Isothermal Amplification Methods for the Detection of Nucleic Acids in Microfluidic Devices

Abstract
:1. Introduction
2. Microfluidics for Nucleic Acids Amplification
3. Isothermal Amplification Methods
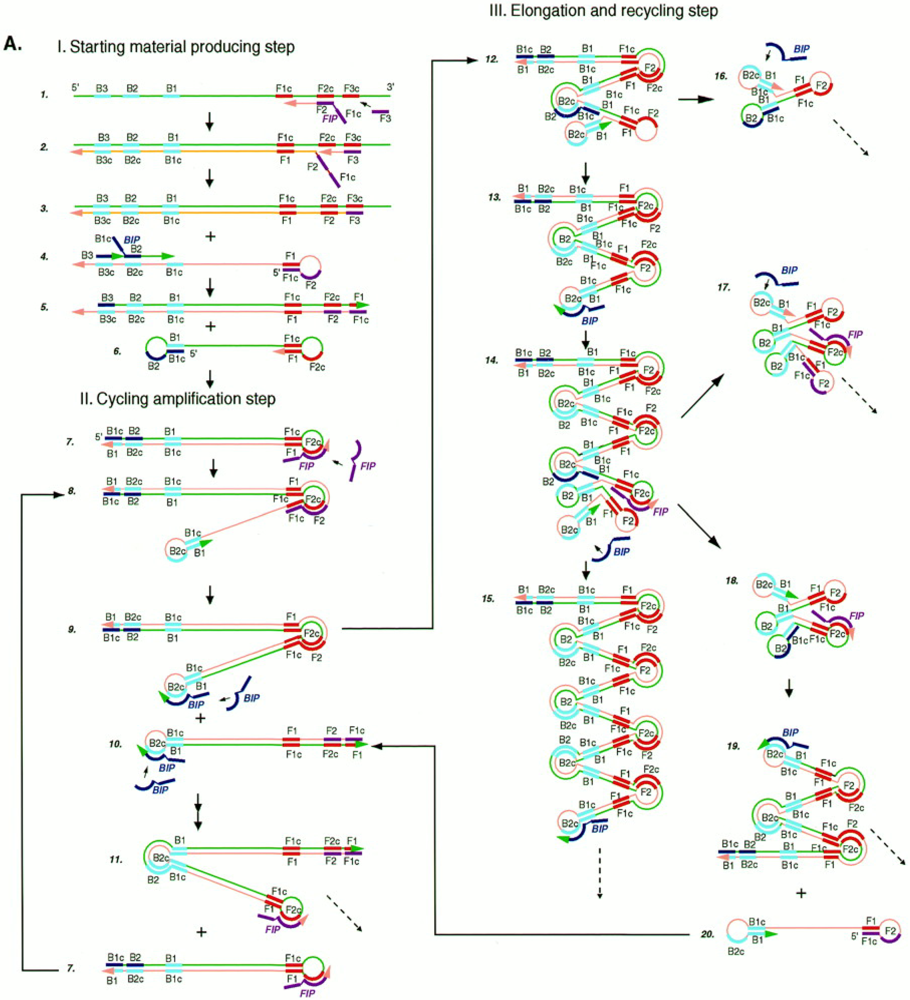
3.1. Loop-Mediated Isothermal Amplification (LAMP)
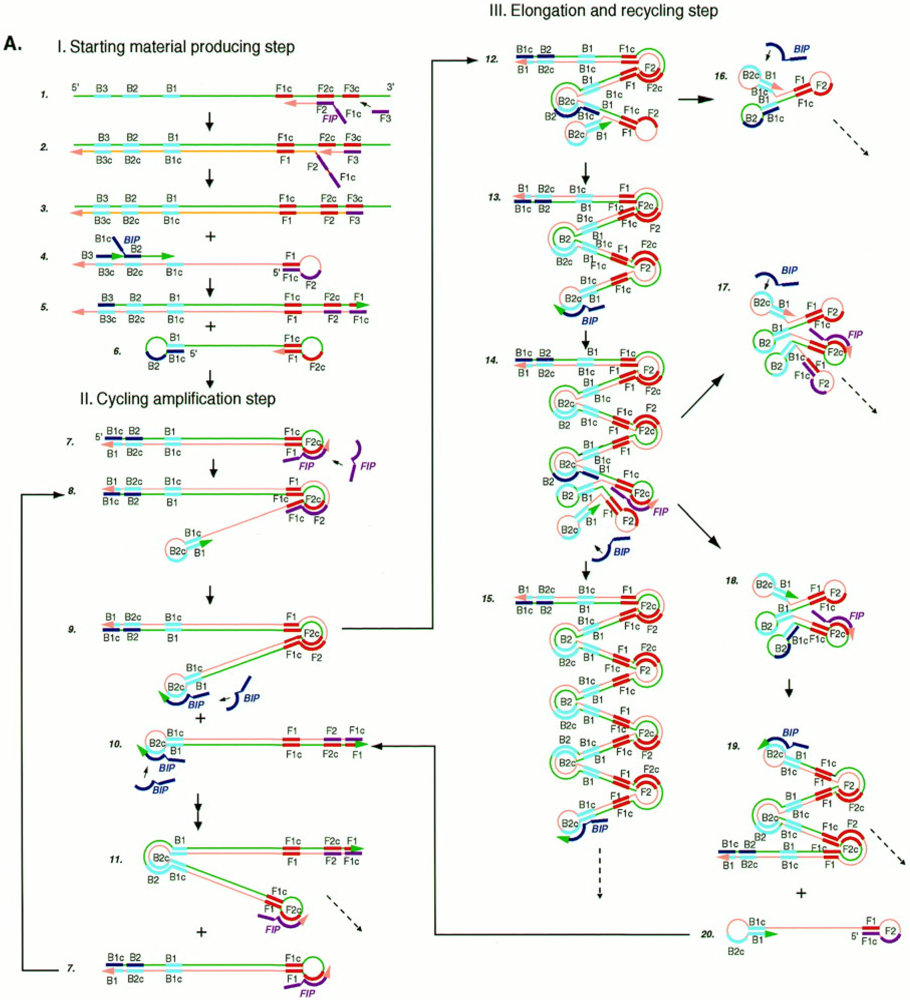
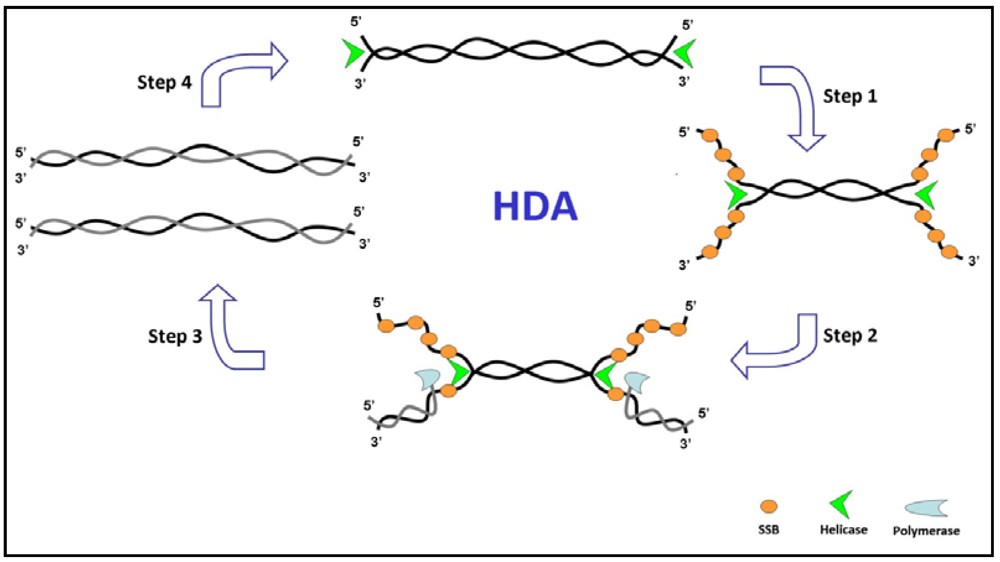
3.2. Helicase-Dependent Amplification (HDA)
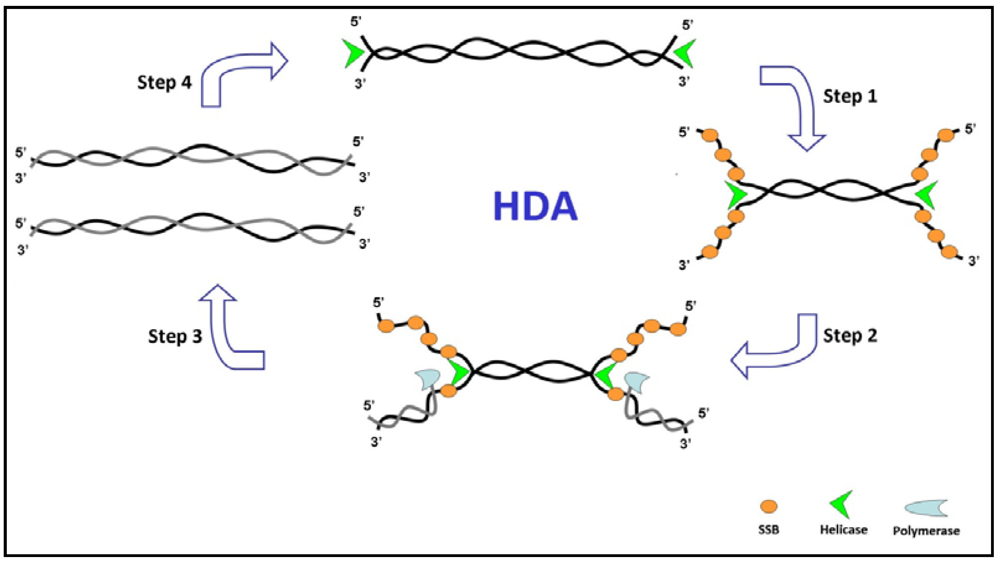
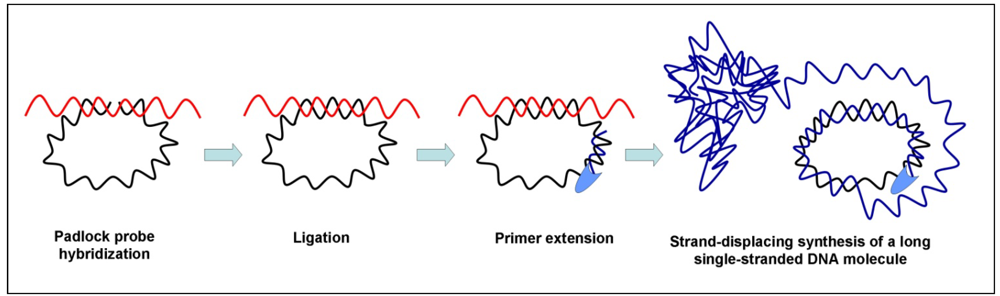
3.3. Rolling Circle Amplification (RCA)
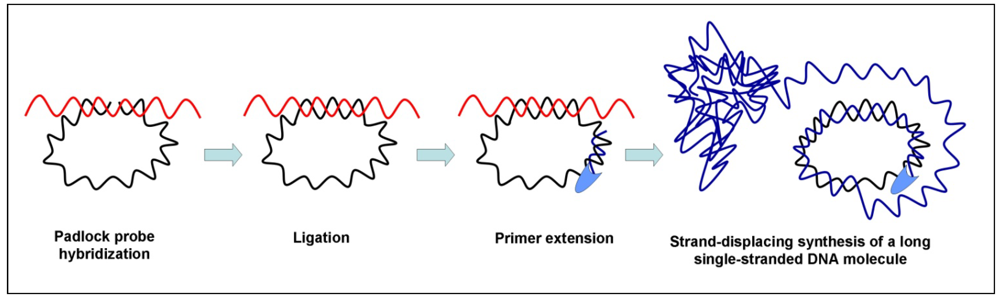
3.4. Multiple Displacement Amplification (MDA)
3.5. Recombinase Polymerase Amplification (RPA)
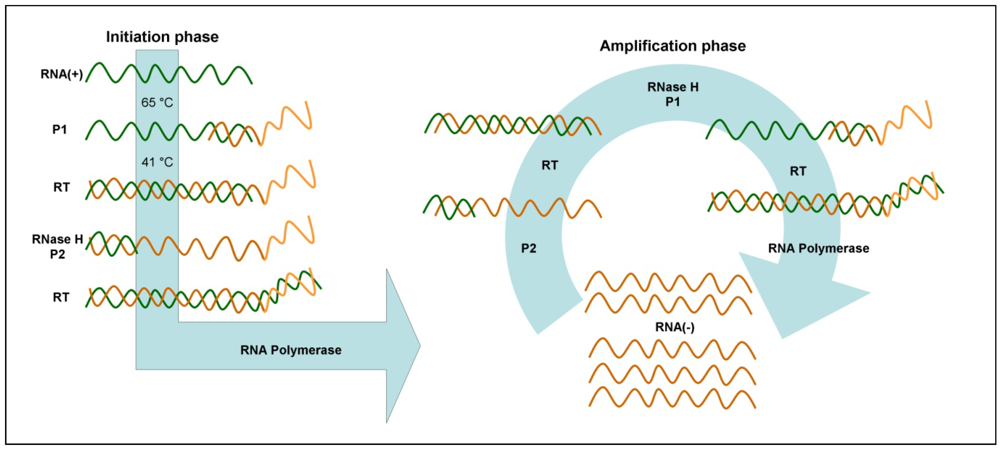
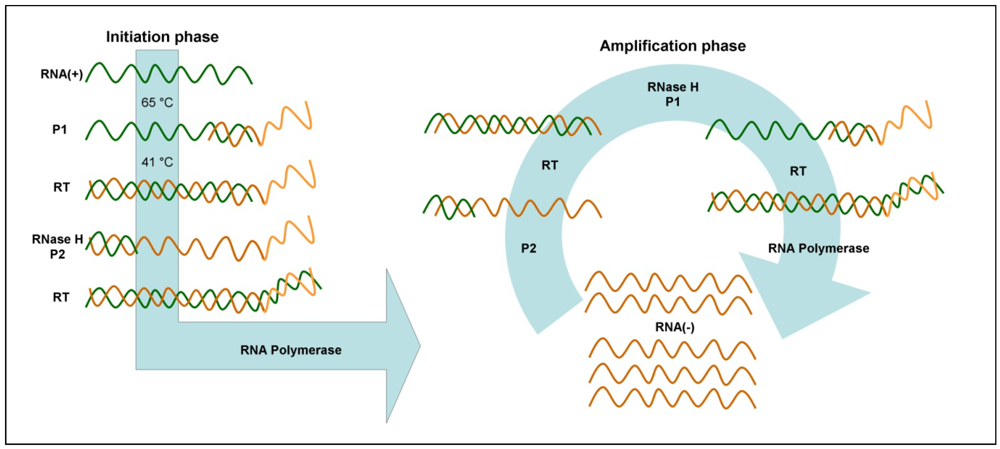
3.6. Nucleic Acid Sequence-Based Amplification (NASBA)
4. Comparison of Isothermal Amplification Methods for Microfluidic Integration

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Amplification time | Reaction volume | Target | Detection limit a | Ref. |
|---|---|---|---|---|---|
| LAMP | within 1 h | 25 µL | hepatitis B virus (HBV) DNA | 50 copies/25 μL | [50,51] |
| within 15 min | 10 µL | prostate-specific antigen gene | 23 fg/μL | [52] | |
| within 1 h | 5 µL | Pseudorabies virus (PRV) DNA | 10 fg | [53] | |
| within 1 h | b | λDNA | two molecule | [54] | |
| 1 h 35 min | 35 µL | E. coli genomic DNA | 24 colony forming units (CFU)/mLl 48 CFU/mL | [55] | |
| HDA | 2 h | 150 µL | N. gonorrhoeae genomic DNA Methicillin resistant S. aureus genomic DNA | 1 ng 250 pg | [74] |
| 0.5 h | ~5 µL/192 nL | BNI-1 fragment of SARS cDNA | 0.01 ng/μL | [76] | |
| 0.5 h | 25 µL | E. coli genomic DNA | 10 CFU | [78] | |
| RCA | within 65 min | 10 µL | Genomic DNA for V. cholerae | 25 ng | [87] |
| 4 h | 2 pL | pIVEX2.2EM-lacZ plasmid | 0.07 pg/μL | [90] | |
| 2.5 h | pL | Human-malaria-causing Plasmodium parasites | less than one parasite/μL | [91] | |
| MDA | 10–16 h | 60 nL | E. coli genomic DNA | b | [99] |
| RPA | within 20 min | 10 µL | mecA gene of Staphylococcus aureus | less than 10 copies | [102] |
| 1 h | 9 nL | Methicillin-resistant Staphylococcus aureus genomic DNA | 300 copies/mL | [106] | |
| NASBA | within 2 h | 10 nL | Human papillomavirus (HPV) | 1.0 ìM | [118] |
| 2,5 h | 80 nL | Artificial human papilloma virus (HPV) 16 sequences SiHa cell line samples | 10−6 ìM 20 cells/μL | [119] | |
| 0,5 h | 2 µL | E. coli tmRNA | 100 cells in 100 ìL | [121] | |
| 2–3 h | 30 ìL | Water pathogens | 105 CFU/mL | [122] |
5. Conclusions
Acknowledgments
References
- D’Agata, R.; Breveglieri, G.; Zanoli, L.M.; Borgatti, M.; Spoto, G.; Gambari, R. Direct detection of point mutations in nonamplified human genomic DNA. Anal. Chem. 2011, 83, 8711–8717. [Google Scholar] [CrossRef]
- Marchelli, R.; Tedeschi, T.; Tonelli, A. DNA analyses in food safety and quality: Current status and expectations. In Detection of Non-Amplified Genomic DNA; Spoto, G., Corradini, R., Eds.; Springer: Dordrecht, The Netherland, 2012; pp. 25–63. [Google Scholar]
- D’Agata, R.; Corradini, R.; Ferretti, C.; Zanoli, L.; Gatti, M.; Marchelli, R.; Spoto, G. Ultrasensitive detection of non-amplified genomic DNA by nanoparticle-enhanced Surface-Plasmon Resonance Imaging. Biosens. Bioelectron. 2010, 25, 2095–2100. [Google Scholar] [CrossRef]
- Shi, L.; Perkins, R.G.; Fang, H.; Tong, W. Reproducible and reliable microarray results through quality control: Good laboratory proficiency and appropriate data analysis practices are essential. Curr. Opin. Biotechnol. 2008, 19, 10–18. [Google Scholar] [CrossRef]
- Spoto, G.; Minunni, M. Surface Plasmon Resonance imaging: What’s next? J. Phys. Chem. Lett. 2012, 3, 2682–2691. [Google Scholar]
- Zanoli, L.M.; D’Agata, R.; Spoto, G. Functionalized gold nanoparticles for the ultrasensitive DNA detection. Anal. Bioanal. Chem. 2012, 402, 1759–1771. [Google Scholar] [CrossRef]
- Asiello, P.J.; Baeumner, A.J. Miniaturized isothermal nucleic acid amplification, a review. Lab Chip 2011, 11, 1420–1430. [Google Scholar] [CrossRef]
- Leng, X.; Zhang, W.; Wang, C.; Cui, L.; Yang, C.J. Agarose droplet microfluidics for highly parallel and efficient single molecule emulsion PCR. Lab Chip 2010, 10, 2841–2843. [Google Scholar] [CrossRef]
- Kojima, T.; Takei, Y.; Ohtsuka, M.; Kawarasaki, Y.; Yamane, T.; Nakano, H. PCR amplification from single DNA molecules on magnetic beads in emulsion: application for high-throughput screening of transcription factor targets. Nucleic Acids Res. 2005, 33. [Google Scholar] [CrossRef]
- Kim, J.; Easley, C.J. Isothermal DNA amplification in bioanalysis: Strategies and applications. Bioanalysis 2011, 3, 227–239. [Google Scholar] [CrossRef]
- Whitesides, G.M. The origins and future of microfluidics. Nature 2006, 422, 368–373. [Google Scholar] [CrossRef]
- Beebe, D.J.; Mensing, G.A.; Walker, G.M. Physics and applications of microfluidics in biology. Annu. Rev. Biomed. Eng. 2002, 4, 261–286. [Google Scholar] [CrossRef]
- Stone, H.A.; Stroock, A.D.; Ajdari, A. Engineering flows in small devices: Microfluidics toward a lab-on-a-chip. Annu. Rev. Fluid Mech. 2004, 36, 381–411. [Google Scholar] [CrossRef]
- Squires, T.M.; Quake, S.R. Microfluidics: Fluid physics at the nanoliter scale. Rev. Mod. Phys. 2005, 77, 977–1026. [Google Scholar] [CrossRef]
- Buchegger, W.; Haller, A.; van den Driesche, S.; Kraft, M.; Lendl, B.; Vellekoop, M. Studying enzymatic bioreactions in a millisecond microfluidic flow mixer. Biomicrofluidics 2012, 6, 12803–128039. [Google Scholar] [CrossRef]
- Buchegger, W.; Wagner, C.; Lendl, B.; Kraft, M.; Vellekoop, M. A highly uniform lamination micromixer with wedge shaped inlet channels for time resolved infrared spectroscopy. Microfluid. Nanofluid. 2011, 10, 889–897. [Google Scholar] [CrossRef]
- Lee, C-Y.; Chang, C-L.; Wang, Y-N.; Fu, L-M. Microfluidic mixing: A review. Int. J. Mol. Sci. 2011, 12, 3263–3287. [Google Scholar] [CrossRef]
- Hartwell, S.K.; Grudpan, K. Flow-based systems for rapid and high-precision enzyme kinetics studies. J. Anal. Methods Chem. 2012. [Google Scholar] [CrossRef]
- Bleul, R.; Ritzi-Lehnert, M.; Höth, J.; Scharpfenecker, N.; Frese, I.; Düchs, D.; Brunklaus, S.; Hansen-Hagge, T.E.; Meyer-Almes, F.J.; Drese, K.S. Compact, cost-efficient microfluidics-based stopped-flow device. Anal. Bioanal. Chem. 2011, 399, 1117–1125. [Google Scholar] [CrossRef]
- Zhang, Y.; Ozdemir, P. Microfluidic DNA amplification—A review. Anal. Chim. Acta. 2009, 638, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Shaw, K.J.; Docker, P.T.; Yelland, J.V.; Dyer, C.E.; Greenman, J.; Greenwaya, G.M.; Haswell, S.J. Rapid PCR amplification using a microfluidic device with integrated microwave heating and air impingement cooling. Lab Chip 2010, 10, 1725–1728. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, C.; Xing, D. Integrated microfluidic reverse transcription-polymerase chain reaction for rapid detection of food- or waterborne pathogenic rotavirus. Anal. Biochem. 2011, 415, 87–96. [Google Scholar]
- Zhang, H.; Jenkins, G.; Zou, Y.; Zhu, Z.; Yang, C.J. Massively parallel single-molecule and single-cell emulsion reverse transcription polymerase chain reaction using Agarose droplet microfluidics. Anal. Chem. 2012, 84, 3599–3606. [Google Scholar]
- Luna-Vera, F.; Alvarez, J.C. Adsorption kinetics of proteins in plastic microfluidic channels: Real-time monitoring of lysozyme adsorption by pulsed streaming potentials. Biosens. Bioelectron. 2010, 25, 1539–1543. [Google Scholar] [CrossRef]
- Christensen, T.B.; Pedersen, C.M.; Gröndahl, K.G.; Jensen, T.G.; Sekulovic, A.; Bang, D.D.; Wolff, A. PCR biocompatibility of lab-on-a-chip and MEMS materials. J. Micromech. Microeng. 2007, 17, 1527–1532. [Google Scholar] [CrossRef]
- Erill, I.; Campoy, S.; Erill, N.; Barbe, J.; Aguilo, J. Biochemical analysis and optimization of inhibition and adsorption phenomena in glass-silicon PCR-chips. Sens. Actuator. B 2003, 96, 685–692. [Google Scholar] [CrossRef]
- Erill, I.; Campoy, S.; Rus, J.; Fonseca, L.; Ivorra, A.; Navarro, Z.; Plaza, J.A.; Aguilo, J.; Barbe, J. Development of a CMOS-compatible PCR chip: Comparison of design and system strategies. J. Micromech. Microeng. 2004, 14, 1558–1568. [Google Scholar] [CrossRef]
- Kricka, L.J.; Wilding, P. Microchip PCR. Anal. Bioanal. Chem. 2003, 377, 820–825. [Google Scholar] [CrossRef]
- Zhang, C.; Xing, D. Miniaturized PCR chips for nucleic acid amplification and analysis: Latest advances and future trends. Nucl. Acids Res. 2007, 35, 4223–4237. [Google Scholar] [CrossRef]
- Felbel, J.; Bieber, I.; Pipper, J.; Kohler, J.M. Investigations on the compatibility of chemically oxidized silicon (SiOx)-surfaces for applications towards chip-based polymerase chain reaction. Chem. Eng. J. 2004, 101, 333–338. [Google Scholar] [CrossRef]
- Panaro, N.J.; Lou, X.J.; Fortina, P.; Kricka, L.J.; Wilding, P. Surface effects on PCR reactions in multichip microfluidic platforms. Biomed. Microdevices. 2004, 6, 75–80. [Google Scholar] [CrossRef]
- Zanoli, L.M.; Licciardello, M.; D’Agata, R.; Lantano, C.; Calabretta, A.; Corradini, R.; Marchelli, R.; Spoto, G. Peptide nucleic acid molecular beacons for the detection of PCR amplicons in droplet-based microfluidic devices. Anal. Bioanal. Chem. 2012. [Google Scholar] [CrossRef]
- Christopher, G.F.; Anna, S.L. Microfluidic methods for generating continuous droplet streams. J. Phys. D Appl. Phys. 2007, 40, R319–R336. [Google Scholar] [CrossRef]
- Griffiths, A.D.; Tawfik, D.S. Miniaturising the laboratory in emulsion droplets. Trends Biotechnol. 2006, 24, 395–402. [Google Scholar] [CrossRef]
- Fair, R.B. Digital microfluidics: Is a true lab-on-a-chip possible? Microfluid. Nanofluid. 2007, 3, 245–281. [Google Scholar] [CrossRef]
- Zeng, Y.; Novak, R.; Shuga, J.; Smith, M.T.; Mathies, R.A. High-performance single cell genetic analysis using microfluidic emulsion generator arrays. Anal. Chem. 2010, 82, 3183–3190. [Google Scholar]
- Kiss, M.M.; Ortoleva-Donnelly, L.; Beer, N.R.; Warner, J.; Bailey, C.G.; Colston, B.W.; Rothberg, J.M.; Link, D.R.; Leamon, J.H. High-throughput quantitative polymerase chain reaction in picoliter droplets. Anal. Chem. 2008, 80, 8975–8981. [Google Scholar]
- Hua, Z.; Rouse, J.L.; Eckhardt, A.E.; Srinivasan, V.; Pamula, V.K.; Schell, W.A.; Benton, J.L.; Mitchell, T.G.; Pollack, M.G. Multiplexed real-time polymerase chain reaction on a digital microfluidic platform. Anal. Chem. 2010, 82, 2310–2316. [Google Scholar] [CrossRef]
- Zhu, Z.; Jenkins, G.; Zhang, W.; Zhang, M.; Guan, Z.; Yang, C.J. Single-molecule emulsion PCR in microfluidic droplets. Anal. Bioanal. Chem. 2012, 403, 2127–2143. [Google Scholar] [CrossRef]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28. [Google Scholar] [CrossRef]
- Curtis, K.A.; Rudolph, D.L.; Owen, S.M. Rapid detection of HIV-1 by reverse-transcription, loop-mediated isothermal amplification (RT-LAMP). J. Virol. Methods 2008, 151, 264–270. [Google Scholar] [CrossRef]
- Hong, T.C.; Mai, Q.L.; Cuong, D.V. Development and evaluation of a novel loop-mediated isothermal amplification method for rapid detection of severe acute respiratory syndrome coronavirus. J. Clin. Microbiol. 2004, 42, 1956–1961. [Google Scholar] [CrossRef]
- Misawa, Y.; Yoshida, A.; Saito, R.; Yoshida, H.; Okuzumi, K.; Ito, N.; Okada, M.; Moriya, K.; Koike, K. Application of loop-mediated isothermal amplifi cation technique to rapid and direct detection of methicillin-resistant Staphylococcus aureus (MRSA) in blood cultures. J. Infect. Chemother. 2007, 13, 134–140. [Google Scholar] [CrossRef]
- Ohtsuka, K.; Yanagawa, K.; Takatori, K.; Hara-Kudo, Y. Detection of Salmonella enterica in naturally contaminated liquid eggs by loop-mediated isothermal amplification, and characterization of salmonella isolates. Appl. Environ. Microbiol. 2005, 71, 6730–6735. [Google Scholar] [CrossRef]
- Liu, C.; Mauk, M.G.; Bau, H.H. A disposable, integrated loop-mediated isothermal amplification cassette with thermally actuated valves. Microfluid. Nanofluid. 2011, 11, 209–220. [Google Scholar] [CrossRef]
- Yoshida, A.; Nagashima, S.; Ansai, T.; Tachibana, M.; Kato, H.; Watari, H.; Notomi, T.; Takehara, T. Loop-mediated isothermal amplification method for rapid detection of the periodontopathic bacteria Porphyromonas gingivalis, Tannerella forsythia, and Treponema denticol. J. Clin. Microbiol. 2005, 43, 2418–2424. [Google Scholar] [CrossRef]
- Mori, Y.; Nagamine, K.; Tomita, N.; Notomi, T. Detection of loop-mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem. Biophys. Res. Commun. 2001, 289, 150–154. [Google Scholar] [CrossRef]
- Nakamura, N.; Fukuda, T.; Nonen, S.; Hashimoto, K.; Azuma, J.; Gemma, N. Simple and accurate determination of CYP2D6 gene copy number by a loop-mediated isothermal amplification method and an electrochemical DNA chip. Clin. Chim. Acta. 2010, 411, 568–573. [Google Scholar] [CrossRef]
- Iwamoto, T.; Sonobe, T.; Hayashi, K. Loop-mediated isothermal amplification for direct detection of mycobacterium tuberculosis complex, M. avium, and M. intracellulare in sputum samples. J. Clin. Microbiol. 2003, 41, 2616–2622. [Google Scholar] [CrossRef]
- Lee, S.Y.; Huang, J.G.; Chuang, T.L.; Sheu, J.C.; Chuang, Y.K.; Holl, M.; Meldrum, D.R.; Lee, C.N.; Lin, C.W. Compact optical diagnostic device for isothermal nucleic acids amplification. Sens. Actuator. B 2008, 133, 493–501. [Google Scholar] [CrossRef]
- Lee, S.Y.; Lee, C.N.; Holl, M.; Meldrum, D.R.; Lin, C.W. Efficient, specific, compact hepatitis B diagnostic device: Optical detection of the hepatitis B virus by isothermal amplification. Sens. Actuator. B 2007, 127, 598–605. [Google Scholar] [CrossRef]
- Hataoka, Y.; Zhang, L.H.; Mori, Y.; Tomita, N.; Notomi, T.; Baba, Y. Analysis of specific gene by integration of isothermal amplification and electrophoresis on poly(methyl methacrylate) microchips. Anal. Chem. 2004, 76, 3689–3693. [Google Scholar] [CrossRef]
- Fang, X.; Liu, Y.; Kong, J.; Jiang, X. Loop-mediated isothermal amplification integrated on microfluidic chips for point-of-care quantitative detection of pathogens. Anal. Chem. 2010, 82, 3002–3006. [Google Scholar] [CrossRef]
- Lam, L.; Sakakihara, S.; Ishizuka, K.; Takeuchi, S.; Arata, H.F.; Fujita, H.; Noji, H. Loop-mediated isothermal amplification of a single DNA molecule in polyacrylamide gel-based microchamber. Biomed. Microdevices 2008, 10, 539–546. [Google Scholar] [CrossRef]
- Safavieh, M.; Ahmed, M.U.; Tolba, M.; Zourob, M. Microfluidic electrochemical assay for rapid detection and quantification of Escherichia coli. Biosens. Bioelectron. 2012, 31, 523–528. [Google Scholar] [CrossRef]
- Nakamura, N.; Ito, K.; Takahashi, M.; Hashimoto, K.; Kawamoto, M.; Yamanaka, M.; Taniguchi, A.; Kamatani, N.; Gemma, N. Detection of six single-nucleotide polymorphisms associated with rheumatoid arthritis by a loop-mediated isothermal amplification method and an electrochemical DNA chip. Anal. Chem. 2007, 79, 9484–9493. [Google Scholar]
- Lee, T.M. Over-the-counter biosensors: Past, present, and future. Sensors 2008, 8, 5535–5559. [Google Scholar] [CrossRef]
- Abdel-Monem, M.; Hoffmann-Berling, H. Enzymic unwinding of DNA. 1: Purification and characterization of a DNA-dependent ATPase from Escherichia coli. Eur. J. Biochem. 1976, 65, 431–440. [Google Scholar] [CrossRef]
- Abdel-Monem, M.; Durwald, H.; Hoffmann-Berling, H. Enzymic unwinding of DNA. 2. Chain separation by an ATP-dependent DNA unwinding enzyme. Eur. J. Biochem. 1976, 65, 441–449. [Google Scholar] [CrossRef]
- Vincent, M.; Xu, Y.; Kong, H. Helicase-dependent isothermal DNA amplification. EMBO Rep. 2004, 5, 795–800. [Google Scholar] [CrossRef]
- Jeong, Y-J.; Park, K.; Kim, D-E. Isothermal DNA amplification in vitro: The helicase-dependent amplification system. Cell. Mol. Life Sci. 2009, 66, 3325–3336. [Google Scholar] [CrossRef]
- Tuteja, N.; Tuteja, R. Unraveling DNA helicases. Motif, structure, mechanism and function. Eur. J. Biochem. 2004, 271, 1849–1863. [Google Scholar] [CrossRef]
- Jankowsky, E. RNA helicases at work: Binding and rearranging. Trends Biochem. Sci. 2011, 36, 19–29. [Google Scholar] [CrossRef]
- Pyle, A.M. Translocation and unwinding mechanisms of RNA and DNA helicases. Annu. Rev. Biophys. 2008, 37, 317–336. [Google Scholar] [CrossRef]
- Hall, M.C.; Matson, S.W. Helicase motifs: The engine that powers DNA unwinding. Mol. Microbiol. 1999, 34, 867–877. [Google Scholar] [CrossRef]
- Runyon, G.T.; Lohman, T.M. Escherichia coli helicase II (uvrD) protein can completely unwind fully duplex linear and nicked circular DNA. J. Biol. Chem. 1989, 264, 17502–17512. [Google Scholar]
- Furukohri, A.; Nishikawa, Y.; Akiyama, M.T.; Maki, H. Interaction between Escherichia coli DNA polymerase IV and single-stranded DNA-binding protein is required for DNA synthesis on SSB-coated DNA. Nucleic Acids Res. 2012, 40, 6039–6048. [Google Scholar] [CrossRef]
- Shereda, R.D.; Kozlov, A.G.; Lohman, T.M.; Cox, M.M.; Keck, J.L. SSB as an organizer/mobilizer of genome maintenance complexes. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 289–318. [Google Scholar] [CrossRef]
- Mechanic, L.E.; Frankel, B.A.; Matson, S.W. Escherichia coli MutL loads DNA helicase II onto DNA. J. Biol. Chem. 2000, 275, 38337–38346. [Google Scholar] [CrossRef]
- Matson, S.W.; Robertson, A.B. The UvrD helicase and its modulation by the mismatch repair protein MutL. Nucleic Acids Res. 2006, 34, 4089–4097. [Google Scholar] [CrossRef]
- An, L.; Tang, W.; Ranalli, T.A.; Kim, H.J.; Wytiaz, J.; Kong, H. Characterization of a thermostable UvrD helicase and its participation in helicase-dependent amplification. J. Biol. Chem. 2005, 280, 28952–28958. [Google Scholar]
- Goldmeyer, J.; Kong, H.; Tang, W. Development of a novel one-tube isothermal reverse transcription thermophilic helicase-dependent amplification platform for rapid RNA detection. J. Mol. Diagn. 2007, 9, 639–644. [Google Scholar] [CrossRef]
- Tong, Y.; Tang, W.; Kim, H.J.; Pan, X.; Ranalli, T.; Kong, H. Development of isothermal TaqMan assays for detection of biothreat organisms. Biotechniques 2008, 45, 543–557. [Google Scholar] [CrossRef]
- Andresen, D.; von Nickisch-Rosenegk, M.; Bier, F.F. Helicase dependent on chip-amplification and its use in multiplex pathogen detection. Clin. Chim. Acta. 2009, 403, 244–248. [Google Scholar] [CrossRef]
- IsoAmp II tHDA Kit; Biohelix Corporation: Beverly, MA, USA. Available online: http://www.biohelix.com/ (accessed on 7 November 2012).
- Ramalingam, N.; San, T.C.; Kai, T.J.; Mak, M.Y.M.; Gong, H-Q. Microfluidic devices harboring unsealed reactors for real-time isothermal helicase-dependent amplification. Microfluid. Nanofluid. 2009, 7, 325–336. [Google Scholar] [CrossRef]
- Tong, Y.; Lemieux, B.; Kong, H. Multiple strategies to improve sensitivity, speed and robustness of isothermal nucleic acid amplification for rapid pathogen detection. BMC Biotechnol. 2011, 11. [Google Scholar] [CrossRef]
- Mahalanabis, M.; Do, J.; ALMuayad, H.; Zhang, J.Y.; Klapperich, C.M. An integrated disposable device for DNA extraction and helicase dependent amplification. Biomed. Microdevices 2010, 12, 353–359. [Google Scholar] [CrossRef]
- Baner, J.; Nilsson, M.; Mendel-Hartvig, M.; Landergren, U. Signal amplification of padlock probes by rolling circle replication. Nucl. Acids Res. 1998, 26, 5073–5078. [Google Scholar] [CrossRef]
- Mothershed, E.A.; Whitney, A.M. Nucleic acid-based methods for the detection of bacterial pathogens: Present and future considerations for the clinical laboratory. Clin. Chim. Acta. 2006, 363, 206–220. [Google Scholar] [CrossRef]
- Nilsson, M.; Malmgren, H.; Samiotaki, M.; Kwiatkowski, M.; Chowdhary, B.P.; Landegren, U. Padlock probes: Circularizing oligonucleotides for localized DNA detection. Science 1994, 265, 2085–2088. [Google Scholar]
- Lizardi, P.M.; Huang, X.H.; Zhu, Z.R.; Bray-Ward, P.; Thomas, D.C.; Ward, D.C. Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nat. Genet. 1998, 19, 225–232. [Google Scholar] [CrossRef]
- Jarvius, J.; Melin, J.; Göransson, J.; Stenberg, J.; Fredriksson, S.; Gonzalez-Rey, C.; Bertilsson, S.; Nilsson, M. Digital quantification using amplified single-molecule detection. Nat. Mater. 2006, 3, 725–727. [Google Scholar] [CrossRef]
- Melin, J.; Jarvius, J.; Gransson, J.; Nilsson, M. Homogeneous amplified single-molecule detection: Characterization of key parameters. Anal. Biochem. 2007, 368, 230–238. [Google Scholar] [CrossRef]
- Johne, R.; Mueller, H.; Rector, A.; van Ranst, M.; Stevens, H. Rolling-circle amplification of viral DNA genomes using phi29 polymerase. Trends Microbiol. 2009, 17, 205–211. [Google Scholar] [CrossRef]
- Hutchison, C.A., III; Smith, H.O.; Pfannkoch, C.; Venter, J.C. Cell-free cloning using phi29 DNA polymerase. Proc. Natl. Acad. Sci. 2005, 102, 17332–17336. [Google Scholar] [CrossRef]
- Mahmoudian, L.; Kaji, N.; Tokeshi, M.; Nilsson, M.; Baba, Y. Rolling circle amplification and circle-to-circle amplification of a specific gene integrated with electrophoretic analysis on a single chip. Anal. Chem. 2008, 80, 2483–2490. [Google Scholar] [CrossRef]
- Dahl, F.; Baner, J.; Gullberg, M.; Mendel-Hartvig, M.; Landegren, U.; Nilsson, M. Circle-to-circle amplification for precise and sensitive DNA analysis. Proc. Natl. Acad. Sci. USA 2004, 101, 4548–4553. [Google Scholar]
- Mahmoudian, L.; Melin, J.; Mohamadi, M.R.; Yamada, K.; Ohta, M.; Kaji, N.; Tokeshi, M.; Nilsson, M.; Baba, Y. Microchip electrophoresis for specific gene detection of the pathogenic bacteria V. cholerae by circle-to-circle amplification. Anal. Sci. 2008, 24, 327–332. [Google Scholar] [CrossRef]
- Mazutis, L.; Araghi, A.F.; Miller, O.J.; Baret, J.C.; Frenz, L.; Janoshazi, A.; Taly, V.; Miller, B.J.; Hutchison, J.B.; Link, D.; Griffiths, A.D.; Ryckelynck, M. Droplet-based microfluidic systems for high-throughput single dna molecule isothermal amplification and analysis. Anal. Chem. 2009, 81, 4813–4821. [Google Scholar]
- Juul, S.; Nielsen, C.J.; Labouriau, R.; Roy, A.; Tesauro, C.; Jensen, P.W.; Harmsen, C.; Kristoffersen, E.L.; Chiu, Y.L.; Frøhlich, R.; Fiorani, P.; Cox-Singh, J.; Tordrup, D.; Koch, J.; Bienvenu, A.L.; Desideri, A.; Picot, S.; Petersen, E.; Leong, K.W.; Ho, Y.P.; Stougaard, M.; Knudsen, B.R. Droplet microfluidics platform for highly sensitive and quantitative detection of malaria-causing plasmodium parasites based on enzyme activity measurement. ACS Nano 2012. [Google Scholar] [CrossRef]
- Dean, F.B.; Hosono, S.; Fang, L.; Wu, X.; Faruqi, A.F.; Bray-Ward, P.; Sun, Z.; Zong, Q.; Du, Y.; Du, J.; Driscoll, M.; Song, W.; Kingsmore, S.F.; Egholm, M.; Lasken, R.S. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. 2002, 99, 5261–5266. [Google Scholar]
- Blanco, L.; Bernad, A.; Lazaro, J.M.; Martin, G.; Garmendia, C.; Salas, M. Highly efficient DNA synthesis by the phage phi 29 DNA polymerase. Symmetrical mode of DNA replication. J. Biol. Chem. 1989, 264, 8935–8940. [Google Scholar]
- Nelson, J.R.; Cai, Y.C.; Giesler, T.L.; Farchaus, J.W.; Sundaram, S.T.; Ortiz-Rivera, M.; Hosta, L.P.; Hewitt, P.L.; Mamone, J.A.; Palaniappan, C.; Fuller, C.W. TempliPhi, phi29 DNA polymerase based rolling circle amplification of templates for DNA sequencing. Biotechniques 2002, 32, S44–S47. [Google Scholar]
- Ling, L.L.; Keohavong, P.; Dias, C.; Thilly, W.G. Optimization of the polymerase chain reaction with regard to fidelity: Modified T7, Taq, and vent DNA polymerases. Genome Res. 1991, 1, 63–69. [Google Scholar] [CrossRef]
- Keohavong, P.; Thilly, W.G. Fidelity of DNA polymerases in DNA amplification. Proc. Natl. Acad. Sci. 1989, 86, 9253–9257. [Google Scholar] [CrossRef]
- Hosono, S.; Faruqi, A.F.; Dean, F.B.; Du, Y.; Sun, Z.; Wu, X.; Du, J.; Kingsmore, S.F.; Egholm, M.; Lasken, R.S. Unbiased whole genome amplification directly from clinical samples. Genome Res. 2003, 13, 954–964. [Google Scholar] [CrossRef]
- Raghunathan, A.; Ferguson, H.R., Jr.; Bornarth, C.J.; Song, W.; Driscoll, M.; Lasken, R.S. Genomic DNA amplification from a single bacterium. Appl. Environ. Microbiol. 2005, 71, 3342–3347. [Google Scholar] [CrossRef]
- Marcy, Y.; Ishoey, T.; Lasken, R.S.; Stockwell, T.B.; Walenz, B.P.; Halpern, A.L.; Beeson, K.Y.; Goldberg, S.M.D.; Quake, S.R. Nanoliter reactors improve multiple displacement amplification of genomes from single cells. PloS Genet. 2007, 3. [Google Scholar] [CrossRef]
- Zhang, C.; Xing, D. Single-Molecule DNA Amplification and analysis using microfluidics. Chem. Rev. 2010, 110, 4910–4947. [Google Scholar] [CrossRef]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA detection using recombination proteins. PloS Biol. 2006, 4. [Google Scholar] [CrossRef]
- Lutz, S.; Weber, P.; Focke, M.; Faltin, B.; Hoffmann, J.; Müller, C.; Mark, D.; Roth, G.; Munday, P.; Armes, N.; Piepenburg, O.; Zengerle, R.; von Stetten, F. Microfluidic lab-on-a-foil for nucleic acid analysis based on isothermal recombinase polymerase amplification (RPA). Lab Chip 2010, 10, 887–893. [Google Scholar]
- Hakenberg, S.; Hügle, M.; Weidmann, M.; Hufert, F.; Dame, G.; Urban, G.A. A phaseguided passive batch microfluidic mixing chamber for isothermal amplification. Lab Chip 2012, 12, 4576–4580. [Google Scholar] [CrossRef]
- Vulto, P.; Podszun, S.; Meyer, P.; Hermann, C.; Manz, A.; Urban, G.A. Phaseguides: A paradigm shift in microfluidic priming and emptying. Lab Chip 2011, 11, 1596–1602. [Google Scholar] [CrossRef]
- Paul, N.; Shum, J.; Le, T. Hot start PCR. Methods Mol. Biol. 2010, 630, 301–318. [Google Scholar] [CrossRef]
- Shen, F.; Davydova, E.K.; Du, W.; Kreutz, J.E.; Piepenburg, O.; Ismagilov, R.F. Digital isothermal quantification of nucleic acids via simultaneous chemical initiation of recombinase polymerase amplification reactions on SlipChip. Anal. Chem. 2011, 83, 3533–3540. [Google Scholar] [CrossRef]
- Compton, J. Nucleic acid sequence-based amplification. Nature 1991, 350, 91–92. [Google Scholar] [CrossRef]
- Deiman, B.; van Aarle, P.; Sillekens, P. Characteristics and applications of nucleic acid sequence-based amplification (NASBA). Mol. Biotechnol. 2002, 20, 163–179. [Google Scholar] [CrossRef]
- Yates, S.; Penning, M.; Goudsmit, J.; Frantzen, I.; van de Weijer, B.; van Strijp, D.; van Gemen, B. Quantitative detection of hepatitis B virus DNA by real-time nucleic acid sequence-based amplification with molecular beacon detection. J. Clin. Microbiol. 2001, 39, 3656–3665. [Google Scholar] [CrossRef]
- van Gemen, B.; van Beuningen, R.; Nabbe, A.; van Strijp, D.; Jurriaans, S.; Lens, P.; Kievits, T. A one-tube quantitative HIV-1 RNA NASBA nucleic acid amplification assay using electrochemiluminiscent (ECL) labelled probes. J. Virol. Methods 1994, 49, 157–168. [Google Scholar] [CrossRef]
- Shan, S.; Ko, L.S.; Collins, R.A.; Wu, Z.; Chen, J.; Chan, K.Y.; Xing, J.; Lau, L.T.; Yu, A.C. Comparison of nucleic acid-based detection of avian influenza H5N1 with virus isolation. Biochem. Biophys. Res. Commun. 2003, 302, 377–383. [Google Scholar] [CrossRef]
- Connelly, J.T.; Nugen, S.R.; Borejsza-Wysocki, W.; Durst, R.A.; Montagna, R.A.; Baeumner, A.J. Human pathogenic Cryptosporidium species bioanalytical detection method with single oocyst detection capability. Anal. Bioanal. Chem. 2008, 391, 487–495. [Google Scholar] [CrossRef]
- Nugen, S.R.; Asiello, P.J.; Connelly, J.T.; Baeumner, A.J. PMMA biosensor for nucleic acids with integrated mixer and electrochemical detection. Biosens. Bioelectron. 2009, 24, 2428–2433. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Jin, J.; Wang, H.; Tang, H.; Yang, R.; Wang, K. Strategy for molecular beacon binding readout: Separating molecular recognition element and signal reporter. Anal. Chem. 2009, 81, 9703–9709. [Google Scholar] [CrossRef]
- Wang, K.; Tang, Z.; Yang, C.J.; Kim, Y.; Fang, X.; Li, W.; Wu, Y.; Medley, C.D.; Cao, Z.; Li, J.; Colon, P.; Lin, H.; Tan, W. Molecular engineering of DNA: Molecular beacons. Angew. Chem. Int. Edit. 2009, 48, 856–870. [Google Scholar] [CrossRef]
- Gore, H.M.; Wakeman, C.A.; Hull, R.M.; McKillip, J.L. Real-time molecular beacon NASBA reveals hblc expression from Bacillus spp. in milk. Biochem. Biophys. Res. Commun. 2003, 311, 386–390. [Google Scholar] [CrossRef]
- Nadal, A.; Coll, A.; Cook, N.; Pla, M. A molecular beacon-based real time NASBA assay for detection of Listeria monocytogenes in food products: Role of target mRNA secondary structure on NASBA design. J. Microbiol. Methods 2007, 68, 623–632. [Google Scholar] [CrossRef]
- Gulliksen, A.; Solli, L.; Karlsen, F.; Rogne, H.; Hovig, E.; Nordstrom, T.; Sirevag, R. Real-time nucleic acid sequence-based amplification in nanoliter volumes. Anal. Chem. 2004, 76, 9–14. [Google Scholar] [CrossRef]
- Gulliksen, A.; Solli, L.A.; Drese, K.S.; Sörensen, O.; Karlsen, F.; Rogne, H.; Hovig, E.; Sirevåg, R. Parallel nanoliter detection of cancer markers using polymer microchips. Lab Chip 2005, 5, 416–420. [Google Scholar] [CrossRef]
- Gulliksen, A.; Keegan, H.; Martin, C.; O’Leary, J.; Solli, L.A.; Falang, I.M.; Grønn, P.; Karlgård, A.; Mielnik, M.M.; Johansen, I.-R.; Tofteberg, T.R.; Baier, T.; Gransee, R.; Drese, K.; Hansen-Hagge, T.; Riegger, L.; Koltay, P.; Zengerle, R.; Karlsen, F.; Ausen, D.; Furuberg, L. Towards a “sample-in, answer-out” point-of-care platformfor nucleic acid extraction and amplification: Using an HPV E6/E7mRNAModel System. J. Oncol. 2012. [Google Scholar] [CrossRef]
- Dimov, I.K.; Garcia-Cordero, J.L.; O’Grady, J.; Poulsen, C.R.; Viguier, C.; Kent, L.; Daly, P.; Lincoln, B.; Maher, M.; O’Kennedy, R.; Smith, T.J.; Ricco, A.J.; Lee, L.P. Integrated microfluidic tmRNA purification and real-time NASBA device for molecular diagnostics. Lab Chip 2008, 8, 2071–2078. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Dong, T.; Yang, Z.; Pires, N.; Hoivik, N. Compatible immuno-NASBA LOC device for quantitative detection of waterborne pathogens: Design and validation. Lab Chip 2012, 12, 602–612. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zanoli, L.M.; Spoto, G. Isothermal Amplification Methods for the Detection of Nucleic Acids in Microfluidic Devices. Biosensors 2013, 3, 18-43. https://doi.org/10.3390/bios3010018
Zanoli LM, Spoto G. Isothermal Amplification Methods for the Detection of Nucleic Acids in Microfluidic Devices. Biosensors. 2013; 3(1):18-43. https://doi.org/10.3390/bios3010018
Chicago/Turabian StyleZanoli, Laura Maria, and Giuseppe Spoto. 2013. "Isothermal Amplification Methods for the Detection of Nucleic Acids in Microfluidic Devices" Biosensors 3, no. 1: 18-43. https://doi.org/10.3390/bios3010018