter core (more clearly seen in
Figure 11A) suggests the formation of hollow spheres, giving the latexes described in this manuscript a range of potential applications besides coatings, including nano-catalyst encapsulation and drug delivery agents. Indeed, upon emulsification in the presence of asolectin, droplets of non-polar monomers are formed. The crosslinking of the droplets’ contents during polymerization ensures that the droplet morphology is maintained even after solvent removal, as confirmed by STEM (
Figure 11). Due to the low monomer concentration in the emulsions originally prepared, it is assumed that most of the original volume of a droplet consists of xylene, which is removed upon drying, leaving a hollow sphere referred to as a “vesicle”.
Despite a few larger particles seen in
Figure 11A, the majority of the particles exhibit diameters on the order of 100 nm (
Figure 11B,C). In order to more accurately determine the vesicles’ particle size, cured emulsions were characterized using a zetasizer.
Table 2 displays average diameter and polydispersity data for samples containing 200 mg of total monomer weight and 40–80 wt.% of tung oil.
From the data in
Table 2, a clear trend of increasing particle size can be observed for increasing tung oil content. Such behavior allows for some level of control on the particle average diameter by simply adjusting the content of tung oil in the monomer mixture. Such aspect confers versatility to the system presented, since it is possible to design vesicles of desired size depending on the targeted application. The average diameter range obtained (170–219 nm,
Table 2) is ideal for the encapsulation of nanomaterials. The polydispersity index (PDI) related to vesicle diameter ranges from 0.2 to 0.4, indicating a reasonably, but consistent broad distribution of particle sizes, with no clear connections with the oil content in the monomer mixture. Despite its polydisperse nature, the emulsions have shown good stability, as previously mentioned in the text, and as observed in similar systems [
20].













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}