A Comparative Study of Impedance versus Optical Label-Free Systems Relative to Labelled Assays in a Predominantly Gi Coupled GPCR (C5aR) Signalling

Abstract
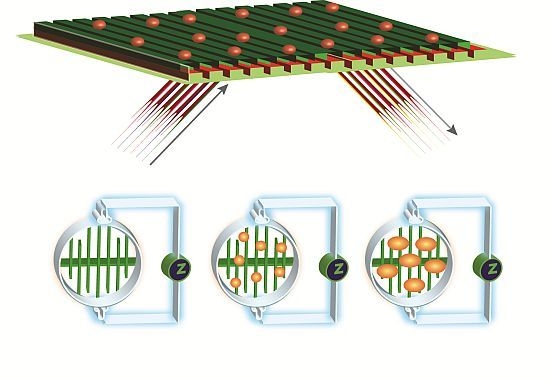
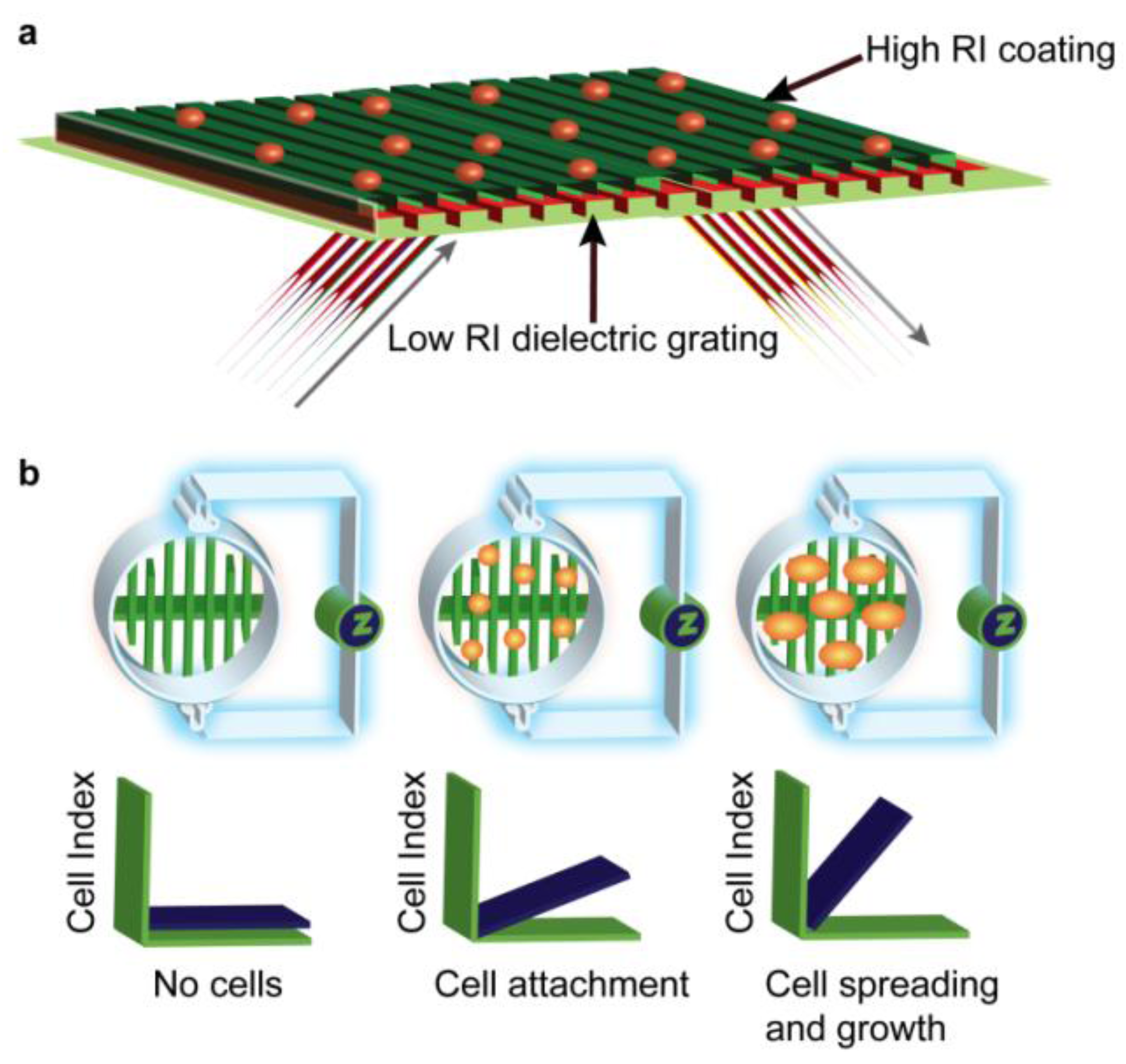
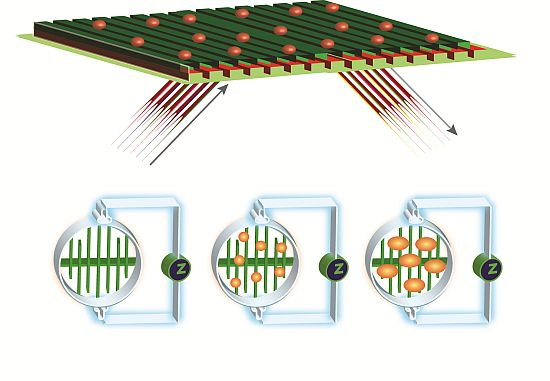
:
1. Introduction
2. Experimental Section
2.1. Ligands
2.2. Cell Culture
2.3. Membrane Preparation
2.4. Receptor Binding Studies
2.5. ERK Phosphorylation Assay
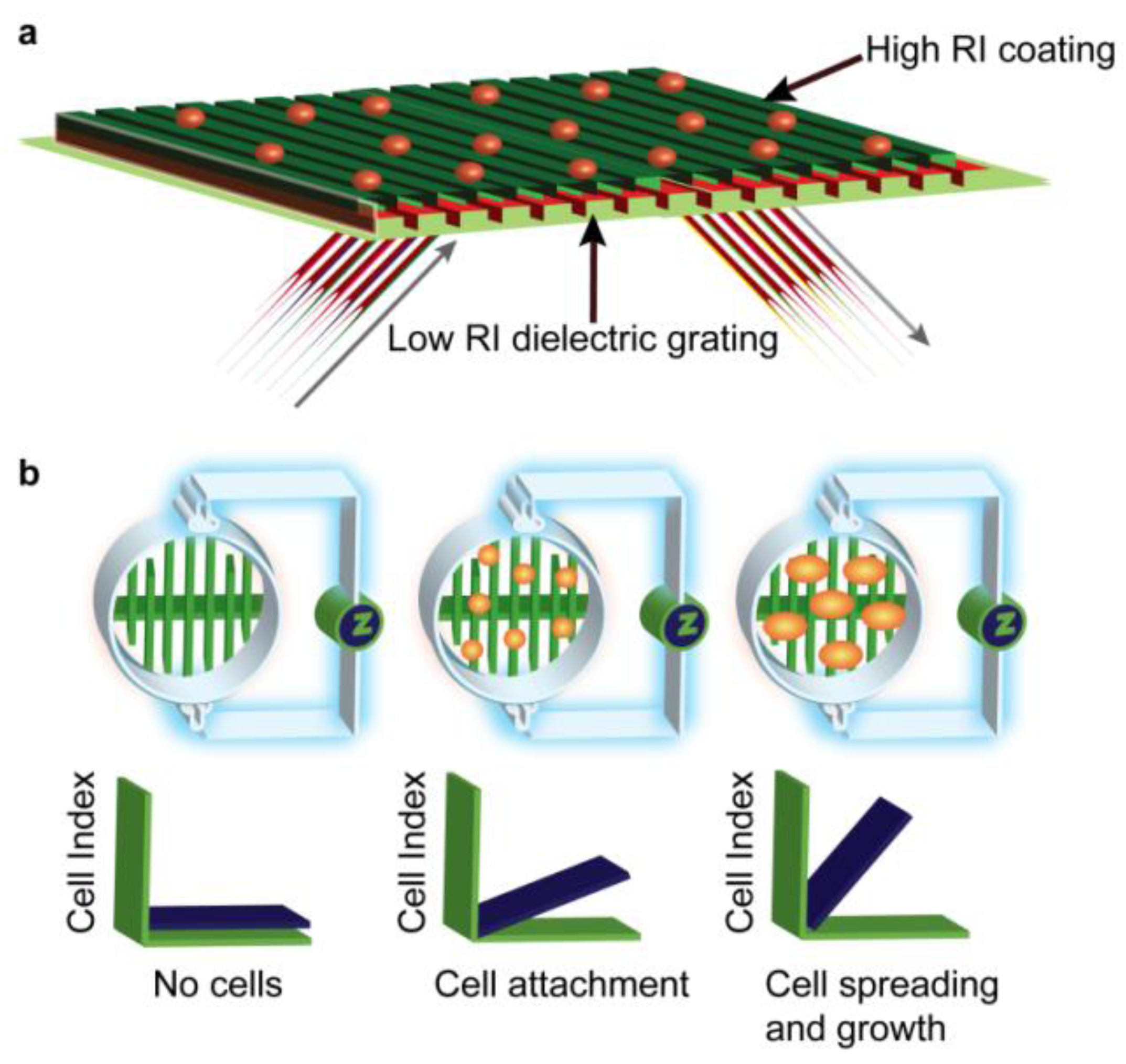
2.6. EPIC® Label-Free Optical System (Corning)
2.7. xCELLigence HT Label-Free Impedance System (Roche)
3. Results and Discussion
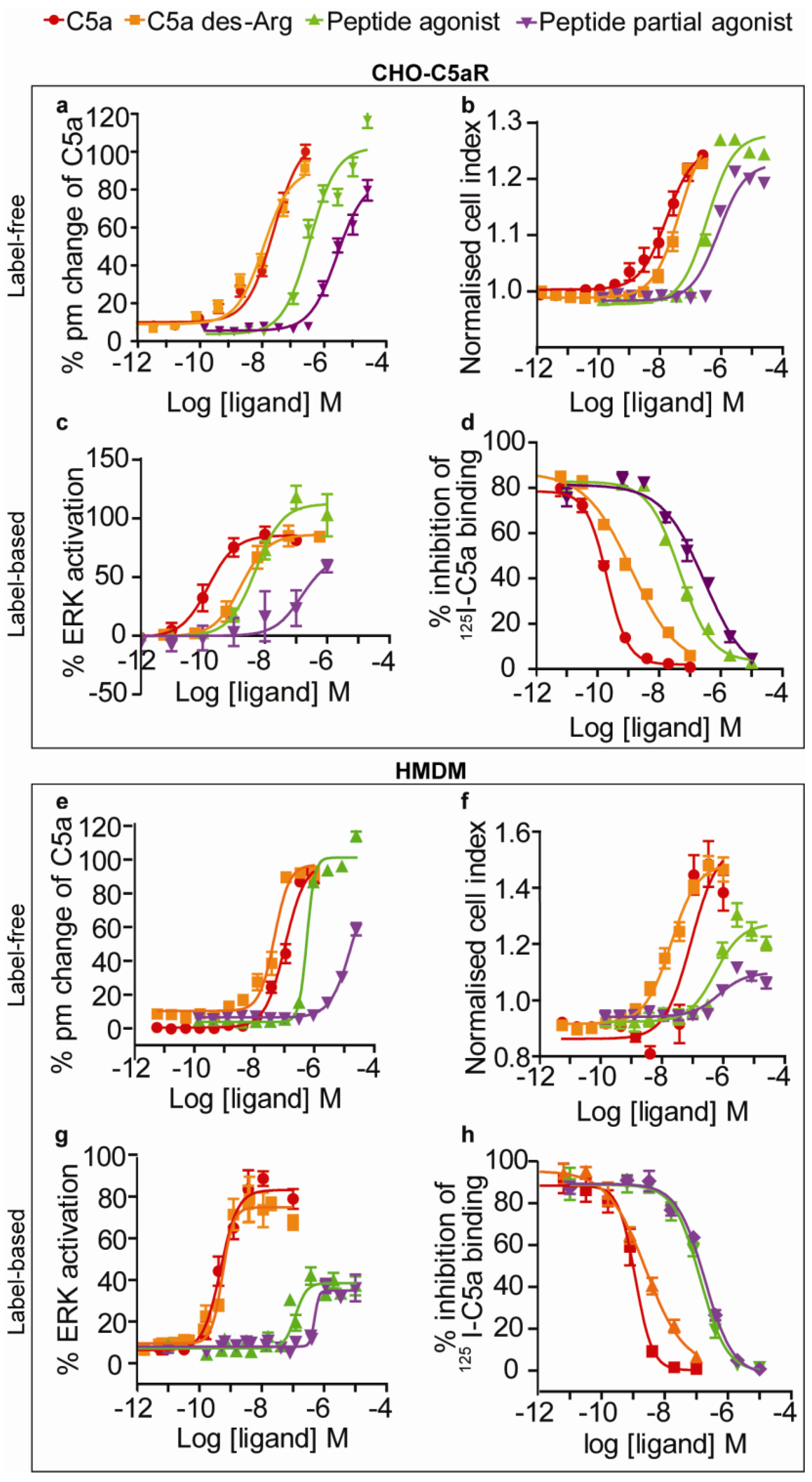
3.1. Receptor Binding Assay

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
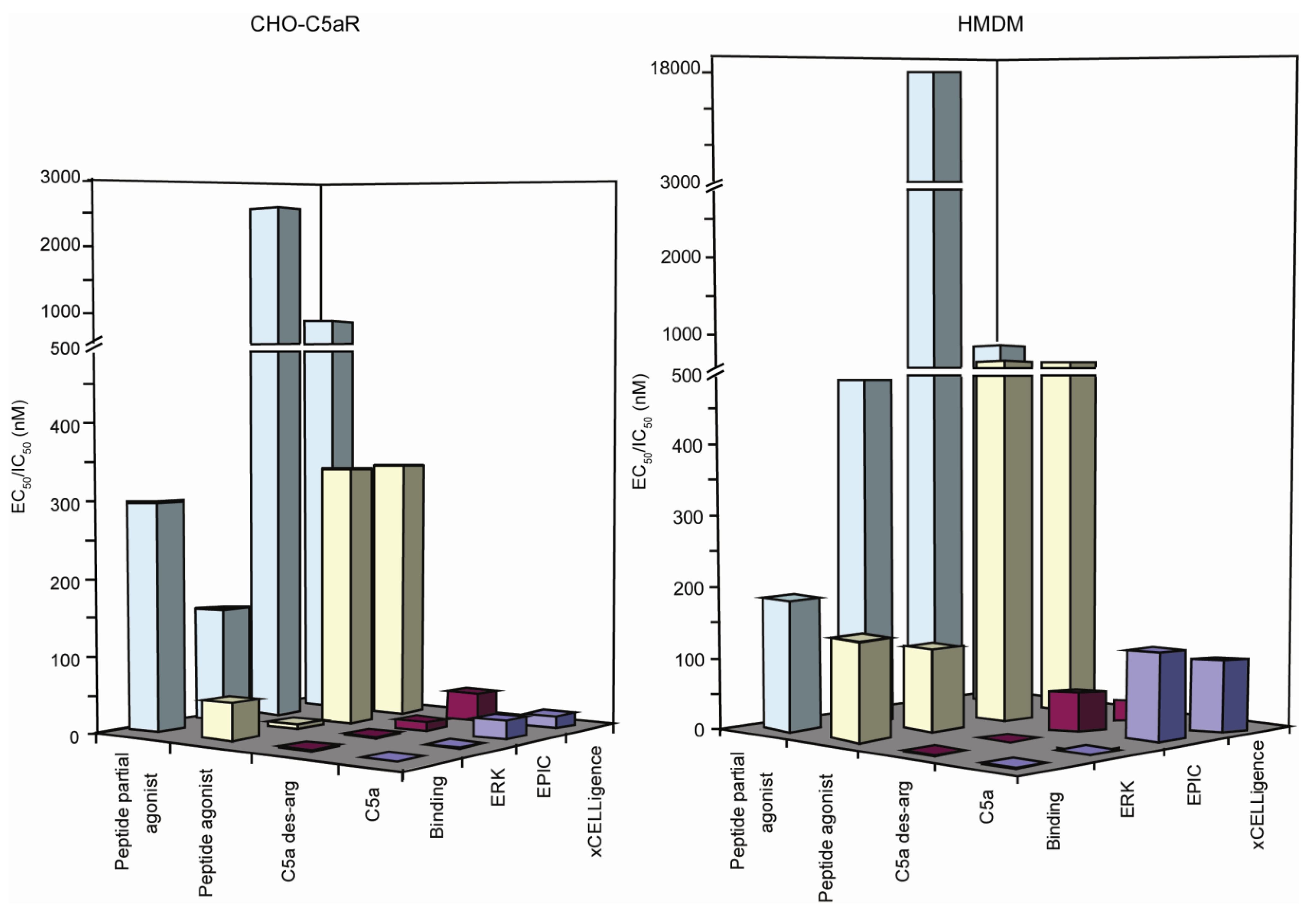
| Binding (IC50) | ERK (EC50) | EPIC® (EC50) | xCELLigence (EC50) | |
|---|---|---|---|---|
| C5a | 1.2 (1–1.4) | 0.47 (0.37–0.59) | 107 (89–129) | 89 (47–169) |
| C5a des-Arg | 2.5 (1.9–3.4) | 0.55 (0.42–0.72) | 47 (38–56) | 20 (15–27) |
| Peptide agonist | 132 (111–157) | 120 (86–167) | 563 (526–603) | 546 (293–1,016) |
| Peptide partial agonist | 185 (141–242) | 492 (227–1,068) | 17380 (6,856–44,070) | 709 (304–1,652) |
| Binding (IC50) | ERK (EC50) | EPIC® (EC50) | xCELLigence (EC50) | |
|---|---|---|---|---|
| C5a | 0.2 (0.17–0.25) | 0.15 (0.08–0.3) | 24 (19–31) | 16 (10–26) |
| C5a des-Arg | 1.23 (0.78–1.95) | 2 (1–3) | 11 (9–15) | 44 (34–57) |
| Peptide agonist | 47 (36–60) | 5 (2–11) | 347 (262–461) | 351 (292–421) |
| Peptide partial agonist | 295 (147–594) | 151 (16–1,382) | 2626 (2,013–3,425) | 819 (674–996) |

3.2. ERK Phosphorylation Assay
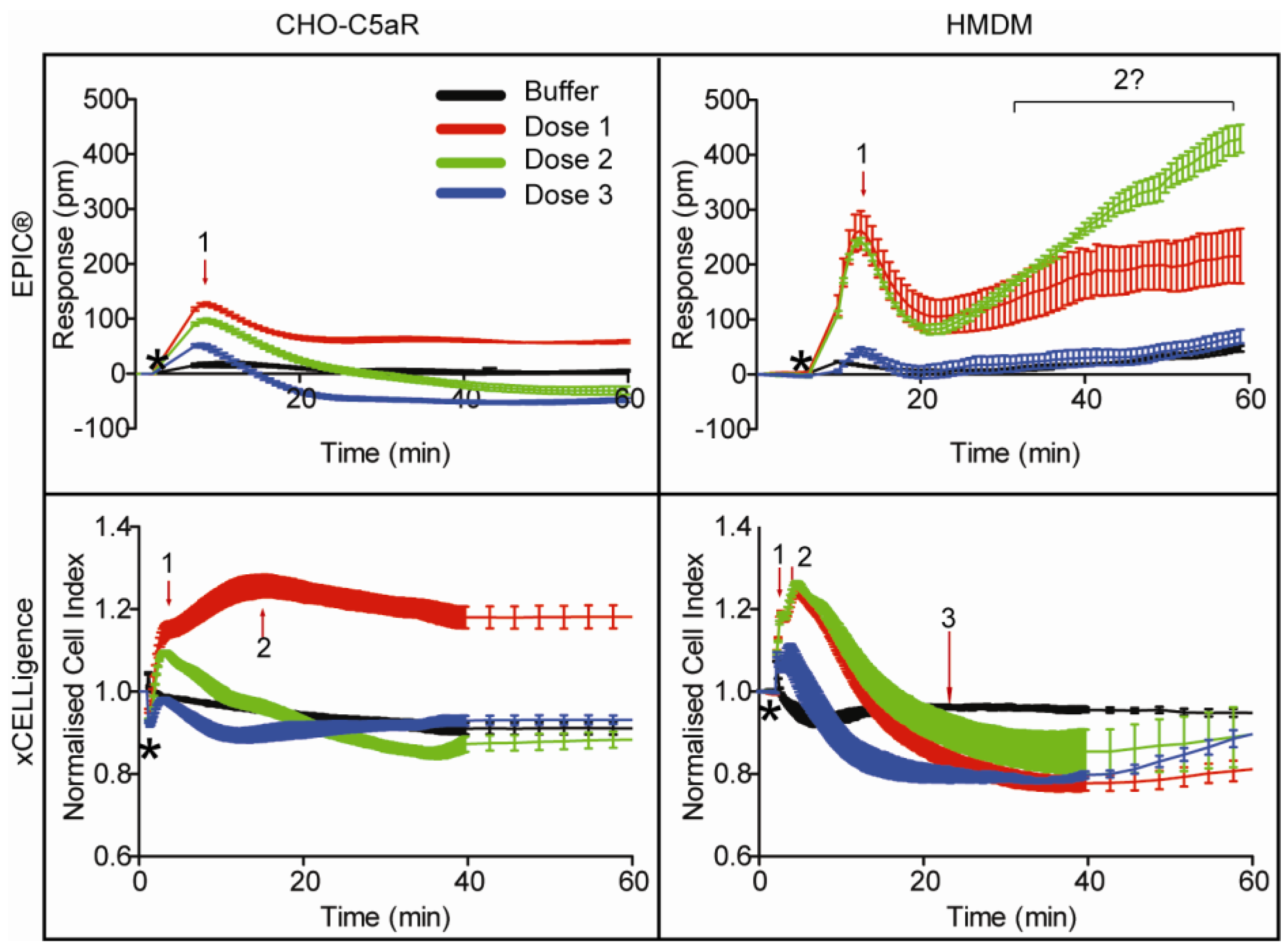
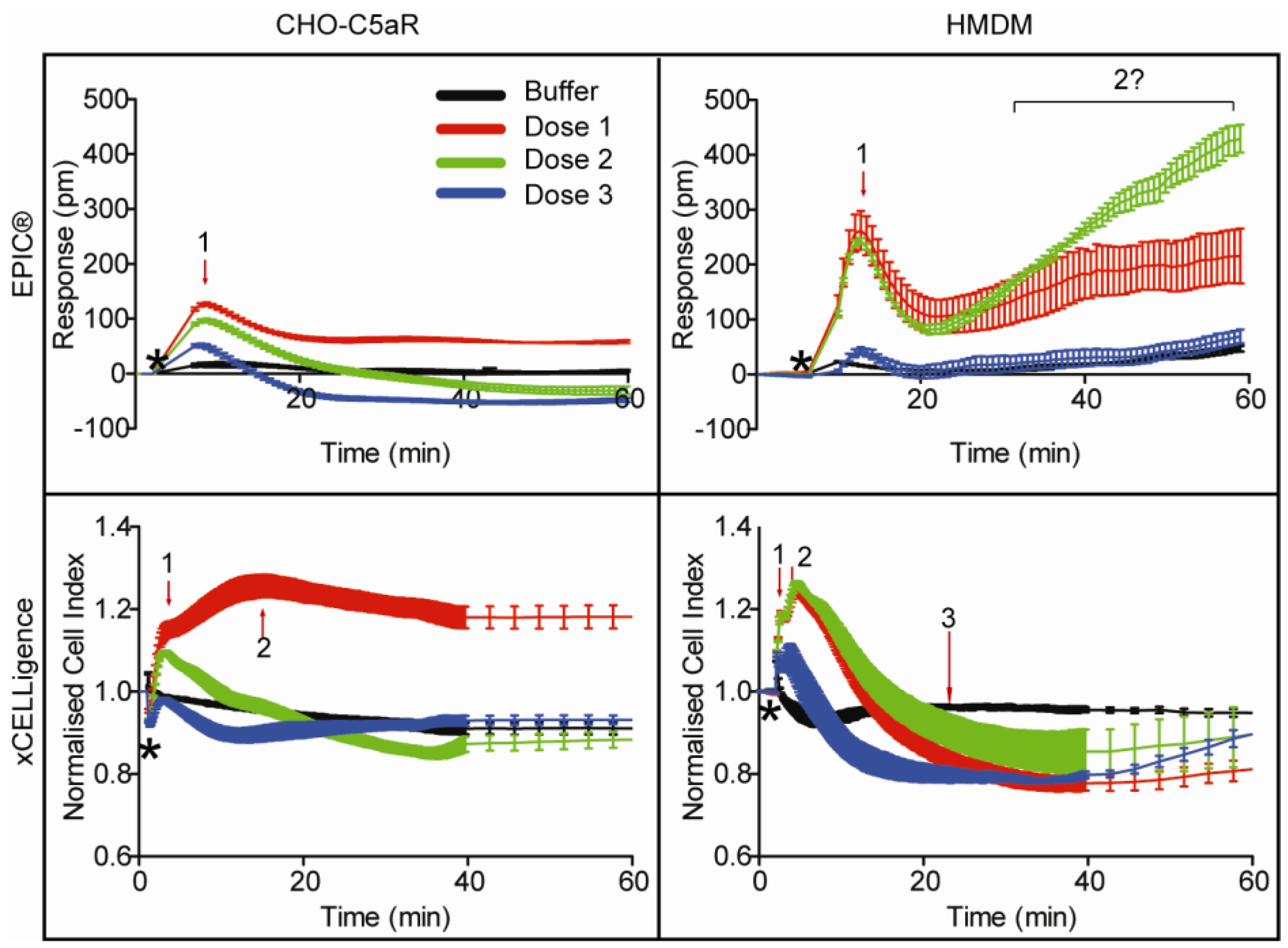
3.3. EPIC® Label-Free Optical System
3.4. xCELLigence Impedance Label-Free System
3.5. Discussion

4. Conclusions
Acknowledgments
References
- Jacoby, E.; Bouhelal, R.; Gerspacher, M.; Seuwen, K. The 7TM G-Protein-coupled receptor target family. Chem. Med. Chem. 2006, 1, 760–782. [Google Scholar]
- Heilker, R.; Wolff, M.; Tautermann, C.S.; Bieler, M. G-Protein-coupled receptor-focused drug discovery using a target class platform approach. Drug Discov. Today 2009, 14, 231–240. [Google Scholar]
- Zwier, J.M.; Roux, T.; Cottet, M.; Durroux, T.; Douzon, S.; Bdioui, S.; Gregor, N.; Bourrier, E.; Oueslati, N.; Nicolas, L.; et al. A fluorescent ligand-binding alternative using Tag-lite® technology. J. Biomol. Screen. 2010, 15, 1248–1259. [Google Scholar] [CrossRef]
- Chambers, C.; Smith, F.; Williams, C.; Marcos, S.; Liu, Z.H.; Hayter, P.; Ciaramella, G.; Keighley, W.; Gribbon, P.; Sewing, A. Measuring intracellular calcium fluxes in high throughput mode. Comb. Chem. High Throughput Screen. 2003, 6, 355–362. [Google Scholar]
- Thomsen, W.; Frazer, J.; Unett, D. Functional assays for screening GPCR targets. Curr. Opin. Biotechnol. 2005, 16, 655–665. [Google Scholar]
- Taouji, S.; Dahan, S.; Bosse, R.; Chevet, E. Current screens based on the AlphaScreen technology for deciphering cell signalling pathways. Curr. Genomics 2009, 10, 93–101. [Google Scholar] [CrossRef]
- Cooper, M.A. Biosensor profiling of molecular interactions in pharmacology. Curr. Opin. Pharmacol. 2003, 3, 557–562. [Google Scholar] [CrossRef]
- Xi, B.; Yu, N.; Wang, X.; Xu, X.; Abassi, Y. The application of cell-based label-free technology in drug discovery. Biotechnol. J. 2008, 3, 484–495. [Google Scholar]
- Scott, C.W.; Peters, M.F. Label-free whole-cell assays: Expanding the scope of GPCR screening. Drug Discov. Today 2010, 15, 704–716. [Google Scholar] [CrossRef]
- Fang, Y. Label-free receptor assays. Drug Discov. Today Technol. 2010, 7, 5–11. [Google Scholar] [CrossRef]
- Shamah, S.M.; Cunningham, B.T. Label-free cell-based assays using photonic crystal optical biosensors. Analyst 2011, 136, 1090–1102. [Google Scholar]
- Jerman, J.C.; Brown, J.; Rocheville, M. Cellular screening for 7TM receptors using label-free detection. In Label-Free Technologies: For Drug Discovery; Cooper, M.A., Mayr, L.M., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2011; pp. 205–222. [Google Scholar]
- Nirschl, M.; Reuter, F.; Voros, J. Review of transducer principles for label-free biomolecular interaction analysis. Biosensors 2011, 1, 70–92. [Google Scholar]
- Li, G.; Ferrie, A.M.; Fang, Y. Label-free profiling of ligands for endogenous GPCRs using a cell-based high-throughput screening technology. JALA 2006, 11, 181–187. [Google Scholar]
- Fang, Y. Label-free cell-based assays with optical biosensors in drug discovery. Assay Drug Dev. Technol. 2006, 4, 583–595. [Google Scholar] [CrossRef]
- Cooper, M.A. Optical biosensors: Where next and how soon? Drug Discov. Today 2006, 11, 1061–1067. [Google Scholar]
- Fang, Y.; Fang, J.; Tran, E.; Xinying, X.; Hallstrom, M.; Frutos, A.G. High-throughput analysis of biomolecular interactions and cellular responses with resonant waveguide grating biosensors. In Label-Free Biosensors: Techniques and Applications; Cooper, M.A., Ed.; Cambridge University Press: Cambridge, UK, 2009; pp. 206–222. [Google Scholar]
- Daniels, J.S.; Pourmand, N. Label-free impedance biosensors: Opportunities and challenges. Electroanalysis 2007, 19, 1239–1257. [Google Scholar] [CrossRef]
- Abassi, Y.A.; Sieler, A.; Watzele, M.; Wang, X.; Xu, X. Dynamic and label-free cell-based assays using the xCELLigence system. In Label-Free Technologies: For Drug Discovery; Cooper, M.A., Mayr, L.M., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2011; pp. 71–81. [Google Scholar]
- Giaever, I.; Keese, C.R. Use of electric fields to monitor the dynamical aspect of cell behavior in tissue culture. IEEE Trans. Biomed. Eng. 1986, 33, 242–247. [Google Scholar]
- Giaever, I.; Keese, C.R. A morphological biosensor for mammalian cells. Nature 1993, 366, 591–592. [Google Scholar]
- Solly, K.; Wang, X.; Xu, X.; Strulovici, B.; Zheng, W. Application of real-time cell electronic sensing (RT-CES) technology to cell-based assays. Assay Drug Dev. Technol. 2004, 2, 363–372. [Google Scholar] [CrossRef]
- McGuinness, R.; Verdonk, E. Electrical impedance technology applied to cell-based assays. In Label-Free Biosensors: Techniques and Applications; Cooper, M.A., Ed.; Cambridge University Press: Cambridge, UK, 2009; pp. 251–277. [Google Scholar]
- Head, B.P.; Patel, H.H.; Roth, D.M.; Murray, F.; Swaney, J.S.; Niesman, I.R.; Farquhar, M.G.; Insel, P.A. Microtubules and actin microfilaments regulate lipid raft/caveolae localization of adenylyl cyclase signaling components. J. Biol. Chem. 2006, 281, 26391–26399. [Google Scholar]
- Szaszi, K.; Kurashima, K.; Kaibuchi, K.; Grinstein, S.; Orlowski, J. Role of the cytoskeleton in mediating cAMP-dependent protein kinase inhibition of the epithelial Na+/H+ exchanger NHE3. J. Biol. Chem. 2001, 276, 40761–40768. [Google Scholar]
- Monk, P.N.; Scola, A.M.; Madala, P.; Fairlie, D.P. Function, structure and therapeutic potential of complement C5a receptors. Br. J. Pharmacol. 2007, 152, 429–448. [Google Scholar]
- Ward, P.A. Role of C5 activation products in sepsis. Sci. World J. 2010, 10, 2395–2402. [Google Scholar] [CrossRef]
- Marc, M.M.; Kristan, S.S.; Rozman, A.; Kern, I.; Flezar, M.; Kosnik, M.; Korosec, P. Complement factor C5a in acute exacerbation of chronic obstructive pulmonary disease. Scand. J. Immunol. 2010, 71, 386–391. [Google Scholar] [CrossRef]
- Manthey, H.D.; Thomas, A.C.; Shiels, I.A.; Zernecke, A.; Woodruff, T.M.; Rolfe, B.; Taylor, S.M. Complement C5a inhibition reduces atherosclerosis in ApoE-/- mice. FASEB J. 2010, 25, 2447–2455. [Google Scholar]
- Yancey, K.B.; Lawley, T.J.; Dersookian, M.; Harvath, L. Analysis of the interaction of human C5a and C5a des Arg with human monocytes and neutrophils: Flow cytometric and chemotaxis studies. J. Invest. Dermatol. 1989, 92, 184–189. [Google Scholar]
- Chenoweth, D.E.; Hugli, T.E. Demonstration of specific C5a receptor on intact human polymorphonuclear leukocytes. Proc. Natl. Acad. Sci. USA 1978, 75, 3943–3947. [Google Scholar] [CrossRef]
- Haviland, D.; McCoy, R.; Whitehead, W.; Akama, H.; Molmenti, E.; Brown, A.; Haviland, J.; Parks, W.; Perlmutter, D.; Wetsel, R. Cellular expression of the C5a anaphylatoxin receptor (C5aR): Demonstration of C5aR on nonmyeloid cells of the liver and lung. J. Immunol. 1995, 154, 1861–1869. [Google Scholar]
- Larsen, G.L.; McCarthy, K.; Webster, R.O.; Henson, J.; Henson, P.M. A differential effect of C5a and C5a des Arg in the induction of pulmonary inflammation. Am. J. Pathol. 1980, 100, 179–192. [Google Scholar]
- Konteatis, Z.; Siciliano, S.; van Riper, G.; Molineaux, C.; Pandya, S.; Fischer, P.; Rosen, H.; Mumford, R.; Springer, M. Development of C5a receptor antagonists. Differential loss of functional responses. J. Immunol. 1994, 153, 4200–4205. [Google Scholar]
- Halili, M.A.; Andrews, M.R.; Labzin, L.I.; Schroder, K.; Matthias, G.; Cao, C.; Lovelace, E.; Reid, R.C.; Le, G.T.; Hume, D.A.; et al. Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. J. Leukoc. Biol. 2010, 87, 1103–1114. [Google Scholar]
- Dutertre, S.; Croker, D.; Daly, N.L.; Andersson, A.; Muttenthaler, M.; Lumsden, N.G.; Craik, D.J.; Alewood, P.F.; Guillon, G.; Lewis, R.J. Conopressin-T from Conus tulipa reveals an antagonist switch in Vasopressin-like peptides. J. Biol. Chem. 2008, 283, 7100–7108. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar]
- Alouani, S. Scintillation proximity binding assay. In Chemokine Protocols; Proudfoot, A.E.I., Wells, T.N.C., Power, C.A., Eds.; Humana Press: Totowa, NJ, USA, 2000; Volume 138, pp. 135–141. [Google Scholar]
- Eglen, R.M.; Reisine, T.; Roby, P.; Rouleau, N.; Illy, C.; Bosse, R.; Bielefeld, M. The use of AlphaScreen technology in HTS: Current status. Curr. Chem. Genomics 2008, 1, 2–10. [Google Scholar]
- Klempner, M.S.; Gallin, J.I.; Balow, J.E.; van Kammen, D.P. The effect of hemodialysis and C5a des Arg on neutrophil subpopulations. Blood 1980, 55, 777–783. [Google Scholar]
- Gerard, C.; Chenoweth, D.E.; Hugli, T.E. Response of human neutrophils to C5a: A role for the oligosaccharide moiety of human C5a des Arg-74 but not of C5a in biologic activity. J. Immunol. 1981, 127, 1978–1982. [Google Scholar]
- Marder, S.R.; Chenoweth, D.E.; Goldstein, I.M.; Perez, H.D. Chemotactic responses of human peripheral blood monocytes to the complement-derived peptides C5a and C5a des Arg. J. Immunol. 1985, 134, 3325–3331. [Google Scholar]
- Barker, M.D.; Jose, P.J.; Williams, T.J.; Burton, D.R. The chemoattractant des-Arg74-C5a regulates the expression of its own receptor on a monocyte-like cell line. Biochem. J. 1986, 236, 621–624. [Google Scholar]
- Senior, R.M.; Griffin, G.L.; Perez, H.D.; Webster, R.O. Human C5a and C5a des Arg exhibit chemotactic activity for fibroblasts. J. Immunol. 1988, 141, 3570–3574. [Google Scholar]
- Bürgi, B.; Brunner, T.; Dahinden, C.A. The degradation product of the C5a anaphylatoxin C5a des Arg retains basophil-activating properties. Eur. J. Immunol. 1994, 24, 1583–1589. [Google Scholar]
- Werfel, T.; Oppermann, M.; Butterfield, J.H.; Begemann, G.; Elsner, J.; Gotze, O.; Zwirner, J. The human mast cell line HMC-1 expresses C5a receptors and responds to C5a but not to C5a(desArg). Scand. J. Immunol. 1996, 44, 30–36. [Google Scholar]
- Crass, T.; Bautsch, W.; Cain, S.A.; Pease, J.E.; Monk, P.N. Receptor activation by human C5a des Arg74 but not intact C5a is dependent on an interaction between Glu199 of the receptor and Lys68 of the ligand. Biochemistry 1999, 38, 9712–9717. [Google Scholar]
- Krasel, C.; Vilardaga, J.P.; Bunemann, M.; Lohse, M.J. Kinetics of G-protein-coupled receptor signalling and desensitization. Biochem. Soc. Trans. 2004, 32, 1029–1031. [Google Scholar]
- Sheth, B.; Banks, P.; Burton, D.R.; Monk, P.N. The regulation of actin polymerization in differentiating U937 cells correlates with increased membrane levels of the pertussis-toxin-sensitive G-protein Gi2. Biochem. J. 1991, 275, 809–811. [Google Scholar]
- Skokowa, J.; Ali, S.R.; Felda, O.; Kumar, V.; Konrad, S.; Shushakova, N.; Schmidt, R.E.; Piekorz, R.P.; Nurnberg, B.; Spicher, K.; et al. Macrophages induce the inflammatory response in the pulmonary arthus reaction through Gαi2 activation that controls C5aR and Fc receptor cooperation. J. Immunol. 2005, 174, 3041–3050. [Google Scholar]
- Oksjoki, R.; Laine, P.; Helske, S.; Vehmaan-Kreula, P.; Mäyränpää, M.I.; Gasque, P.; Kovanen, P.T.; Pentikäinen, M.O. Receptors for the anaphylatoxins C3a and C5a are expressed in human atherosclerotic coronary plaques. Atherosclerosis 2007, 195, 90–99. [Google Scholar]
- Koch, W.J.; Hawes, B.E.; Allen, L.F.; Lefkowitz, R.J. Direct evidence that Gi-coupled receptor stimulation of mitogen-activated protein kinase is mediated by G beta gamma activation of p21ras. Proc. Natl. Acad. Sci. USA 1994, 91, 12706–12710. [Google Scholar]
- Mochizuki, N.; Ohba, Y.; Kiyokawa, E.; Kurata, T.; Murakami, T.; Ozaki, T.; Kitabatake, A.; Nagashima, K.; Matsuda, M. Activation of the ERK/MAPK pathway by an isoform of rap1GAP associated with Gαi. Nature 1999, 400, 891–894. [Google Scholar]
- Watson, J.; Collin, L.; Ho, M.; Riley, G.; Scott, C.; Selkirk, J.V.; Price, G.W. 5-HT(1A) receptor agonist-antagonist binding affinity difference as a measure of intrinsic activity in recombinant and native tissue systems. Br. J. Pharmacol. 2000, 130, 1108–1114. [Google Scholar]
- Shapira, M.; Vogel, Z.; Sarne, Y. Opioid and cannabinoid receptors share a common pool of GTP-binding proteins in cotransfected cells, but not in cells which endogenously coexpress the receptors. Cell Mol. Neurobiol. 2000, 20, 291–304. [Google Scholar]
- Prinster, S.C.; Hague, C.; Hall, R.A. Heterodimerization of G protein-coupled receptors: Specificity and functional significance. Pharmacol. Rev. 2005, 57, 289–298. [Google Scholar]
- Okinaga, S.; Slattery, D.; Humbles, A.; Zsengeller, Z.; Morteau, O.; Kinrade, M.B.; Brodbeck, R.M.; Krause, J.E.; Choe, H.-R.; Gerard, N.P.; et al. C5L2, a nonsignaling C5a binding protein. Biochemistry 2003, 42, 9406–9415. [Google Scholar]
- Fang, Y.; Ferrie, A.M.; Fontaine, N.H.; Yuen, P.K. Characteristics of dynamic mass redistribution of epidermal growth factor receptor signaling in living cells measured with label-free optical biosensors. Anal. Chem. 2005, 77, 5720–5725. [Google Scholar]
- Fang, Y. Non-invasive optical biosensor for probing cell signaling. Sensors 2007, 7, 2316–2329. [Google Scholar]
- Schroder, R.; Janssen, N.; Schmidt, J.; Kebig, A.; Merten, N.; Hennen, S.; Muller, A.; Blattermann, S.; Mohr-Andra, M.; Zahn, S.; et al. Deconvolution of complex G protein-coupled receptor signaling in live cells using dynamic mass redistribution measurements. Nat. Biotechnol. 2010, 28, 943–949. [Google Scholar]
- Dodgson, K.; Gedge, L.; Murray, D.C.; Coldwell, M. A 100K well screen for a muscarinic receptor using the Epic label-free system-a reflection on the benefits of the label-free approach to screening seven-transmembrane receptors. J. Recept. Signal Transduct. Res. 2009, 29, 163–172. [Google Scholar]
- Schuster, D.; Laggner, C.; Langer, T. Why drugs fail-a study on side effects in new chemical entitie. Curr. Pharm. Des. 2005, 11, 3545–3559. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Halai, R.; Croker, D.E.; Suen, J.Y.; Fairlie, D.P.; Cooper, M.A. A Comparative Study of Impedance versus Optical Label-Free Systems Relative to Labelled Assays in a Predominantly Gi Coupled GPCR (C5aR) Signalling. Biosensors 2012, 2, 273-290. https://doi.org/10.3390/bios2030273
Halai R, Croker DE, Suen JY, Fairlie DP, Cooper MA. A Comparative Study of Impedance versus Optical Label-Free Systems Relative to Labelled Assays in a Predominantly Gi Coupled GPCR (C5aR) Signalling. Biosensors. 2012; 2(3):273-290. https://doi.org/10.3390/bios2030273
Chicago/Turabian StyleHalai, Reena, Daniel E. Croker, Jacky Y. Suen, David P. Fairlie, and Matthew A. Cooper. 2012. "A Comparative Study of Impedance versus Optical Label-Free Systems Relative to Labelled Assays in a Predominantly Gi Coupled GPCR (C5aR) Signalling" Biosensors 2, no. 3: 273-290. https://doi.org/10.3390/bios2030273