Simple Screening of Listeria monocytogenes Based on a Fluorescence Assay via a Laminated Lab-On-Paper Chip

Abstract
:
1. Introduction
2. Materials and Methods
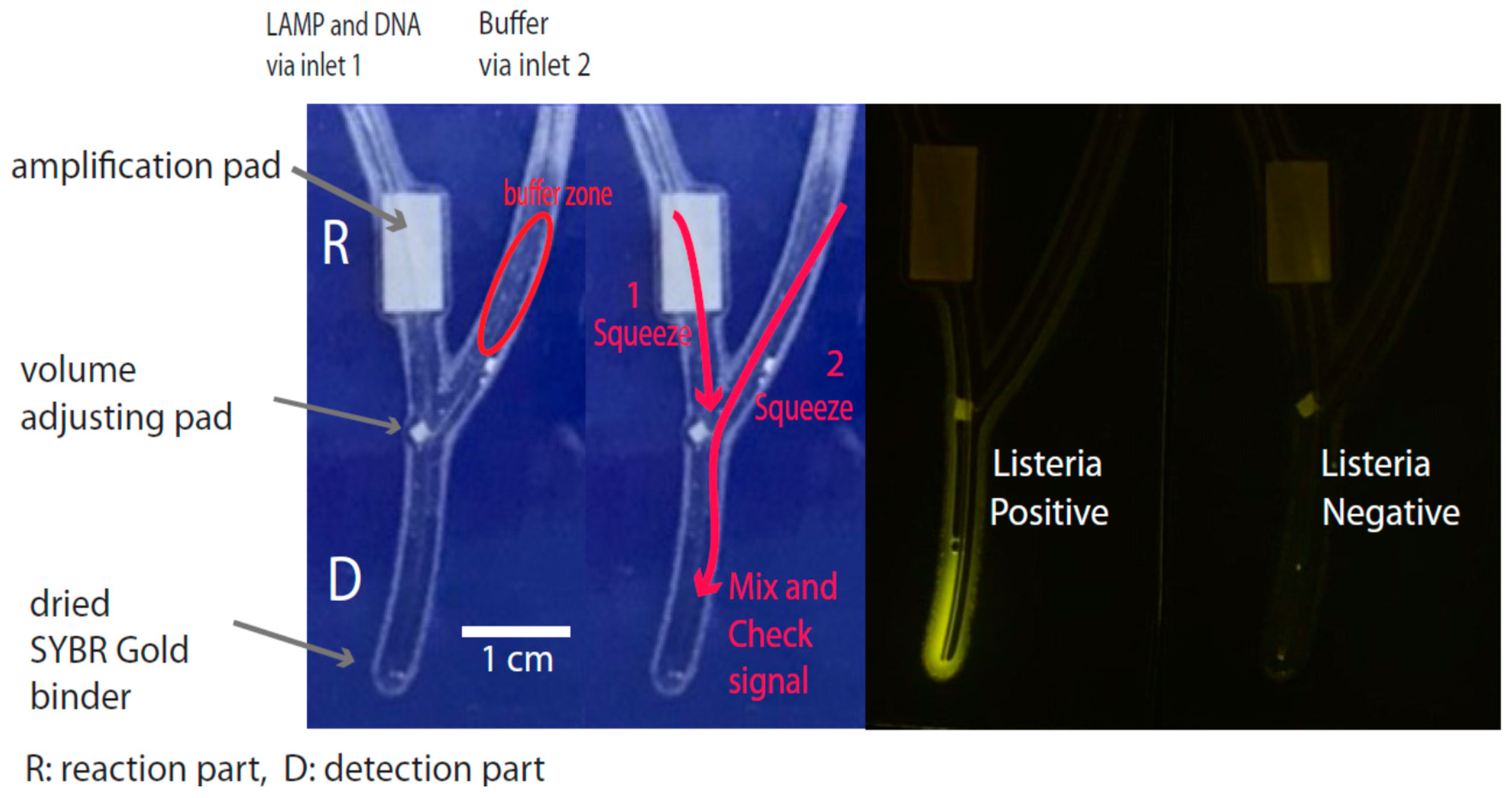
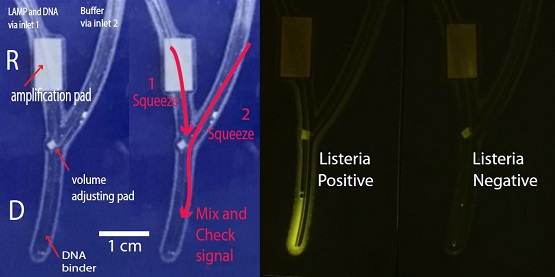
2.1. Chip Design and Fabrication
2.2. Establishment of Nucleic Acid Amplification
2.3. Test Specimens
2.4. Signal Detection via Fluorescence Assay
2.5. Routine Operation of the Chip
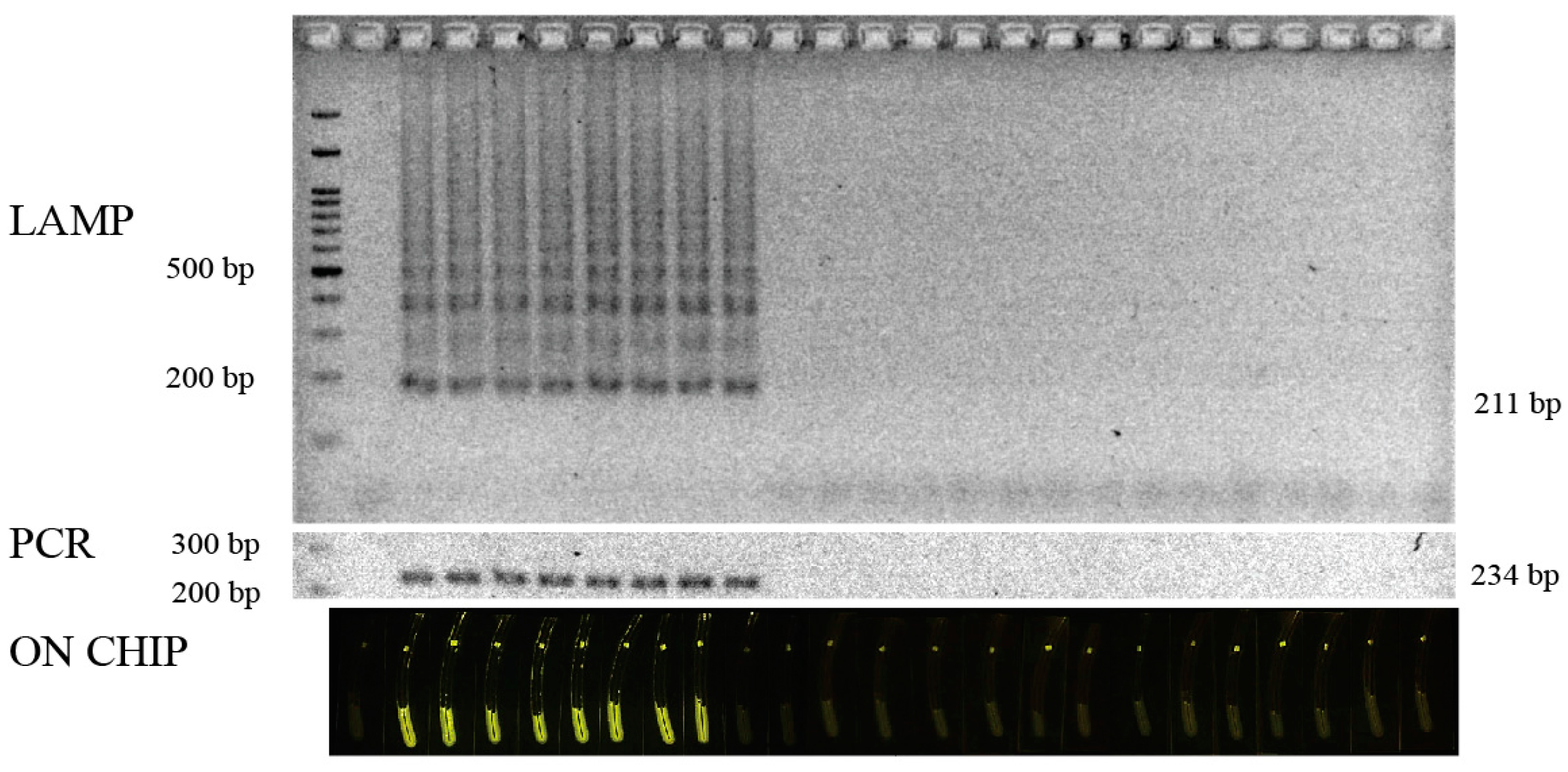
2.6. The Specificity and Sensitivity of LAMP Based on the Fluorescence Signal
3. Results and Discussion
3.1. Chip and Chip Fabrication
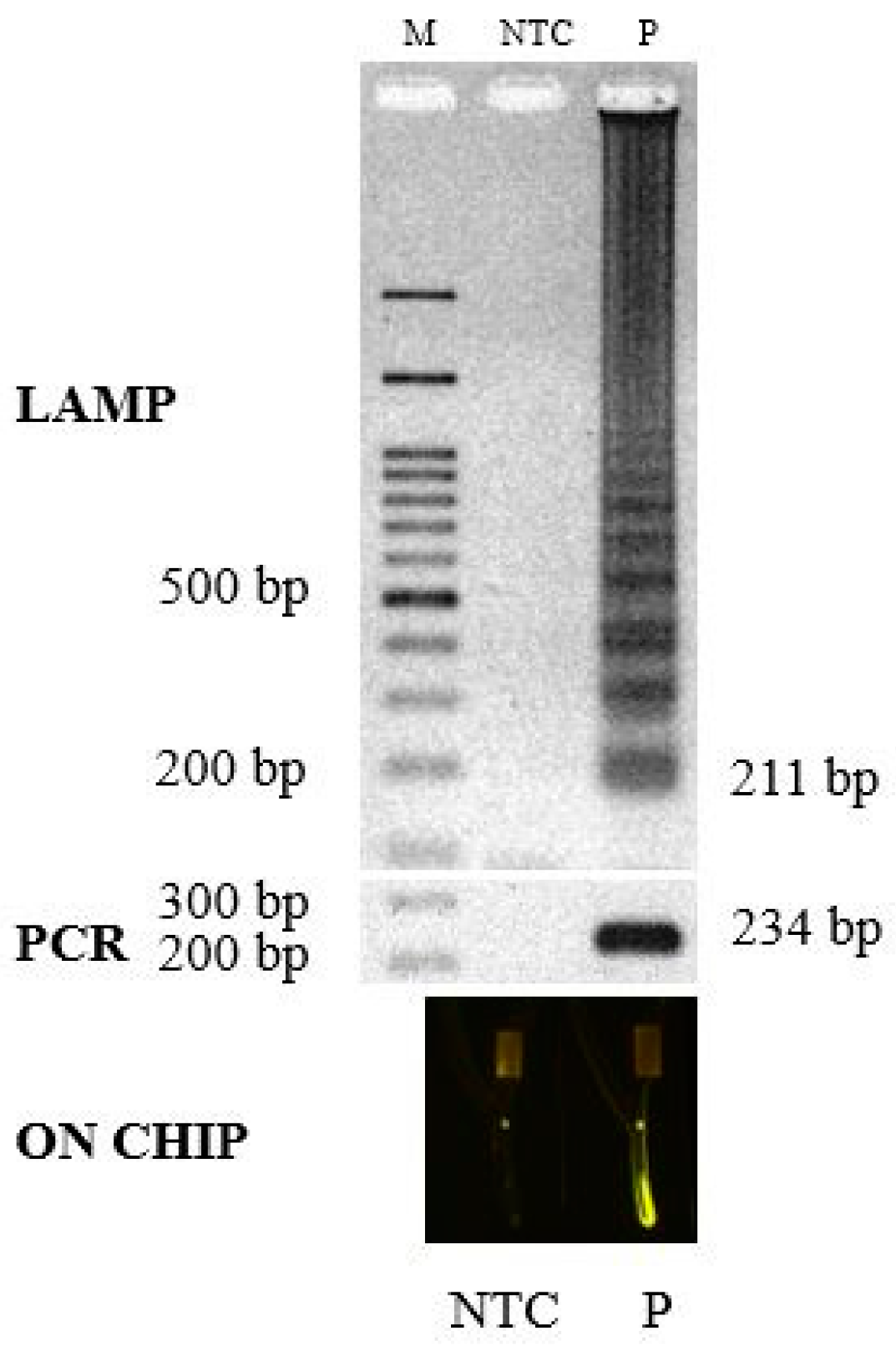
3.2. Amplification Platform
3.3. Signal Detection Platform
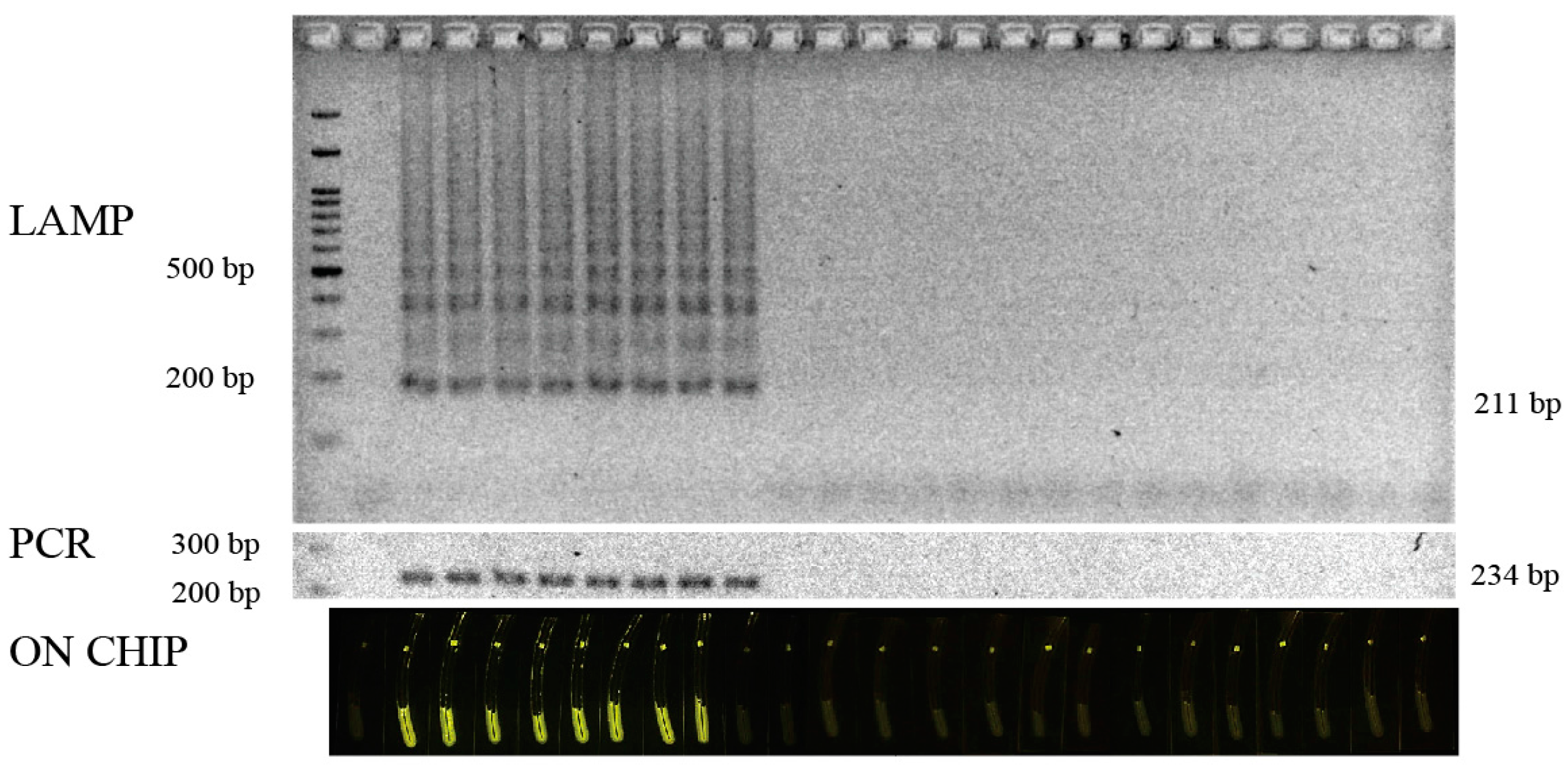
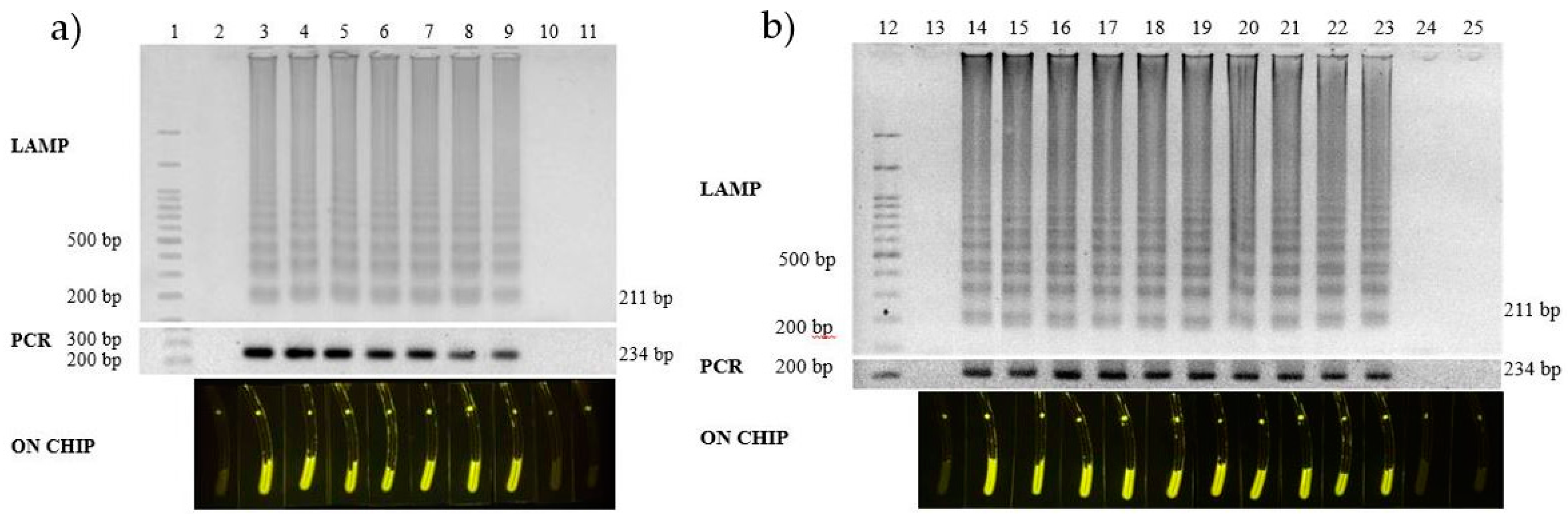
3.4. Validation of the Test
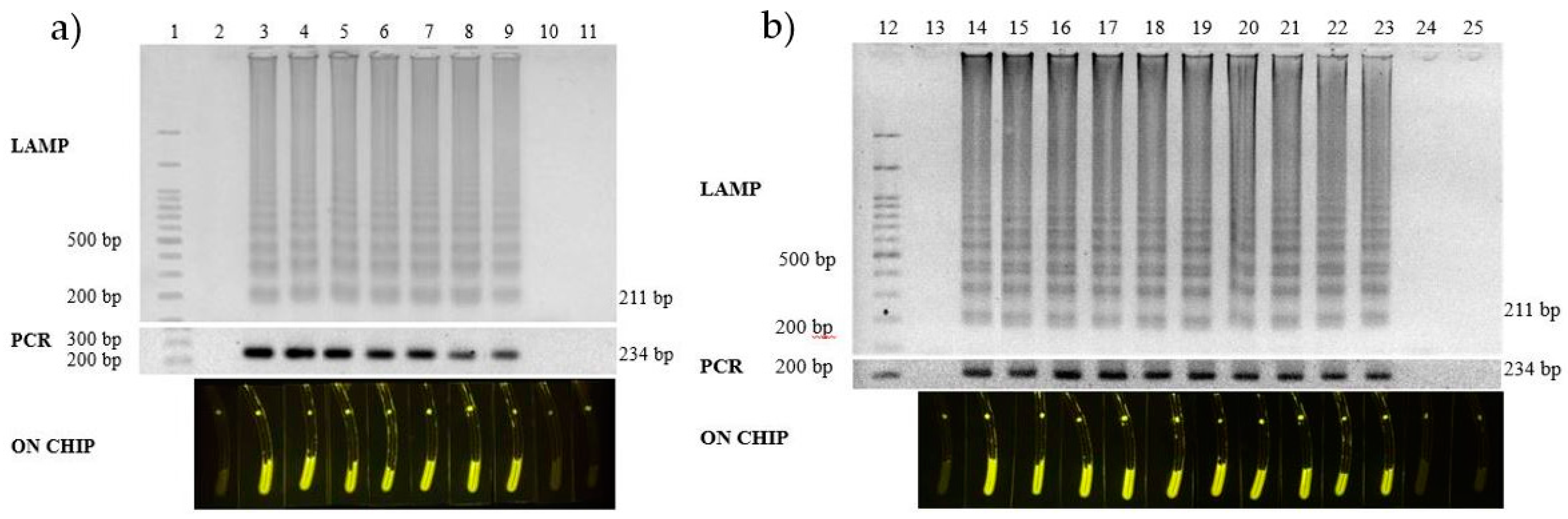
3.5. Field Monitoring Application
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lund, B.M.; O’Brien, S.J. The Occurrence and Prevention of Foodborne Disease in Vulnerable People. Foodborne Pathog. Dis. 2011, 8, 961–973. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization [WHO], Initiative to Estimate the Global Burden of Foodborne Diseases: Information and Publications. Available online: http://www.who.int/foodsafety/publications/foodborne_disease/fergreport/en/ (accessed on 26 November 2016).
- Jørgensen, L.V.; Huss, H.H. Prevalence and growth of Listeria monocytogenes in naturally contaminated seafood. Int. J. Food Microbiol. 1998, 42, 127–131. [Google Scholar] [CrossRef]
- Andras, S.C.; Power, J.B.; Cocking, E.C.; Davey, M.R. Strategies for signal amplification in nucleic acid detection. Mol. Biotechnol. 2001, 19, 29–44. [Google Scholar] [CrossRef]
- Compton, J. Nucleic acid sequence-based amplification. Nature 1991, 350, 91–92. [Google Scholar] [CrossRef] [PubMed]
- Gravani, R. Incidence and control of Listeria in food-processing facilities. In Listeria, Listeriosis, and Food Safety, 2nd ed.; Ryser, E.T., Marh, E.H., Eds.; Marcel Decker: New York, NY, USA, 1999; pp. 657–709. ISBN 0-8247-0235-2. [Google Scholar]
- Nørrung, B.; Adensen, J.K.; Schlundt, J. Incidence and control of Listeria monocytogenes in foods in Denmark. Int. J. Food Microbiol. 1999, 53, 195–203. [Google Scholar] [CrossRef]
- Vázquez-Boland, J.A.; Kuhn, M.; Berche, P.; Chakraborty, T.; Dominguez-Bernal, G.; Goebel, W.; Gonzaiez-Zorn, B.; Wehland, J.; Kreft, J. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 2001, 14, 584–640. [Google Scholar] [CrossRef] [PubMed]
- Bronze, M.S. Listeria monocytogenes: Epidemiology, human disease, and mechanisms of brain invasion. FEMS Immunol. Med. Microbiol. 2008, 53, 151–165. [Google Scholar] [CrossRef]
- Bickley, J.; Short, J.K.; McDowell, D.G.; Parkes, H.C. Polymerase chain reaction (PCR) detection of Listeria monocytogenes in diluted milk and reversal of PCR inhibition caused by calcium ions. Lett. Appl. Microbiol. 1996, 22, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Border, P.M.; Howard, J.J.; Plastow, G.S.; Siggens, K.W. Detection of Listeria species and Listeria monocytogenes using polymerase chain reaction. Lett. Appl. Microbiol. 1990, 11, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.J.; King, R.K.; Burchak, J.; Gannon, V.P. Sensitive and specific detection of Listeria monocytogenes in milk and ground beef with the polymerase chain reaction. Appl. Environ. Microbiol. 1991, 57, 2576–2580. [Google Scholar] [PubMed]
- Jeong, Y.J.; Park, K.; Kim, D.E. Isothermal DNA amplification in vitro: The helicase-dependent amplification system. Cell Mol. Life Sci. 2009, 66, 3325–3326. [Google Scholar] [CrossRef] [PubMed]
- Dou, M.; Sanjay, S.T.; Benhabib, M.; Xu, F.; Li, X. Low-cost bioanalysis on paper-based and its hybrid microfluidic platforms. Talanta 2015, 145, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.T.; Fraiser, M.S.; Schram, J.L.; Little, M.C.; Nadeau, J.G.; Malinowski, D.P. Strand displacement amplification-An isothermal in vitro DNA amplification Technique. Nucleic Acids. Res. 1992, 20, 1691–1696. [Google Scholar] [CrossRef] [PubMed]
- Chaumpluk, P.; Plubcharoensook, P.; Prasongsuk, S. Rapid detection of aflatoxigenic Aspergillus sp. in herbal specimens by a simple, bendable, paper-based lab-on-a-chip. Biotechnol. J. 2016, 11, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Phillips, S.; Butte, M.; Whitesides, G. Patterned paper as a platform for inexpensive, low-volume, portable bioassays. Angew. Chem. Int. Ed. Engl. 2007, 46, 1318–1320. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.W.; Phillips, S.T.; Whitesides, G.M. Three-dimensional microfluidic devices fabricated in layered paper and tape. Proc. Natl. Acad. Sci. USA 2008, 105, 19606–19611. [Google Scholar] [CrossRef] [PubMed]
- Bruzewicz, D.A.; Reches, M.; Whitesides, G.M. Low-cost printing of poly (dimethylsiloxane) barriers to define microchannels in paper. Anal. Chem. 2008, 80, 3387–3392. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Shi, W.; Jiang, L.; Qin, J.; Lin, B. Rapid prototyping of paper-based microfluidics with wax for low-cost, portable bioassay. Electrophoresis 2009, 30, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Shi, W.; Qin, J.; Lin, B. Fabrication and characterization of paper-based microfluidics prepared in nitrocellulose membrane by wax printing. Anal. Chem. 2010, 82, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Cassano, C.L.; Fan, Z.H. Laminated paper-based analytical devices (LPAD): Fabrication, characterization, and assays. Microfluid. Nanofluid. 2013, 15, 173–181. [Google Scholar] [CrossRef]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, e63. [Google Scholar] [CrossRef] [PubMed]
- Muangchuen, A.; Chaumpluk, P.; Suriyasomboon, A.; Ekgasit, S. Colorimetric Detection of Ehrlichia canis via nucleic acid hybridization in gold nano-colloids. Sensors 2014, 14, 14472–14487. [Google Scholar] [CrossRef] [PubMed]
- Chaumpluk, P.; Chaiprasart, P.; Vilaivan, T. Postharvest nondestructive determination of fruits: A model on fruit maturity assay via biosensor based on colorimetric change of gold nanoparticles. Acta Hortic. 2012, 945, 205–212. [Google Scholar] [CrossRef]
- Cossart, P. The Listeriolysin O Gene: A Chromosomal Locus Crucial for the Virulence of Listeria monocytogenes. Infection 1988, 16, S157–S159. [Google Scholar] [CrossRef] [PubMed]
- Higgins, D.G. CLUSTAL V: Multiple Alignment of DNA and Protein Sequences. Methods. Mol. Biol. 1994, 25, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Nørrung, B. Microbiological criteria for Listeria monocytogenes in foods under special consideration of risk assessment approaches. Int. J. Food Microbiol. 2000, 62, 217–221. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kibbe, W.A. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res. 2007, 35, W43–W46. [Google Scholar] [CrossRef] [PubMed]
- Furrer, B.; Candrian, U.; Hoefelein, C.; Luethy, J. Detection and identification of Listeria monocytogenes in cooked sausage products and in milk by in vitro amplification of haemolysin gene fragments. J. Appl. Bacteriol. 1991, 70, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Manonmani, H.K.; Anand, S.; Chandrashekar, A.; Rati, E.R. Detection of aflatoxigenic fungi in selected food commodities by PCR. Process Biochem. 2005, 40, 2859–2864. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989; Volume 1, ISBN 978-0879693091. [Google Scholar]
- Kuribara, H.; Shindo, Y.; Matsuoka, T.; Takubo, K.; Futo, S.; Aoki, N.; Hirao, T.; Akiyama, H.; Goda, Y.; Toyoda, M.; et al. Novel reference molecules for quantitation of genetically modified maize and soybean. J. AOAC Int. 2002, 85, 1077–1089. [Google Scholar] [PubMed]
- Fawcett, T. An introduction to ROC analysis. Pattern Recognit. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]
- Chaumpluk, P.; Chaiprasart, P. Fluorescence biosensor based on N-(2-Aminoethyl) glycine peptide nucleic acid for a simple and rapid detection of Escherichia coli in fresh-cut mango. Acta Hortic. 2013, 992, 551–560. [Google Scholar] [CrossRef]
- Creating Standard Curves with Genomic DNA or Plasmid DNA Templates for Use in Quantitative PCR. Available online: http://www3.appliedbiosystems.com/cms/groups/mcb_marketing/documents/generaldocuments/cms_042486.pdf (accessed on 20 February 2015).
- Yano, A.; Ishimaru, R.; Hujikata, R. Rapid and sensitive detection of heat-labile I and heat-stable I enterotoxin genes of enterotoxigenic Escherichia coli by Loop-Mediated Isothermal Amplification. J. Microbiol. Methods 2007, 68, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Rodríguez, F.; Valero, A.; Carrasco, E.; García, R.M.; Zurera, G. Understanding and modelling bacterial transfer to foods: A review. Trends Food Sci. Technol. 2008, 19, 131–144. [Google Scholar] [CrossRef]
- Lorber, B. Listeriosis, in Listeria monocytogenes; Pathogenesis and Host Response; Goldfine, H., Shen, H., Eds.; Springer: New York, NY, USA, 2007; pp. 13–32. ISBN 978-0-387-49376-3. [Google Scholar]
- Aznar, R.; Alarcon, B. On the specificity of PCR detection of Listeria monocytogenes in food: A comparison of published primers. Syst. Appl. Microbiol. 2002, 25, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Qu, L.; Wimbrow, A.N.; Jiang, X.; Sun, Y. Rapid detection of Listeria monocytogenes by nanoparticle-based immunomagnetic separation and real-time PCR. Int. J. Food Microbiol. 2007, 118, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Kurosawa, Y.; Furui, S.; Kerman, K.; Kobayashi, M.; Rao, S.R.; Yonezawa, Y.; Nakano, K.; Hino, A.; Yamamura, S.; et al. Circumventing air bubbles in microfluidic systems and quantitative continuous-flow PCR applications. Anal. Bioanal. Chem. 2006, 386, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Parida, M.; Sannarangaiah, S.; Dash, P.K.; Rao, P.V.; Morita, K. Loop mediated isothermal amplification (LAMP): A new generation of innovative gene amplification technique; perspectives in clinical diagnosis of infectious diseases. Rev. Med. Virol. 2008, 18, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Cossart, P.; Mengaud, J. Listeria monocytogenes. A model system for the molecular study of intracellular parasitism. Mol. Biol. Med. 1989, 6, 463–474. [Google Scholar] [PubMed]
- Churchill, R.L.T.; Lee, H.; Hall, J.C. Detection of Listeria monocytogenes and the toxin listeriolysin O in food. J. Microbiol. Methods 2006, 64, 141–170. [Google Scholar] [CrossRef] [PubMed]
- Jallewar, P.K.; Kalorey, D.R.; Kurkure, N.V.; Pandeb, V.V.; Barbuddhe, S.B. Genotypic characterization of Listeria spp. isolated from fresh water fish. Int. J. Food Microbiol. 2007, 114, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.J.; Zhou, S.; Zhang, X.Y.; Pu, J.H.; Ge, Q.L.; Tang, X.J.; Gao, Y.S. Rapid and sensitive detection of Listeria monocytogenes by loop-mediated isothermal amplification. Curr. Microbiol. 2011, 63, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Liu, Y.; Kong, J.; Jiang, X. Loop-mediated isothermal amplification integrated on microfluidic chips for point-of-care quantitative detection of pathogens. Anal. Chem. 2010, 82, 3002–3006. [Google Scholar] [CrossRef] [PubMed]
- Sher, M.; Zhuang, R.; Demirci, U.; Asghar, W. Paper-based analytical devices for clinical diagnosis: Recent advances in the fabrication techniques and sensing mechanisms. Expert Rev. Mol. Diagn. 2017, 17, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Shindy, H.A. Basics, Mechanisms and Properties in the Chemistry of Cyanine Dyes: A Review Paper. Mini-Rev. Org. Chem. 2012, 9, 352. [Google Scholar] [CrossRef]
- Bugno, A.; Almodovar, A.A.B.; Pereira, T.C.; Pinto, T.D.J.A.; Sabino, M. Occurrence of toxigenic fungi in herbal drugs. Braz. J. Microbiol. 2006, 37, 47–51. [Google Scholar] [CrossRef]
- Egholm, M.; Buchardt, O.; Christensen, L.; Behrens, C.; Freier, S.M.; Driver, D.A.; Berg, R.H.; Kim, S.K.; Norden, B.; Nielsen, P.E. PNA hybridizes to complementary oligonucleotides obeying the Watsoncrick hydrogen-bonding rules. Nature 1993, 365, 566–568. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E.; Haaima, G. Peptide nucleic acid (PNA). A DNA mimic with a pseudopeptide backbone. Chem. Soc. Rev. 1997, 26, 73–78. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, G.; Lu, C.; Deng, R.; Zhi, A.; Guo, J.; Zhao, D.; Xu, Z. Rapid detection of Listeria monocytogenes in raw milk with loop-mediated isothermal amplification and chemosensor. J. Food Sci. 2011, 76, M611–M615. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Zhang, Y.; Zhang, Z.; Chen, M.; Su, Y.; Yuan, Y.; Alam, M.J.; Yan, H.; Shi, L. Rapid detection of food-borne Listeria monocytogenes by real-time quantitative loop-mediated isothermal amplification. Food Sci. Biotechnol. 2012, 21, 1. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5′ to 3′) | Reference Position * |
|---|---|---|
| F3 | ACAATGTATTAGTATACCACGGA | 218-240 |
| B3 | TCTGGTTGATTTTCTACTAATTCC | 405-428 |
| FIP | GGATTTCTCTTTTTCTCCACAACGTTTTGATGCAGTGACAAATGTGC | (297-321)–(241-259) |
| BIP | GCAGACATCCAAGTTGTAAATGCTTTTCGCTTTTACGAGAGCACC | (337-359)–(382-399) |
| PCR Forward Primer ** | CGGAGGTTCCGCAAAAGATG | 1044-1036 |
| PCR Reverse Primer ** | CCTCCAGAGTGATCGATGTT | 1258-1277 |
| No. | Bacteria | Strain |
|---|---|---|
| 1 | Listeria monocytogenes | DMST * 17303 |
| 2 | L. monocytogenes | DMST * 1327 |
| 3 | L. monocytogenes | DMST * 20093 |
| 4 | L. monocytogenes | DMST * 23145 |
| 5 | L. monocytogenes | DMST * 31802 |
| 6 | L. monocytogenes | FRTL ** 1299 |
| 7 | L. monocytogenes | FRTL ** 1401 |
| 8 | L. innocua | DMST * 9011 |
| 9 | L. innocua | FRTL ** 1265 |
| 10 | L. innocua | FRTL ** 1445 |
| 11 | L. innocua | FRTL ** 1446 |
| 12 | L. ivanovii | FRTL ** 1243 |
| 13 | L. ivanovii | DMST * 9012 |
| 14 | L. welshimeri | DMST * 20559 |
| 15 | Vibrio cholera | FRTL ** 1322 |
| 16 | V. parahaemolyticus | FRTL ** 0886 |
| 17 | Salmonella enteritidis | DMST * 15676 |
| 18 | Escherichia coli O157:H7 | DMST * 12743 |
| 19 | E. coli (ETEC) | DMST * 30543 |
| 20 | E. coli (EPEC) | DMST * 30546 |
| 21 | Pseudomonas putida | DMST * 16074 |
| 22 | Shigella flexneri | DMST * 4423 |
| Specimens | PCR | LAMP on Chip |
|---|---|---|
| Positive | 40 | 41 |
| Negative | 60 | 59 |
| False positive | - | 1 |
| False negative | - | 0 |
| Sensitivity A | 100.00 | |
| Specificity B | 98.33 |
| Number | Sample | Collecting Location | Result | |
|---|---|---|---|---|
| on PCR | on Chip | |||
| 1 | Salmon filet portion sashimi | Supermarket | N | N |
| 2 | Raw salmon meat (block) | Supermarket | N | N |
| 3 | Raw salmon meat | Supermarket | N | N |
| 4 | Smoked salmon | Supermarket | N | N |
| 5 | Salmon | Supermarket | N | N |
| 6 | Fresh cut salmon | Supermarket | N | N |
| 7 | Frozen peeled shrimp | Supermarket | N | N |
| 8 | Fresh shrimp sashimi | Supermarket | N | N |
| 9 | Raw tuna sashimi | Supermarket | N | N |
| 10 | Raw tuna block for sashimi | Supermarket | N | N |
| 11 | Raw salmon meat for sashimi | Supermarket | N | N |
| 12 | Raw salmon halve dressing | Supermarket | N | N |
| 13 | Fresh cut salmon (disc) | Supermarket | N | N |
| 14 | Frozen salmon sashimi | Supermarket | N | N |
| 15 | Repacked smoked salmon | Supermarket | N | N |
| 16 | Raw salmon for sashimi | Local market | N | N |
| 17 | Raw salmon sushi | Local market | N | N |
| 18 | Fresh shrimp | Local market | N | N |
| 19 | Frozen shrimp peeled clean tail on | Local market | N | N |
| 20 | Uncooked shrimp easy peeled tail on | Local market | N | N |
| 21 | Ready-to-eat salmon sushi | Local market | P | P |
| 22 | Repacked smoked salmon | Local market | N | N |
| 23 | Raw salmon for sushi | Local market | N | N |
| 24 | Raw salmon for sushi | Local market | N | N |
| 25 | Frozen salmon block | Local market | N | N |
| 26 | Raw shrimp peeled tail on | Local market | N | N |
| 27 | Raw salmon block | Local market | N | N |
| 28 | Fresh shrimp for raw serve | Local market | N | N |
| 29 | Fresh shrimp for raw serve | Local market | N | N |
| 30 | Salmon for sushi | Local market | N | N |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pisamayarom, K.; Suriyasomboon, A.; Chaumpluk, P. Simple Screening of Listeria monocytogenes Based on a Fluorescence Assay via a Laminated Lab-On-Paper Chip. Biosensors 2017, 7, 56. https://doi.org/10.3390/bios7040056
Pisamayarom K, Suriyasomboon A, Chaumpluk P. Simple Screening of Listeria monocytogenes Based on a Fluorescence Assay via a Laminated Lab-On-Paper Chip. Biosensors. 2017; 7(4):56. https://doi.org/10.3390/bios7040056
Chicago/Turabian StylePisamayarom, Kankanit, Annop Suriyasomboon, and Piyasak Chaumpluk. 2017. "Simple Screening of Listeria monocytogenes Based on a Fluorescence Assay via a Laminated Lab-On-Paper Chip" Biosensors 7, no. 4: 56. https://doi.org/10.3390/bios7040056