High-Throughput Sequencing Analysis of the Actinobacterial Spatial Diversity in Moonmilk Deposits

 , ,
, ,
Abstract
:1. Introduction
2. Results
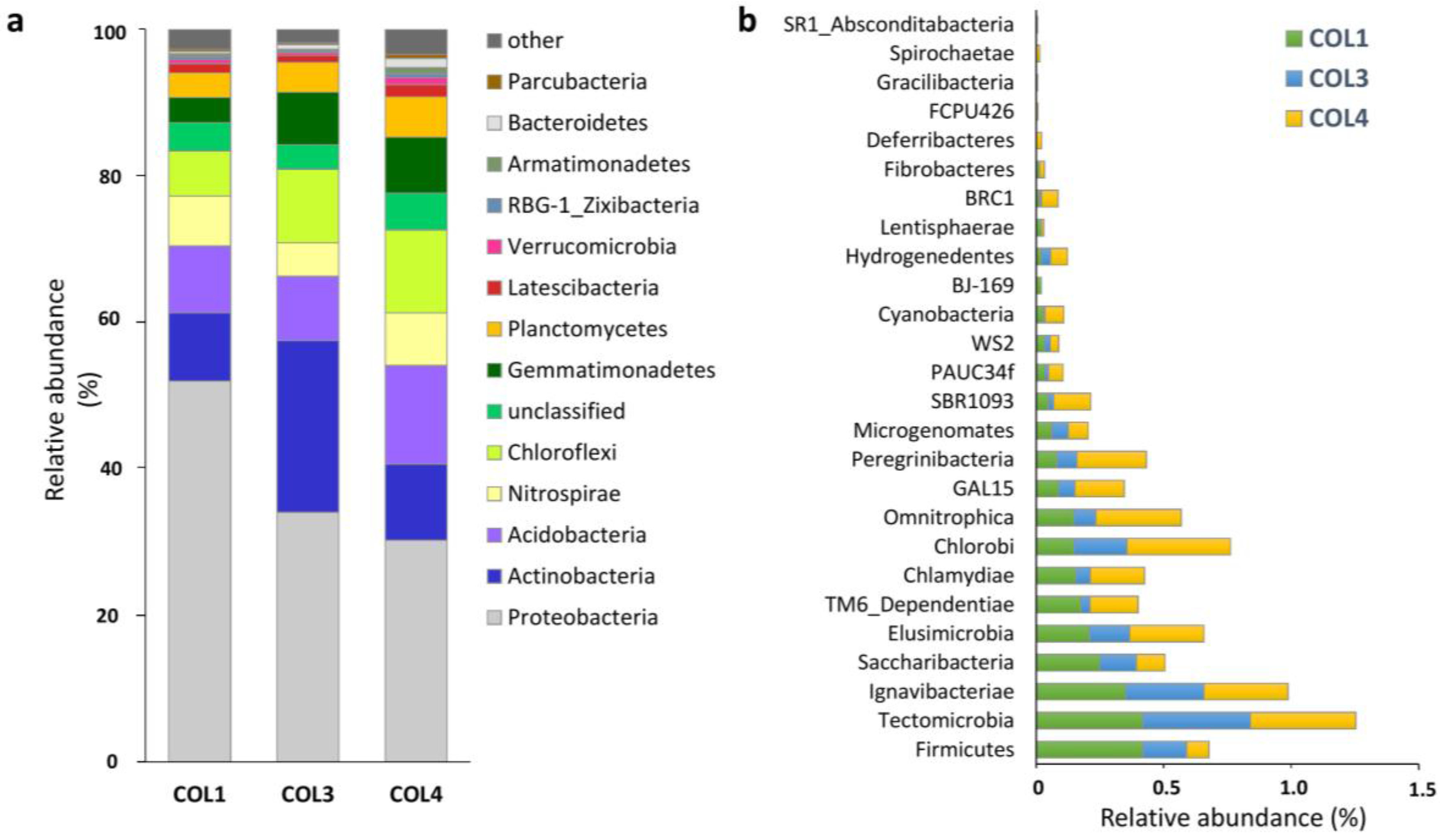
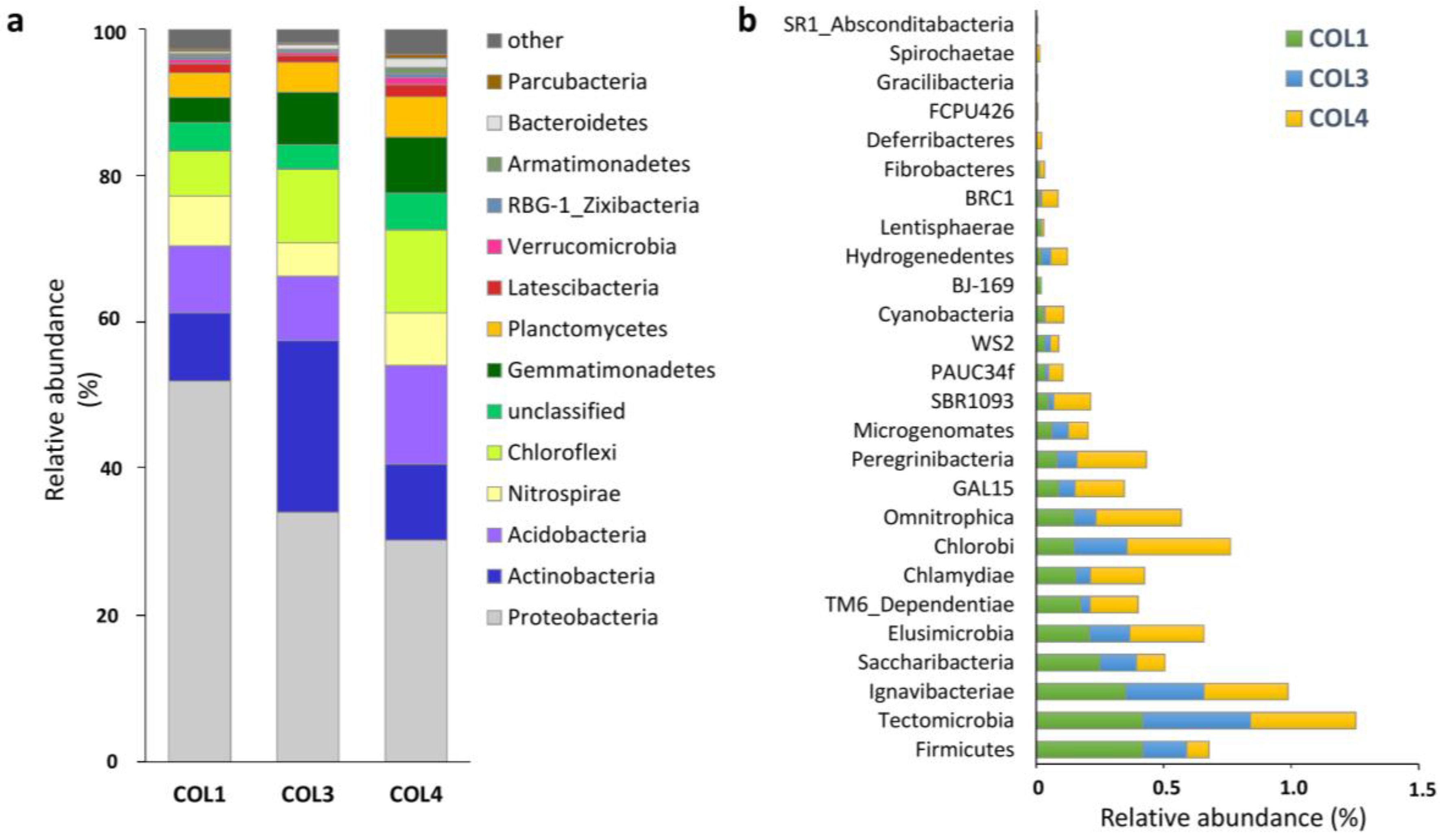
2.1. Actinobacterial Abundance within the Whole Moonmilk Bacterial Microbiome
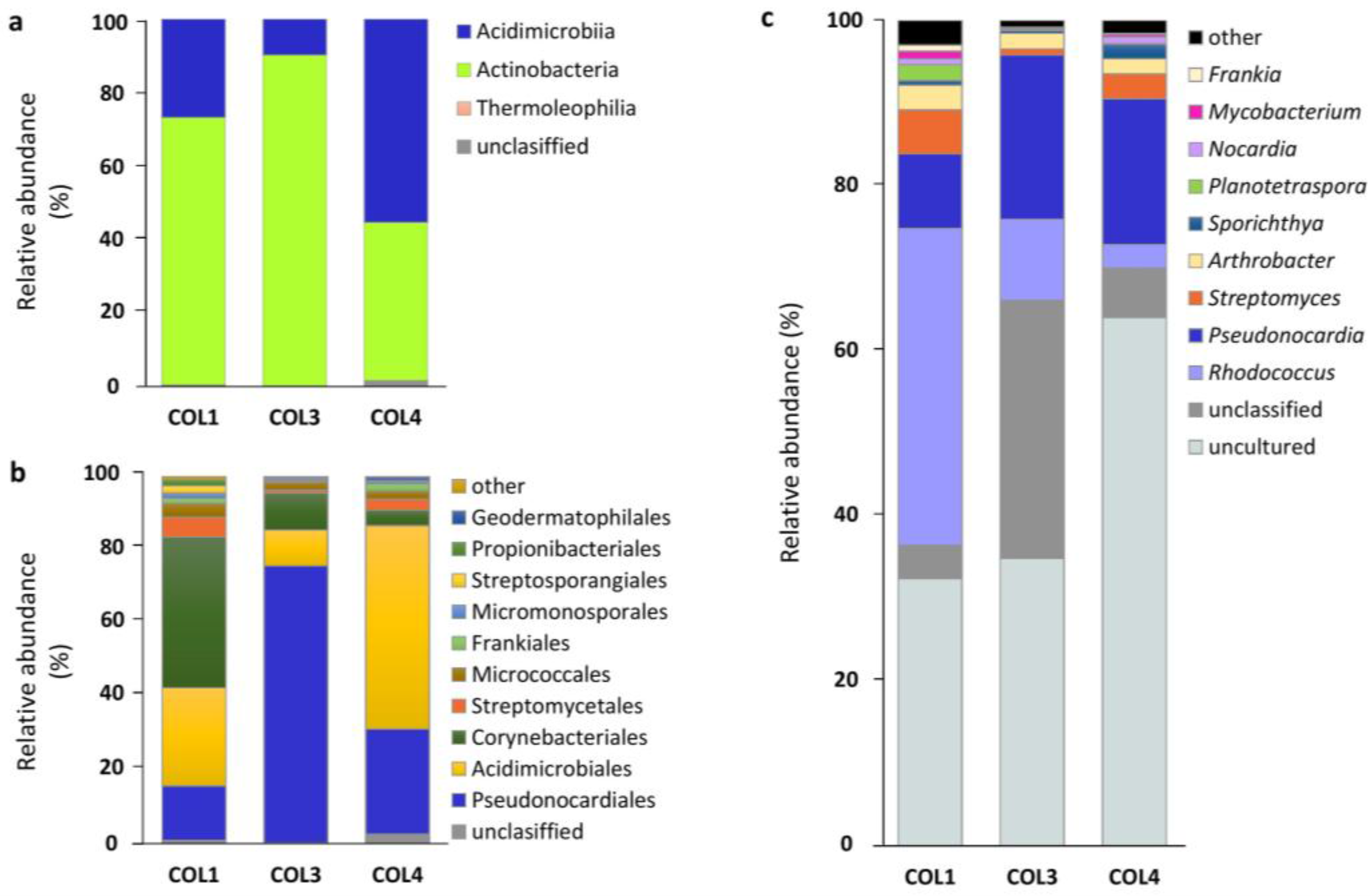
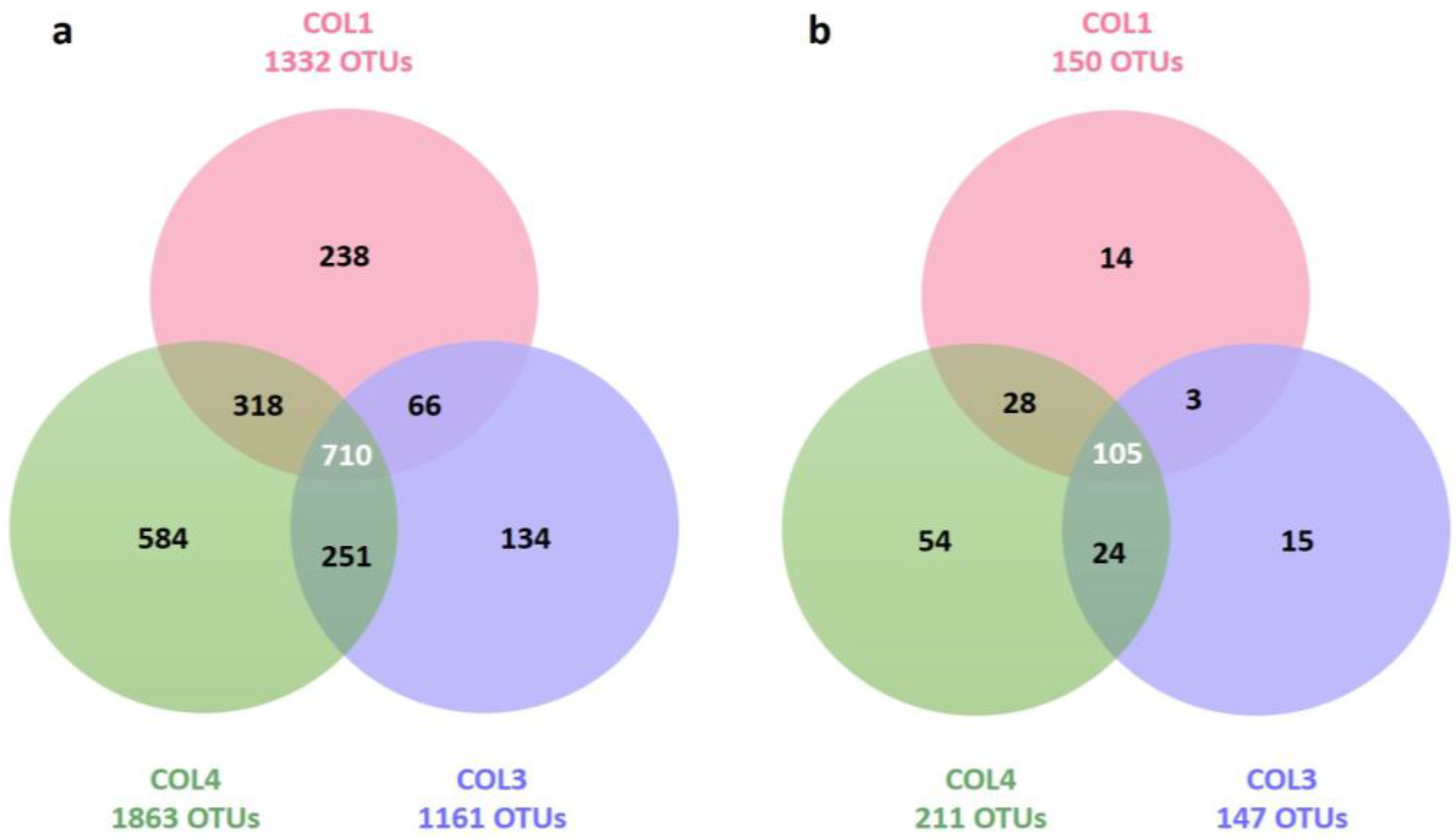
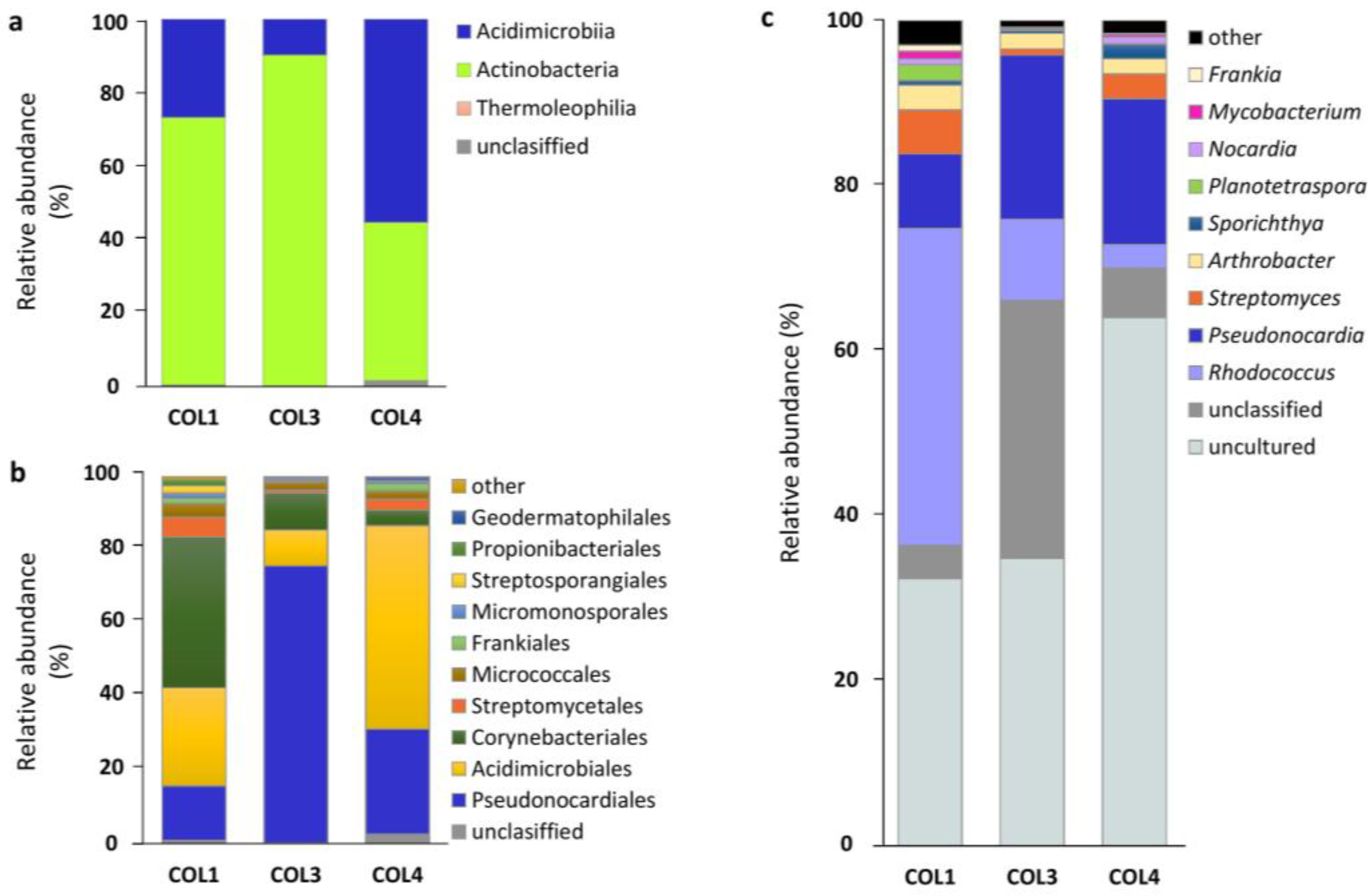
2.2. Actinobacterial Diversity in Moonmilk Deposits
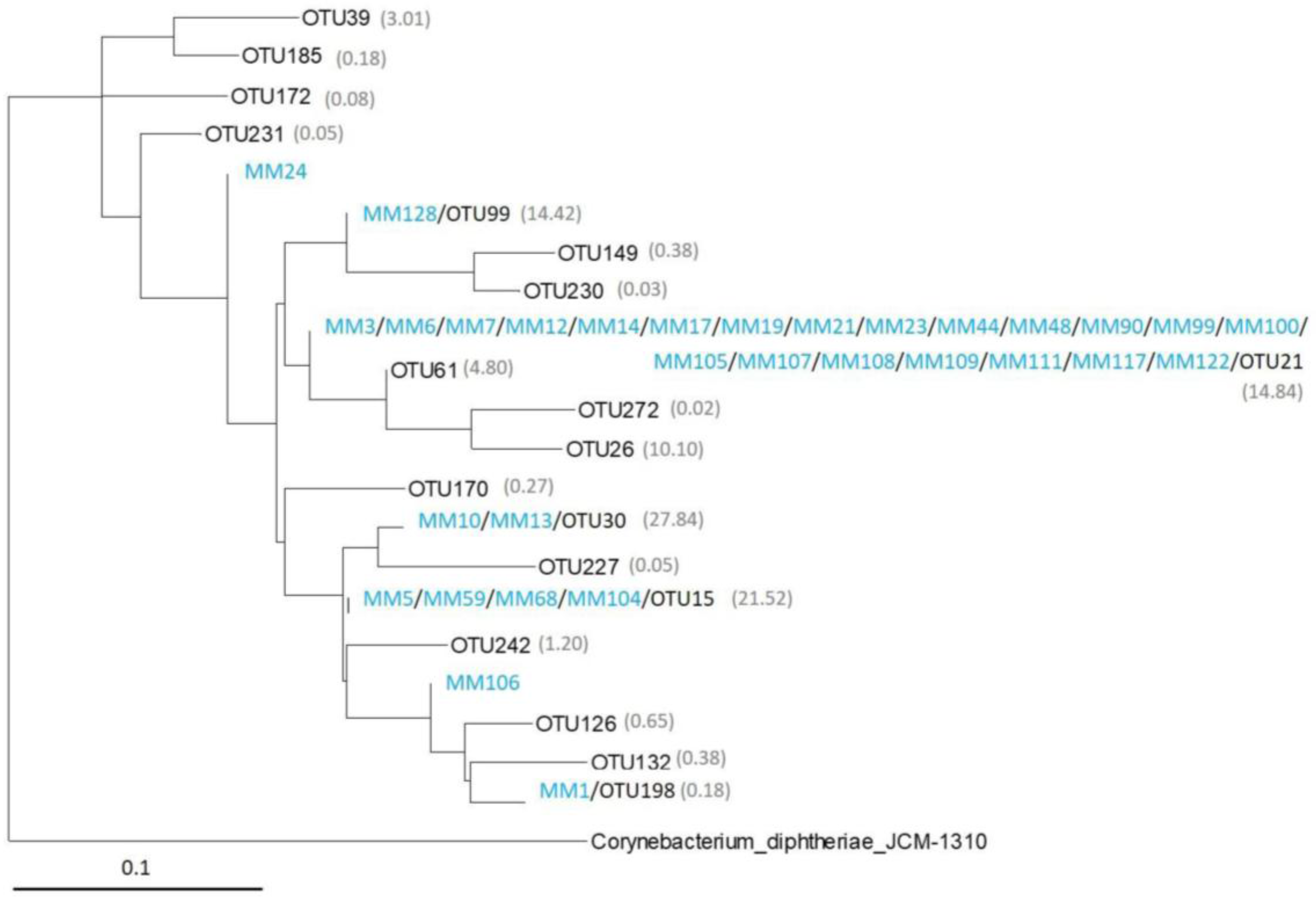
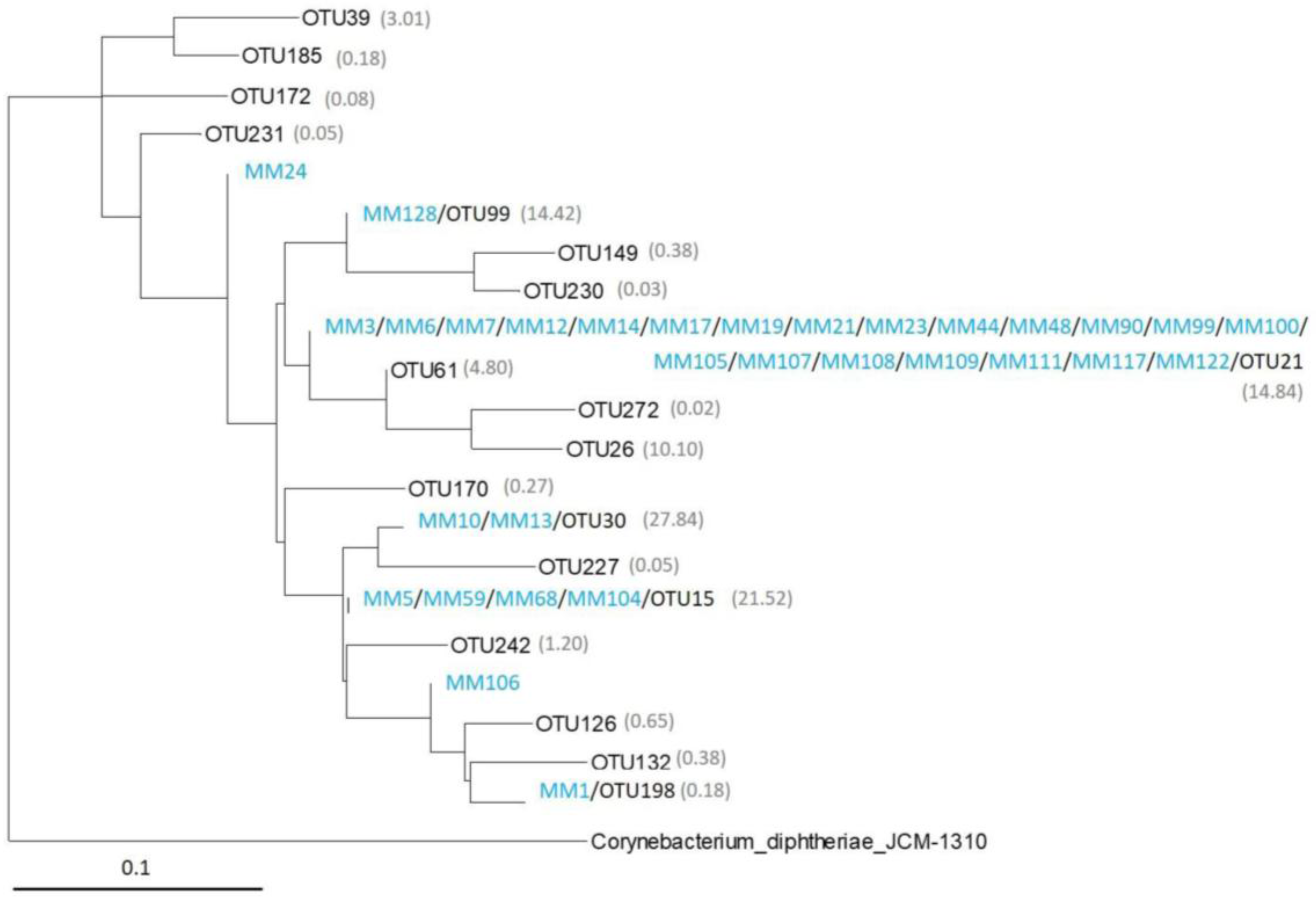
2.3. Analysis of the Most Abundant Actinobacterial OTUs
2.4. Comparison of Moonmilk Streptomyces OTUs and Streptomyces Strains Isolated via the Culture-Dependent Approach
3. Discussion
3.1. New Insights into Moonmilk Bacterial Diversity Revealed by High-Throughput Sequencing
3.2. Moonmilk Deposits as Appealing Source of Novel Producers of Bioactive Compounds
4. Materials and Methods
4.1. Site description and Sampling
4.2. Total DNA Extraction and 16S rRNA Gene Amplicon High-Throughput Sequencing
4.3. 16S rRNA Amplicon Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Engel, A.S. Microbial diversity of cave ecosystems. In Geomicrobiology: Molecular and Environmental Perspective; Springer: Berlin, Germany, 2010; ISBN 9789048192038. [Google Scholar]
- Barton, H.A.; Northup, D.E. Geomicrobiology in cave environments: Past, current and future perspectives. J. Cave Karst Stud. 2007, 69, 163–178. [Google Scholar]
- Groth, I.; Vettermann, R.; Schuetze, B.; Schumann, P.; Saiz-Jimenez, C. Actinomycetes in Karstic caves of northern Spain (Altamira and Tito Bustillo). J. Microbiol. Methods 1999, 36, 115–122. [Google Scholar] [CrossRef]
- Jurado, V.; Kroppenstedt, R.M.; Saiz-Jimenez, C.; Klenk, H.P.; Mouniée, D.; Laiz, L.; Couble, A.; Pötter, G.; Boiron, P.; Rodríguez-Nava, V. Hoyosella altamirensis gen. nov., sp. nov., a new member of the order Actinomycetales isolated from a cave biofilm. Int. J. Syst. Evol. Microbiol. 2009, 59, 3105–3110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurado, V.; Groth, I.; Gonzalez, J.M.; Laiz, L.; Saiz-Jimenez, C. Agromyces salentinus sp. nov. and Agromyces neolithicus sp. nov. Int. J. Syst. Evol. Microbiol. 2005, 55, 153–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejewska, M.; Pessi, I.S.; Arguelles-Arias, A.; Noirfalise, P.; Luis, G.; Ongena, M.; Barton, H.; Carnol, M.; Rigali, S. Streptomyces lunaelactis sp. nov., a novel ferroverdin A-producing Streptomyces species isolated from a moonmilk speleothem. Antonie van Leeuwenhoek, Int. J. Gen. Mol. Microbiol. 2015, 107, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.D.; Kang, S.O.; Hah, Y.C. Hongia gen, nov., a new genus of the order Actinomycetales. Int. J. Syst. Evol. Microbiol. 2000, 50, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, M.; Neilson, J.W.; Nelson, W.M.; Legatzki, A.; Byrne, A.; Yu, Y.; Wing, R.A.; Soderlund, C.A.; Pryor, B.M.; Pierson, L.S.; et al. Profiling Bacterial Diversity and Taxonomic Composition on Speleothem Surfaces in Kartchner Caverns, AZ. Microb. Ecol. 2013, 65, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.S.; Paoletti, M.G.; Beggio, M.; Dorigo, L.; Pamio, A.; Gomiero, T.; Furlan, C.; Brilli, M.; Dreon, A.L.; Bertoni, R.; et al. Comparative microbial community composition from secondary carbonate (moonmilk) deposits: Implications for the Cansiliella servadeii cave hygropetric food web. Int. J. Speleol. 2013, 42, 181–192. [Google Scholar] [CrossRef]
- Maciejewska, M.; Adam, D.; Martinet, L.; Naômé, A.; Calusinska, M.; Smargiasso, N.; De Pauw, E.; Barton, H.; Carnol, M.; Hanikenne, M.; et al. A Phenotypic and Genotypic Analysis of the Antimicrobial Potential of Cultivable Streptomyces isolated from Cave Moonmilk Deposits. Front. Microbiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Cañaveras, J.C.; Cuezva, S.; Sanchez-Moral, S.; Lario, J.; Laiz, L.; Gonzalez, J.M.; Saiz-Jimenez, C. On the origin of fiber calcite crystals in moonmilk deposits. Naturwissenschaften 2006, 93, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Cañaveras, J.C.; Hoyos Gómez, M.; Sánchez-Moral, S.; Sanz Rubio, E.; Bedoya, J.; Hoyos, V.; Groth, I.; Schumann, P.; Laiz Trobajo, L. Microbial Communities Associated with Hydromagnesite and Needle-Fiber Aragonite Deposits in a Karstic Cave (Altamira, Northern Spain). Geomicrobiol. J. 1999, 16, 9–25. [Google Scholar]
- Rooney, D.C.; Hutchens, E.; Clipson, N.; Baldini, J.; McDermott, F. Microbial Community Diversity of Moonmilk Deposits at Ballynamintra Cave, Co. Waterford, Ireland. Microb. Ecol. 2010, 60, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Portillo, M.C.; Gonzalez, J.M. Moonmilk Deposits Originate from Specific Bacterial Communities in Altamira Cave (Spain). Microb. Ecol. 2011, 61, 182–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reitschuler, C.; Lins, P.; Schwarzenauer, T.; Spotl, C.; Wagner, A.O.; Illmer, P. New Undescribed Lineages of Non-extremophilic Archaea Form a Homogeneous and Dominant Element within Alpine Moonmilk Microbiomes. Geomicrobiol. J. 2015, 32. [Google Scholar] [CrossRef]
- Reitschuler, C.; Spötl, C.; Hofmann, K.; Wagner, A.O.; Illmer, P. Archaeal Distribution in Moonmilk Deposits from Alpine Caves and Their Ecophysiological Potential. Microb. Ecol. 2016, 71, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Reitschuler, C.; Lins, P.; Wagner, A.O.; Illmer, P. Cultivation of moonmilk-born non-extremophilic Thaum and Euryarchaeota in mixed culture. Anaerobe 2014, 29, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Axenov-Gibanov, D.V.; Voytsekhovskaya, I.V.; Tokovenko, B.T.; Protasov, E.S.; Gamaiunov, S.V.; Rebets, Y.V.; Luzhetskyy, A.N.; Timofeyev, M.A. Actinobacteria isolated from an underground lake and moonmilk speleothem from the biggest conglomeratic karstic cave in Siberia as sources of novel biologically active compounds. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Dhami, N.K.; Mukherjee, A.; Watkin, E. Characterisation of Mineralogical-Mechanical-Microbial properties of calcitic speleothems and the in vitro biomineralization potential of associated microbial communities. Front. Microbiol. 2018, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Bindschedler, S.; Milliere, L.; Cailleau, G.; Job, D.; Verrecchia, E.P. Calcitic nanofibres in soils and caves: A putative fungal contribution to carbonatogenesis. Geol. Soc. Lond. Spec. Publ. 2010, 336, 225–238. [Google Scholar] [CrossRef]
- Maciejewska, M.; Adam, D.; Naômé, A.; Martinet, L.; Tenconi, E.; Calusinska, M.; Delfosse, P.; Hanikenne, M.; Baurain, D.; Compère, P.; et al. Assessment of the potential role of Streptomyces in cave moonmilk formation. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Nimaichand, S.; Devi, A.M.; Tamreihao, K.; Ningthoujam, D.S.; Li, W.J. Actinobacterial diversity in limestone deposit sites in Hundung, Manipur (India) and their antimicrobial activities. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Tan, L.; Liu, W.; Wang, B.; Wang, J.; Cai, Y.; Lin, X. Profiling bacterial diversity in a limestone cave of the western Loess Plateau of China. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Northup, D.E.; Melim, L.A.; Spilde, M.N.; Hathaway, J.J.M.; Garcia, M.G.; Moya, M.; Stone, F.D.; Boston, P.J.; Dapkevicius, M.L.N.E.; Riquelme, C. Lava Cave Microbial Communities within Mats and Secondary Mineral Deposits: Implications for Life Detection on Other Planets. Astrobiology 2011, 11, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, C.; Hathaway, J.J.M.; Dapkevicius, M.d.L.N.E.; Miller, A.Z.; Kooser, A.; Northup, D.E.; Jurado, V.; Fernandez, O.; Saiz-Jimenez, C.; Cheeptham, N. Actinobacterial diversity in volcanic caves and associated geomicrobiological interactions. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Cheeptham, N.; Sadoway, T.; Rule, D.; Watson, K.; Moote, P.; Soliman, L.C.; Azad, N.; Donkor, K.K.; Horne, D. Cure from the cave: Volcanic cave actinomycetes and their potential in drug discovery. Int. J. Speleol. 2013, 42, 35–47. [Google Scholar] [CrossRef]
- Tebo, B.M.; Davis, R.E.; Anitori, R.P.; Connell, L.B.; Schiffman, P.; Staudigel, H. Microbial communities in dark oligotrophic volcanic ice cave ecosystems of Mt. Erebus, Antarctica. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Chater, K.F.; Biró, S.; Lee, K.J.; Palmer, T.; Schrempf, H. The complex extracellular biology of Streptomyces. FEMS Microbiol. Rev. 2010, 34, 171–198. [Google Scholar] [CrossRef] [PubMed]
- Castelle, C.J.; Hug, L.A.; Wrighton, K.C.; Thomas, B.C.; Williams, K.H.; Wu, D.; Tringe, S.G.; Singer, S.W.; Eisen, J.A.; Banfield, J.F. Extraordinary phylogenetic diversity and metabolic versatility in aquifer sediment. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Schabereiter-Gurtner, C.; Saiz-Jimenez, C.; Pinar, G.; Lubitz, W.; Rolleke, S. Phylogenetic diversity of bacteria associated with Paleolithic paintings and surrounding rock walls in two Spanish caves (Llonin and La Garma). FEMS Microbiol. Ecol. 2004, 47, 235–247. [Google Scholar] [CrossRef]
- Pedersen, K.; Arlinger, J.; Ekendahl, S.; Hallbeck, L. 16S rRNA gene diversity of attached and unattached bacteria in boreholes along the access tunnel to the Äspö hard rock laboratory, Sweden. FEMS Microbiol. Ecol. 1996, 19, 249–262. [Google Scholar] [CrossRef]
- Zhou, J.; Gu, Y.; Zou, C.; Mo, M. Phylogenetic diversity of bacteria in an earth-cave in Guizhou Province, Southwest of China. J. Microbiol. 2007, 45, 105–112. [Google Scholar] [PubMed]
- De Mandal, S.; Chatterjee, R.; Kumar, N.S. Dominant bacterial phyla in caves and their predicted functional roles in C and N cycle. BMC Microbiol. 2017, 17, 90. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, K.; Gupta, R.K. Rare actinomycetes: A potential storehouse for novel antibiotics. Crit. Rev. Biotechnol. 2012, 32, 108–132. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Kim, H.J.; Lee, H.S.; Kim, P.; Kim, E.S. Genome mining of rare actinomycetes and cryptic pathway awakening. Process Biochem. 2015, 50, 1184–1193. [Google Scholar] [CrossRef]
- Rigali, S.; Anderssen, S.; Naômé, A.; van Wezel, G.P. Cracking the regulatory code of biosynthetic gene clusters as a strategy for natural product discovery. Biochem. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Norris, P. Class II. Acidimicrobiia class. nov. In Bergey’s Manual of Systematic Bacteriology; Whitman, W., Goodfellow, M., Kämpfer, P., Busse, H.-J., Trujillo, M., Ludwig, W., Suzuki, K.-I., Parte, A., Eds.; Springer: New York, NY, USA, 2012; Volume 5, p. 1968. [Google Scholar]
- Bull, A.T. Actinobacteria of the extremobiosphere. In Extremophiles Handbook; Springer: Tokyo, Japan, 2011; pp. 1203–1240. ISBN 9784431538981. [Google Scholar]
- Stefani, F.O.P.; Bell, T.H.; Marchand, C.; de la Providencia, I.E.; El Yassimi, A.; St-Arnaud, M.; Hijri, M. Culture-Dependent and -Independent Methods Capture Different Microbial Community Fractions in Hydrocarbon-Contaminated Soils. PLoS ONE 2015, 10, e0128272. [Google Scholar] [CrossRef] [PubMed]
- Vaz-Moreira, I.; Egas, C.; Nunes, O.C.; Manaia, C.M. Culture-dependent and culture-independent diversity surveys target different bacteria: A case study in a freshwater sample. Antonie van Leeuwenhoek, Int. J. Gen. Mol. Microbiol. 2011, 100, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, J.; Jäckel, U.; Kämpfer, P. Development of a new PCR primer system for selective amplification of Actinobacteria. FEMS Microbiol. Lett. 2010, 311, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Goux, X.; Calusinska, M.; Fossépré, M.; Benizri, E.; Delfosse, P. Start-up phase of an anaerobic full-scale farm reactor—Appearance of mesophilic anaerobic conditions and establishment of the methanogenic microbial community. Bioresour. Technol. 2016, 212, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Peplies, J.; Glöckner, F.O. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 2012, 28, 1823–1829. [Google Scholar] [CrossRef] [PubMed]
- Boratyn, G.M.; Schäffer, A.A.; Agarwala, R.; Altschul, S.F.; Lipman, D.J.; Madden, T.L. Domain enhanced lookup time accelerated BLAST. Biol. Direct 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Adam, D.; Maciejewska, M.; Naômé, A.; Martinet, L.; Coppieters, W.; Karim, L.; Baurain, D.; Rigali, S. Isolation, Characterization, and Antibacterial Activity of Hard-to-Culture Actinobacteria from Cave Moonmilk Deposits. Antibiotics 2018, in press. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target group | Site | Total OTUs (Richness) | Unique OTUs (Specificity) | Inverse Simpson Index (Diversity) | Simpson Index (Evenness) |
|---|---|---|---|---|---|
| Bacteria | COL1 | 1332 | 238 (17.9%) | 13.23 | 0.01 |
| COL3 | 1161 | 134 (11.6%) | 58.94 | 0.05 | |
| COL4 | 1863 | 584 (31.3%) | 155.31 | 0.08 | |
| Actinobacteria | COL1 | 150 | 14 (9.3%) | 6.21 | 0.04 |
| COL3 | 147 | 15 (10.2%) | 7.74 | 0.05 | |
| COL4 | 211 | 54 (25.6%) | 24.13 | 0.11 |
| Target group | COL1 and COL3 | COL1 and COL4 | COL4 and COL3 |
|---|---|---|---|
| Bacteria | 776/2493 (31.1%) | 1028/3195 (32.2%) | 961/3024 (31.8%) |
| Actinobacteria | 108/297 (36.4%) | 133/361 (36.9%) | 129/358 (36.0%) |
| Genus | COL1 | COL3 | COL4 | TOTAL | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Seq. | % | OTUs | Seq. | % | OTUs | Seq. | % | OTUs | Seq. | Av. % | Diff. OTUs | |
| Uncultured | 46,346 | 32.38 | 65 | 58,903 | 34.80 | 63 | 92,444 | 63.98 | 90 | 197,693 | 43.7 | 96 |
| Rhodococcus | 54,920 | 38.37 | 3 | 16,636 | 9.83 | 3 | 4102 | 2.84 | 3 | 75,658 | 17.0 | 4 |
| Pseudonocardia † | 12,913 | 9.02 | 7 | 33,640 | 19.87 | 8 | 25,545 | 17.68 | 9 | 72,098 | 15.5 | 9 |
| Unclassified | 5762 | 4.03 | 18 | 52,908 | 31.26 | 20 | 8668 | 6.00 | 26 | 67,338 | 13.8 | 32 |
| Streptomyces † | 7628 | 5.33 | 10 | 1292 | 0.76 | 11 | 4347 | 3.01 | 18 | 13,267 | 3.0 | 19 |
| Arthrobacter | 4323 | 3.02 | 4 | 3214 | 1.90 | 4 | 2649 | 1.83 | 5 | 10,186 | 2.3 | 5 |
| Sporichthya * | 771 | 0.54 | 1 | 556 | 0.33 | 2 | 2515 | 1.74 | 2 | 3842 | 0.9 | 2 |
| Planotetraspora * | 2769 | 1.93 | 1 | 294 | 0.17 | 1 | 113 | 0.08 | 1 | 3176 | 0.7 | 1 |
| Nocardia | 1023 | 0.71 | 2 | 157 | 0.09 | 2 | 1212 | 0.84 | 2 | 2392 | 0.5 | 2 |
| Mycobacterium † | 1299 | 0.91 | 2 | 127 | 0.08 | 2 | 440 | 0.30 | 3 | 1866 | 0.4 | 3 |
| Frankia * | 1067 | 0.75 | 2 | 77 | 0.05 | 1 | 126 | 0.09 | 3 | 1270 | 0.3 | 3 |
| Luedemannella * | 555 | 0.39 | 2 | 212 | 0.13 | 2 | 276 | 0.19 | 2 | 1043 | 0.2 | 2 |
| Longispora * | 371 | 0.26 | 1 | 374 | 0.22 | 1 | 110 | 0.08 | 1 | 855 | 0.2 | 1 |
| Agromyces * | 310 | 0.22 | 2 | 136 | 0.08 | 2 | 355 | 0.25 | 2 | 801 | 0.2 | 2 |
| Actinoplanes * | 591 | 0.41 | 2 | 85 | 0.05 | 2 | 111 | 0.08 | 2 | 787 | 0.2 | 2 |
| Nakamurella * | 416 | 0.29 | 1 | 114 | 0.07 | 1 | 101 | 0.07 | 1 | 631 | 0.1 | 1 |
| Nocardioides † | 360 | 0.25 | 4 | 52 | 0.03 | 3 | 158 | 0.11 | 8 | 570 | 0.1 | 9 |
| Geodermatophilus † | 78 | 0.05 | 1 | 70 | 0.04 | 2 | 374 | 0.26 | 2 | 522 | 0.1 | 2 |
| Catellatospora * | 95 | 0.07 | 3 | 70 | 0.04 | 2 | 141 | 0.10 | 4 | 306 | 0.07 | 4 |
| Kribbella † | 120 | 0.08 | 1 | 36 | 0.02 | 1 | 135 | 0.09 | 2 | 291 | 0.07 | 2 |
| Kocuria * | 261 | 0.18 | 1 | 23 | 0.01 | 1 | - | - | - | 284 | 0.1 | 2 |
| Actinomyces * | 247 | 0.17 | 5 | 1 | 0.001 | 1 | 6 | 0.004 | 1 | 254 | 0.06 | 5 |
| Corynebacterium * | 151 | 0.11 | 1 | 32 | 0.02 | 3 | 10 | 0.01 | 2 | 193 | 0.04 | 5 |
| Rhizocola * | - | - | - | 27 | 0.02 | 1 | 145 | 0.10 | 1 | 172 | 0.06 | 1 |
| Microbacterium * | 108 | 0.08 | 1 | 48 | 0.03 | 1 | 5 | 0.003 | 1 | 161 | 0.04 | 1 |
| Iamia * | 107 | 0.07 | 1 | - | - | - | 31 | 0.02 | 1 | 138 | 0.05 | 1 |
| Pseudoclavibacter * | 138 | 0.10 | 1 | - | - | - | - | - | - | 138 | 0.1 | 1 |
| Lentzea * | - | - | - | - | - | - | 111 | 0.08 | 1 | 111 | 0.08 | 1 |
| Aeromicrobium † | 73 | 0.05 | 1 | - | - | - | 28 | 0.02 | 2 | 101 | 0.04 | 2 |
| Amycolatopsis | 86 | 0.06 | 1 | - | - | - | 5 | 0.003 | 1 | 91 | 0.03 | 2 |
| Cryptosporangium * | - | - | - | 71 | 0.04 | 1 | 10 | 0.01 | 1 | 81 | 0.02 | 1 |
| Glycomyces * | 35 | 0.02 | 1 | - | - | - | 45 | 0.03 | 1 | 80 | 0.03 | 1 |
| Streptosporangium * | 61 | 0.04 | 1 | - | - | - | 19 | 0.01 | 1 | 80 | 0.03 | 1 |
| Smaragdicoccus * | 43 | 0.03 | 1 | - | - | - | 34 | 0.02 | 1 | 77 | 0.03 | 1 |
| Propionibacterium | 57 | 0.04 | 1 | - | - | - | - | - | 0 | 57 | 0.04 | 1 |
| Kineosporia * | 44 | 0.03 | 1 | - | - | - | 8 | 0.01 | 1 | 52 | 0.02 | 1 |
| Jatrophihabitans * | - | - | - | 21 | 0.01 | 1 | 24 | 0.02 | 1 | 45 | 0.01 | 1 |
| Promicromonospora * | - | - | - | 22 | 0.01 | 1 | 21 | 0.01 | 1 | 43 | 0.01 | 2 |
| Millisia * | - | - | - | 22 | 0.01 | 1 | 6 | 0.004 | 1 | 28 | 0.01 | 1 |
| Rothia * | 2 | 0.001 | 1 | 13 | 0.01 | 1 | 7 | 0.005 | 1 | 22 | 0.005 | 1 |
| Tessaracoccus * | - | - | - | 17 | 0.01 | 1 | - | - | - | 17 | 0.01 | 1 |
| Acidothermus * | - | - | - | - | - | - | 16 | 0.01 | 1 | 16 | 0.01 | 1 |
| Marmoricola * | - | - | - | - | - | - | 14 | 0.01 | 2 | 14 | 0.01 | 2 |
| Dermacoccus * | - | - | - | - | - | - | 11 | 0.008 | 1 | 11 | 0.01 | 1 |
| Ponticoccus * | - | - | - | 8 | 0.005 | 1 | - | - | - | 8 | 0.005 | 1 |
| Stackebrandtia * | - | - | - | - | - | - | 8 | 0.006 | 1 | 8 | 0.01 | 1 |
| Umezawaea * | - | - | - | - | - | - | 2 | 0.001 | 1 | 2 | 0.001 | 1 |
| Actinospica * | - | - | - | - | - | - | 1 | 0.001 | 1 | 1 | 0.001 | 1 |
| Propionimicrobium * | - | - | - | - | - | - | 1 | 0.001 | 1 | 1 | 0.001 | 1 |
| OTU | COL1 | COL3 | COL4 | Av. % | Class | Family | Genus |
|---|---|---|---|---|---|---|---|
| OTU1 | 38.28 | 9.68 | 2.74 | 16.90 | Actinobacteria | Nocardiaceae | Rhodococcus |
| OTU2 | 0.31 | 28.89 | 2.13 | 10.44 | Actinobacteria | Pseudonocardiaceae | unclassified |
| OTU8 | 0.75 | 13.96 | 0.58 | 5.10 | Actinobacteria | Pseudonocardiaceae | uncultured |
| OTU4 | 3.35 | 1.13 | 11.47 | 5.32 | Acidimicrobiia | uncultured | uncultured |
| OTU3 | 0.52 | 7.80 | 4.87 | 4.40 | Actinobacteria | Pseudonocardiaceae | Pseudonocardia |
| OTU262 | 3.45 | 4.12 | 5.89 | 4.49 | Actinobacteria | Pseudonocardiaceae | Pseudonocardia |
| OTU6 | 7.53 | 1.00 | 3.98 | 4.17 | Acidimicrobiia | uncultured | uncultured |
| OTU12 | 2.97 | 6.61 | 1.82 | 3.80 | Actinobacteria | Pseudonocardiaceae | uncultured |
| OTU5 | 3.53 | 1.70 | 5.87 | 3.70 | Acidimicrobiia | uncultured | uncultured |
| OTU13 | 0.46 | 4.91 | 3.65 | 3.01 | Actinobacteria | Pseudonocardiaceae | Pseudonocardia |
| OTU98 | 0.99 | 1.07 | 6.94 | 3.00 | Acidimicrobiia | uncultured | uncultured |
| OTU203 | 1.73 | 0.31 | 5.92 | 2.66 | Acidimicrobiia | uncultured | uncultured |
| OTU432 | 3.68 | 1.40 | 1.66 | 2.25 | Actinobacteria | Pseudonocardiaceae | Pseudonocardia |
| OTU142 | 0.72 | 1.91 | 2.35 | 1.66 | Actinobacteria | Pseudonocardiaceae | unclassified |
| OTU7 | 0 | 1.70 | 3.31 | 1.67 | Actinobacteria | Pseudonocardiaceae | uncultured |
| OTU19 | 2.06 | 0.52 | 1.65 | 1.41 | Actinobacteria | Micrococcaceae | Arthrobacter |
| OTU190 | 0.46 | 0.31 | 2.84 | 1.20 | Acidimicrobiia | uncultured | uncultured |
| OTU10 | 0.81 | 0.29 | 1.68 | 0.93 | Acidimicrobiia | Acidimicrobiaceae | uncultured |
| OTU9 | 0.24 | 0.15 | 2.39 | 0.93 | Acidimicrobiia | uncultured | uncultured |
| OTU251 | 0.77 | 0.65 | 1.12 | 0.85 | Actinobacteria | Pseudonocardiaceae | Pseudonocardia |
| OTU14 | 0.54 | 0.31 | 1.73 | 0.86 | Actinobacteria | Sporichthyaceae | Sporichthya |
| OTU30 | 2.11 | 0.13 | 0.31 | 0.85 | Actinobacteria | Streptomycetaceae | Streptomyces |
| OTU360 | 0.40 | 0.17 | 1.68 | 0.75 | Acidimicrobiia | uncultured | uncultured |
| OTU24 | 1.93 | 0.17 | 0.08 | 0.73 | Actinobacteria | Streptosporangiaceae | Planotetraspora |
| OTU11 | 0.38 | 0.03 | 1.72 | 0.71 | Acidimicrobiia | uncultured | uncultured |
| OTU20 | 0.71 | 0.47 | 0.81 | 0.67 | Acidimicrobiia | Iamiaceae | uncultured |
| OTU47 | 0.75 | 0.26 | 0.98 | 0.66 | Acidimicrobiia | uncultured | uncultured |
| OTU15 | 0.30 | 0.17 | 1.48 | 0.65 | Actinobacteria | Streptomycetaceae | Streptomyces |
| OTU192 | 0.01 | 1.65 | 0.01 | 0.56 | Actinobacteria | Pseudonocardiaceae | uncultured |
| OTU16 | 0.24 | 0.51 | 1.11 | 0.62 | Acidimicrobiia | uncultured | uncultured |
| OTU50 | 0.88 | 0.54 | 0.37 | 0.60 | Actinobacteria | Pseudonocardiaceae | uncultured |
| OTU22 | 1.61 | 0.16 | 0.06 | 0.61 | Actinobacteria | Propionibacteriaceae | unclassified |
| OTU23 | 0.27 | 1.19 | 0.10 | 0.52 | Actinobacteria | Micrococcaceae | Arthrobacter |
| OTU54 | 0.13 | 0.92 | 0.46 | 0.50 | Actinobacteria | Pseudonocardiaceae | Pseudonocardia |
| OTU18 | 0.48 | 0.12 | 1.04 | 0.54 | Acidimicrobiia | uncultured | uncultured |
| OTU25 | 0.56 | 0.09 | 0.83 | 0.49 | Actinobacteria | Nocardiaceae | Nocardia |
| OTU21 | 0.80 | 0.19 | 0.34 | 0.45 | Actinobacteria | Streptomycetaceae | Streptomyces |
| OTU99 | 1.05 | 0.09 | 0.17 | 0.44 | Actinobacteria | Streptomycetaceae | Streptomyces |
| OTU36 | 0.22 | 0.04 | 0.96 | 0.41 | Acidimicrobiia | uncultured | uncultured |
| OTU44 | 0.80 | 0.14 | 0.26 | 0.40 | Acidimicrobiia | uncultured | uncultured |
| OTU32 | 0.22 | 0.03 | 0.92 | 0.39 | Actinobacteria | unclassified | unclassified |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maciejewska, M.; Całusińska, M.; Cornet, L.; Adam, D.; Pessi, I.S.; Malchair, S.; Delfosse, P.; Baurain, D.; Barton, H.A.; Carnol, M.; et al. High-Throughput Sequencing Analysis of the Actinobacterial Spatial Diversity in Moonmilk Deposits. Antibiotics 2018, 7, 27. https://doi.org/10.3390/antibiotics7020027
Maciejewska M, Całusińska M, Cornet L, Adam D, Pessi IS, Malchair S, Delfosse P, Baurain D, Barton HA, Carnol M, et al. High-Throughput Sequencing Analysis of the Actinobacterial Spatial Diversity in Moonmilk Deposits. Antibiotics. 2018; 7(2):27. https://doi.org/10.3390/antibiotics7020027
Chicago/Turabian StyleMaciejewska, Marta, Magdalena Całusińska, Luc Cornet, Delphine Adam, Igor S. Pessi, Sandrine Malchair, Philippe Delfosse, Denis Baurain, Hazel A. Barton, Monique Carnol, and et al. 2018. "High-Throughput Sequencing Analysis of the Actinobacterial Spatial Diversity in Moonmilk Deposits" Antibiotics 7, no. 2: 27. https://doi.org/10.3390/antibiotics7020027