New Insight on Hydrogen Evolution Reaction Activity of MoP2 from Theoretical Perspective


1
Institute of Atomic and Molecular Physics, Sichuan University, Chengdu610065, China
2
School of Chemical Engineering, Sichuan University, Chengdu 610065, China
3
School of Aeronautics and Astronautics, Sichuan University, Chengdu 610065, China
*
Author to whom correspondence should be addressed.
Nanomaterials 2019, 9(9), 1270; https://doi.org/10.3390/nano9091270
Submission received: 22 July 2019
/
Revised: 31 August 2019
/
Accepted: 4 September 2019
/
Published: 5 September 2019
(This article belongs to the Special Issue Computational Materials Design for Renewable Energy Applications)
Abstract
:We systematically investigated the hydrogen evolution reaction (HER) of six facets of based on the periodic density functional theory (DFT). The calculated values of Gibbs free energy of hydrogen adsorption () indicated that the (111) facet has a good HER activity for a large range of hydrogen coverages. The zigzagged patterns before 75% hydrogen coverage suggest a facilitation among Mo1, P1 and Mo2 sites, which are attributed to repeat occupancy sites of H atoms. From ab initial atomistic thermodynamics analysis of hydrogen coverage, we gained that the most stable coverage of hydrogen is 18.75% at 1 atm and 298 K. Finally, the doping effects on HER activity were investigated and found that catalytic performance can be improved by substituting P with an S or N atom, as well as substituting the Mo atom with an Fe atom, respectively. We hope this work can provide new insights on further understanding of HER for and give instructions for the experimental design and synthesis of transition metal phosphides (TMPs)-based high-performance catalysts.
1. Introduction
Hydrogen has been regarded as a promising energy carrier and future sustainable high energy fuel, due to its properties of non-pollution, energy conversion and gravimetric energy density [1,2]. The energetic efficiency, environmental sustainability and carbon-free alterative of hydrogen generations come from the interconversion between water and hydrogen. Electrolytic water-splitting [3] is one of the most reliable and effective methods to produce hydrogen in various hydrogen productions, which usually involves the hydrogen evolution reaction (HER). Therefore, it is very important to explore a stable, low cost, economically and environmentally friendly and high efficiency HER catalyst for realizing large-scale industrial electrocatalytic hydrogen production [4].
Platinum and its alloys are considered to be the best active electrocatalyst to facilitate HER in acid condition because of its small overpotential and accelerating reaction rates [5], which can lead to more efficient water-splitting [6]. However, the high cost and low abundance of platinum limits its widespread application in practice [7,8]. Thus, it is highly necessary to explore earth-abundant and non-noble-metal HER electrocatalysts to substitute for platinum. Recently, many promising HER electrocatalysts have been unveiled, such as anatase [9], metal dioxides [10], carbides [11], selenides [12], sulfides [13], phosphides [14] and nitrides [15]. Among potential materials considered thus far, the family of transition metal phosphides (TMPs) has drawn a wide range of interests as a promising alternative catalyst for HER due to its high catalytic activities [16,17]. For instance, TMPs, including W-P [18,19], Fe-P [20,21], Ni-P [22,23] and Mo-P [24,25], are well known for their high catalytic activity and durability. Experimentally, many approaches were used to synthesize TMPs, such as the solvothermal method [26], hypophosphite thermal decomposition [27,28], high vacuum solid-state reaction method [28], metal-organic framework phosphatized method [29] and so on. Recently, researchers found TMPs with higher phosphorous component holds a higher catalytic activity for HER [29,30,31].
As a typical TMP with higher phosphorous component, great stability and good electronical conductivity, has attracted tremendous attention from academia [32,33,34]. Experimentally, a facile two-step strategy was used to fabricate nanoparticle films on a metal Mo plate showing superior HER catalytic activity at all pH values [35]. Through a phosphidation reaction based on an nanosheet array on carbon cloth, an nanosheet was fabricated by Zhu et al. [36]. They found that the nanosheet delivers superior catalytic activity and stability with an overpotential as low as 58 mV to drive a catalytic current density of 10 mA cm−2. In addition, Wu et al. [28] synthesized nanoparticles with a high vacuum solid-state reaction method and carried out electrochemical tests. It was revealed nanoparticles show superior HER performance with small overpotentials of 38 mV and low Tafel slopes of 52 mV dev−1. More recently, Gao et al. [37] synthesized nanosheets using nanosheets as a precursor by high temperature solid state reaction with phosphorus under vacuum conditions. It was found that nanosheets exhibit very promising HER activity, considerably better than bulk and non-exfoliated . To our best knowledge, though there are many exciting experimental findings, insights into for the systematical mechanism of hydrogen absorptions are lacking, which is of critical importance for further enhancing the activities and stabilities of HER electrocatalysts.
Gibbs free energy of hydrogen adsorption is usually used as a descriptor to predict HER activity of the electrocatalyst [38]. Nørskov et al. [39,40] reported that the maximum of HER activity is obtainable with the value of closing to zero. More negative values of mean the binding between hydrogen atom and facet is stronger, which is impedimentary for desorption of . On the contrary, more positive values of stand for the weaker blinding between hydrogen atom and facet. The step of proton/electron-transfer can be hindered with the positive value. Therefore, the best-performing and ideal electrocatalyst for HER should have a value of close to zero.
To figure out HER mechanisms of , the adsorption structures and energetics of atomic hydrogen have been systematically calculated using periodic density functional theory (DFT). HER activities of the surface were predicted by the value of . Ab initio atomistic thermodynamics was employed to determine the most stable phase at 1 atm pressure and 298 K. Bader analysis [41] and different charge density [42] analysis were employed to further understand the interaction between Mo and P atoms. Finally, the doping effects on HER activity were investigated by substituting P with an S or N atom, as well as Mo atom with an Fe, Cu, Cr, Co, Mn or Ni atom, respectively. We hope our results can shed new light on further understanding of HER for and designing transition phosphide HER electrocatalysts.
2. Computational Methods
The spin-polarized periodic DFT calculations were performed by the plane-wave basis set with the projector augmented plan wave (PAW) method for treating core and valence electronics [43,44]. Perdew-Burke-Ernzerh (PBE) functional with generalized gradient approximation (GGA) [45] was carried to deal with the electronic exchange-correlation interaction. All calculations were implemented in the Vienna Ab initio Simulation Package (VASP) [46]. The plane-wave cutoff energy was tested using a well-known variable-controlling approach and set to 400 eV. The Gaussian smearing method was employed to represent the total energy with a smearing width of 0.02 eV. The low Miller-index facets were modeled by cleaved with the optimized structure of bulk MoP2, with a four-atom-layer 1 × 2 × 1 supercell for (100) facet and four-atom-layer 2 × 1 × 1 supercells for (111), (110), (101), (001) and (011) facets. To avoid the interaction among the adjacent images, 15 Å of vacuum along the perpendicular catalyst facets was added. The Monkhorst-Pack method with a centered k-point mesh of 3 × 3 × 1 was used to sample the Brillouin zone. The top half of the layers together with the added hydrogen atoms was relaxed during the geometry optimization in the whole calculations. During the structure optimization, the residual forces have converged to less than 0.025eV Å−1 and the total energy converged to less than 1.0×10−6 eV. To obtain a deep understanding of HER, the partial atomic charges were investigated using Bader charge analysis developed by Henkelman et al. [41]
The surface energies are determined as:
where and are the total energy of the surface slab and the bulk, respectively. N is the number of formula unit in the slab and A is the facet area of the optimized slab. The differential hydrogen adsorption energy is defined as:
where and are the total energy of the system with n and (n-1) hydrogen atoms adsorbed on the surface.
is the total energy of molecule in the gas phase. A negative value of suggests beneficial absorption. The differential Gibbs free energy of the adsorbed hydrogen atom is calculated by:
here, is the adsorption energy of the hydrogen atom determined by Equation (2). and stand for the zero-point energy correction and entropy change between adsorbed hydrogen and hydrogen under standard conditions at the temperature of . can be obtained by ; here, is the entropy of an isolated hydrogen molecular in gas phase at standard condition. The value of is approximately equal to −0.2 eV. is defined as:
where , and are the zero-point energy of n and (n-1) hydrogen atoms adsorbed on the surface, as well as a gas phase hydrogen molecular, respectively. The represents the zero-point energy of hydrogen molecule in gas phase. The zero-point energy can be calculated by:
where h represents the Plank constant and is the vibrational frequency. The calculated value of the vibrational frequency is 4301 cm−1 for a single hydrogen molecular, which is consistent with the experimental finding of 4395 cm−1 [47]. The calculated values of vibrational frequencies are 1744.53 cm−1, 608.71 cm−1 and 548.19 cm−1 for hydrogen adsorbed on the (111) facet. Using Equation (4) and Equation (5), the values of 0.04 eV for was obtained for the (111) facet. Thus, the expression of can be represented as for the (111) facet.
To further identity the most stable phase, the Ab initio atomistic thermodynamics [48] was employed to determine at a specific temperature and pressure denoted by , which can be obtained by:
where is the total energy of the system with the adsorbed hydrogen atoms on the surface, is the total energy of the clean facet, is the total energy of molecule in gas phase and is the hydrogen chemical potential at a given temperature and pressure. Clearly, is a function of for a surface at a concrete hydrogen coverage. based on the specific condition can be determined by:
here, is the hydrogen chemical potential at the standard pressure and is Boltzmann constant.
To investigate the kinetic mechanism of HER, the activation energies were calculated based on the transition theory. The activation energy barrier () and reaction energy ( of the elemental reaction are obtained by the following formulas:
where , and represent the total energy of initial state, transition state and final state, respectively.
3. Results and Discussion
3.1. Electronic Structures of Bulk
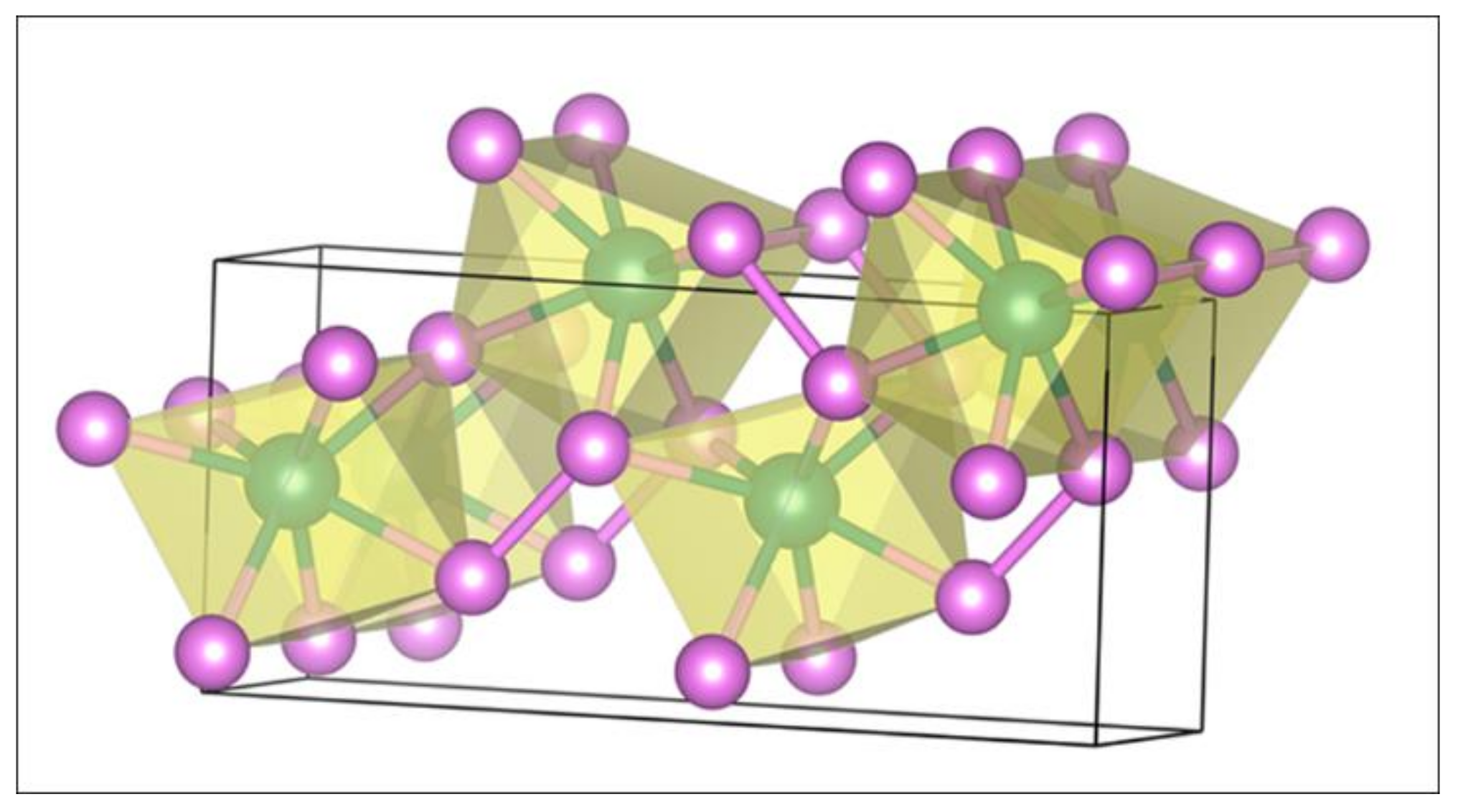
Figure 1 shows the bulk , which has an orthorhombic structure with a space group of Cmc21 (36). A Mo atom is seven-coordinated by six P atoms at ends of the prism and one P atom at outside of the rectangular facet of the prism, which shows five ring-pore structure and the zigzagging layered with a relatively large free volume. Table 1 lists the optimized lattice parameters that are in good agreement with the available experimental findings [28,49] and recent theoretical results [50]. The maximum error is less than 0.06 Å between the calculated and experimental findings.
In surface science studies, priority will be given to low Miller-index facets due to their high durability. For orthorhombic , the low Miller-index facets can be cleaved to (100), (110), (111), (011), (101) and (001) examples. The optimized structures of these facets are plotted in Figure S1 and corresponding surface energies are summarized in Table 2. Generally, the most stable facet has the lowest surface energy and can be synthesized in experiments [51,52,53,54]. From computing surface energy, it can be found the thermodynamic stability of the clean facets follows the order of (111) > (110) > (101) > (011) > (100) > (001). Thus, the most stable facet is (111), which agrees with the recent experimental finding where the (111) facet has been synthesized [54]. In addition, surface packing densities (SPD) are usually used to identify the stability. A larger value of SPD means that it is more stable. Table 2 also lists the calculated data of SPD for each facet. Likewise, the (111) facet is recognized as the most stable one. Therefore, the (111) facet is further examined.
To further understand the electronic structure of the (111) facet, Figure 2 sketches the calculated total and project electronic density of states (TDOS and PDOS, respectively) data. Obviously, the facet shows metallic behavior with none-zero TDOS data at the Fermi level. It is advantageous for good conductivity which is beneficial for HER activity. The main contributions of TDOS are from Mo-d orbitals.
3.2. Hydrogen Adsorption
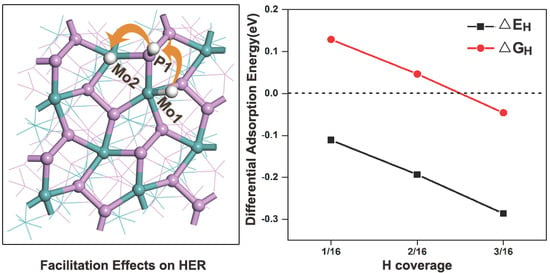
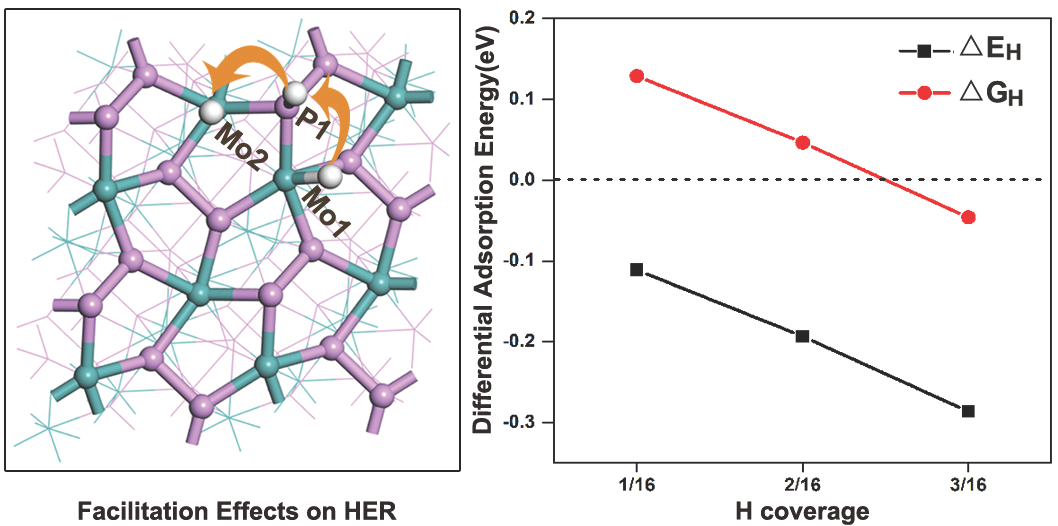
To investigate HER activity, all possible adsorption sites on the (111) facet were carefully tested. It was revealed that there are six favorable adsorption sites located at Mo1, P1, bridge (Mo1-P3), Mo2, bridge (Mo1-P2) and P2, as shown in Figure S2. The absorption energies at each site were listed in Table S1, which follows the trend: Mo1 < P1 < bridge (Mo1-P3) < Mo2 < bridge (Mo1-P2) < P2. With the assumption that hydrogen atoms easily spread over the facet at the lowest energy sites for each type of hydrogen coverage, we depicted the global minimum of hydrogen adsorption for each type of hydrogen coverage on the (111) facet. Figure 3 shows the optimized structures with different hydrogen coverages (here, 100% coverage means adsorbed 16 H atoms on the (111) facet). It is usually thought hydrogen coverage should first occupy all sites with the lowest adsorption energies, i.e., all Mo1 sites on the (111) facet. Strangely, when the first H atom takes over one Mo1 site on the (111) facet, the second H atom do not go to the other Mo1 site. It occupies one P1 site and the third H atom occupies an adjacent Mo2 site, as shown in Figure 3a. Subsequently, occupancy sites of H atoms will repeat the procedure until reaching 75% coverage. For the last 75–100% coverage, additional H atoms prefer to adsorb on the bridge (Mo1-P3). Interestingly, there are no H atoms adsorbed on bridge (Mo1-P2) and P2 sites during the processes of hydrogen coverage. To further understand these phenomena, the adoption energies and charge density differences were investigated.
Figure 4 describes the calculated values of and for H adsorption on the (111) facet with different hydrogen coverages. The absolute values of are less than 0.2 eV, which is a favorable value for HER. Therefore, it is indicated that the (111) facet can favor HER over a large hydrogen coverage range. The zigzagged patterns before 75% hydrogen coverage suggest a synergy among Mo1, P1 and Mo2 sites, which are attributed to repeating occupancy sites of H atoms.
To obtain a deep understanding, herein, the charge density difference induced by the adsorbed H atoms on the (111) facet were analyzed. The charge density differences for H atoms adsorbed on the surface are defined by , where , and are the charge densities of the catalytic surface adsorbed H atoms system, the clean surface and H atoms, respectively.
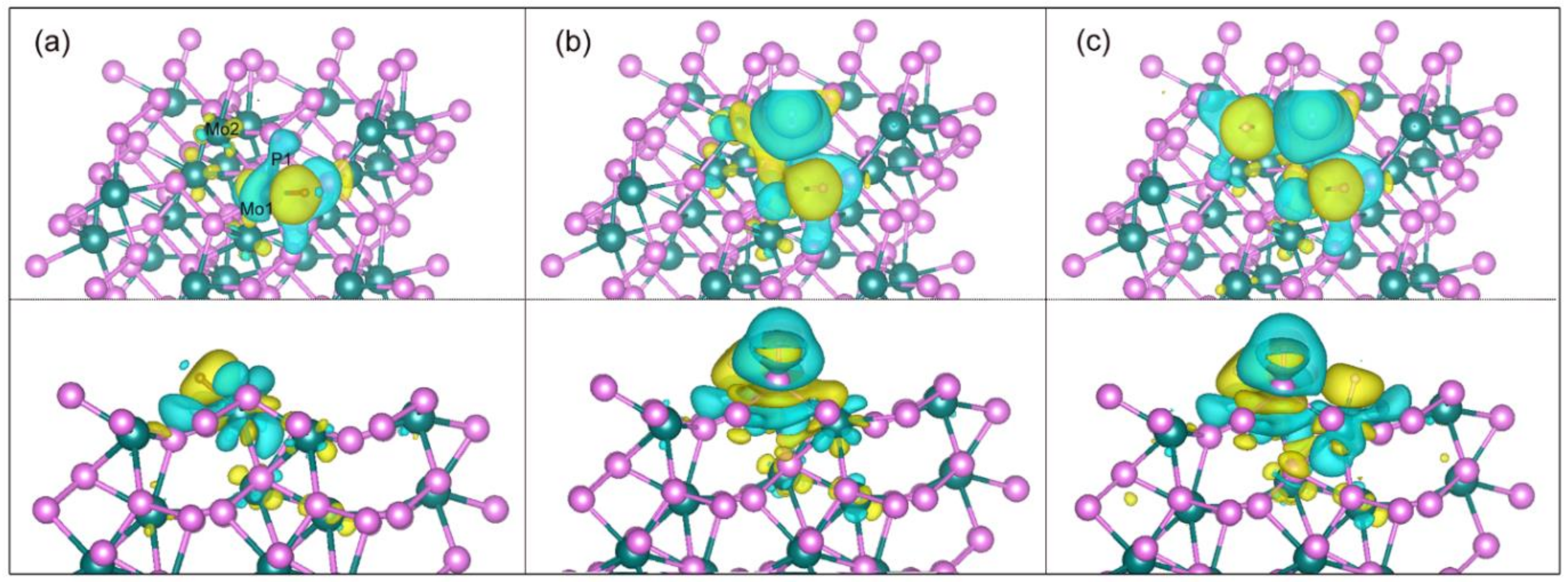
Figure 5 shows the charge density differences for the system made up of the (111) facet and the adsorbed H atoms with H coverage of 1/16, 2/16 and 3/16. For 1/16 H coverage at Mo1 site, there are significant electron accumulation (yellow) and depletion (light blue) around the adsorbed H atom and the connected Mo atom, respectively. The election transfer mainly exists between H and Mo atoms, indicating a strong interaction between them. However, no electron depletion is found around the H atom, revealing that many electrons transferred from the Mo to the H atom. Interesting, a small amount of the electron depletion and accumulation are found around Mo1-P1 and P1-Mo2 bonds, respectively, which predict that the bond length of Mo1-P1 will increase, while that of P1-Mo2 will decrease compared with those in a clean facet, agreeing with the analysis of bond lengths from the geometry optimization in Table 3. Meanwhile, the small electron depletion of P atom at P1 site and accumulation of Mo atom at Mo2 site are in line with that from the Bader analysis in Table 3. Small amounts of charge density differences on the P1 site due to the occupation of the Mo1 site will affect the adsorption energy on the P1 site compared with that of only H coverage at the P1 site in Table 4, in which the adsorption energy will decrease. In other words, H adsorption at Mo1 site facilitates hydrogen adsorption at the P1 site, agreeing with a zigzag pattern with H coverage discussed above. For 2/16 H coverage, where the adsorbed H atoms are at both Mo1 and P1 sites, an accumulation (yellow) forms between H and P atoms due to a bonding generated between them. In addition, a depletion of the electron density outside the adsorbed H atom connected P atom occurs because of the rearrangement of electron toward the bond of H and P. When H coverage increase to 3/16, the phenomenon of electron accumulation and depletion around the H atom and connected Mo atom at Mo2 site are analogous to that of 1/16 H coverage.
Overall, H adsorption will lead to a rearrangement of electrons and change the bond length of Mo-P on the (111) facet shown in Table 3. Meanwhile, H adsorption at one site will facilitate hydrogen adsorption at the other site shown in Table 4.
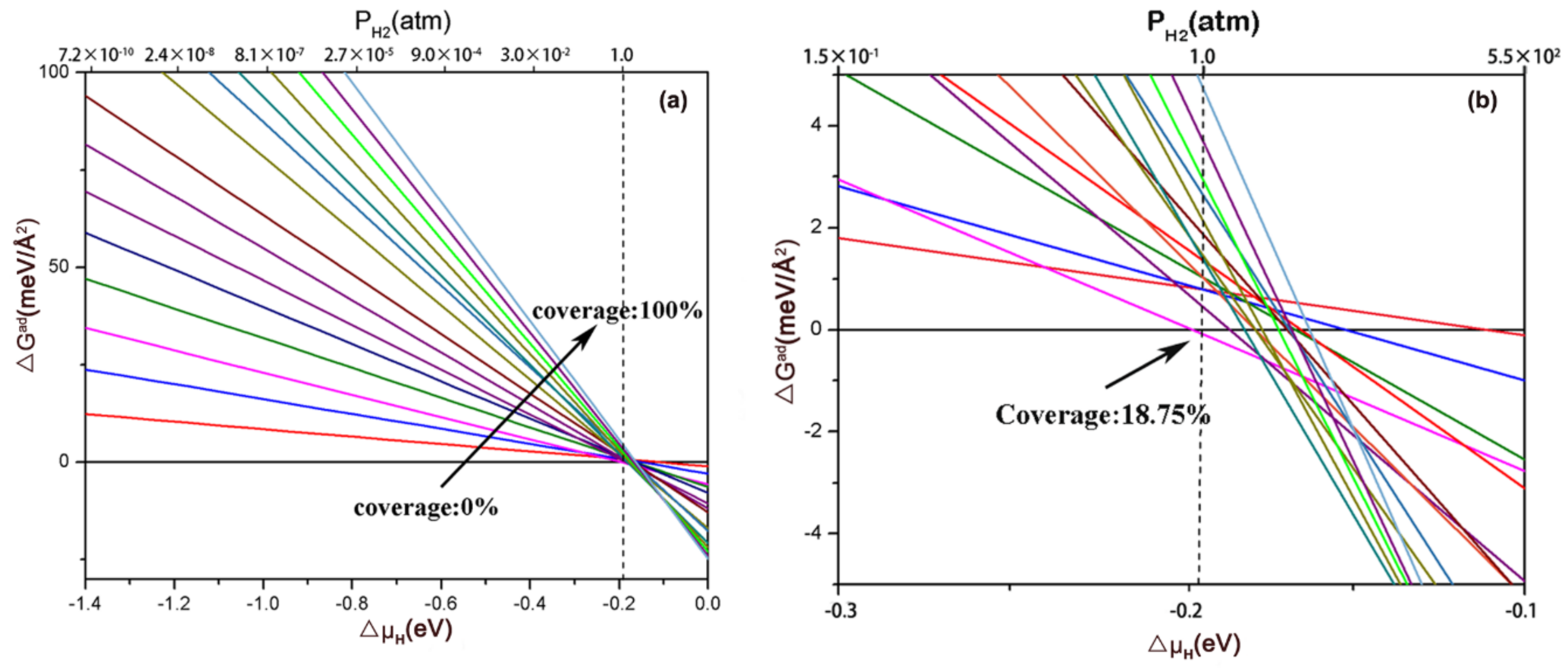
Furthermore, to estimate the most stable coverage of H on the (111) facet at 1 atm and 298K, ab initio atomistic thermodynamics was employed to identify the by the hydrogen chemical potential and pressure illustrated in Figure 6. Each line represents a given H coverage. Obviously, the most stable phase at 1 atm and 298 K is 18.75% H coverage, in which the active sites for HER is the Mo2 site. As shown in the Figure S3, the kinetic energy barrier of the Tafel pathway was considered by calculating the kinetic energy barrier profiles of the formation of Hydrogen, from which the HER activity of electrocatalysts can be known. The kinetic energy barrier profiles for the Tafel path at 18.75% H coverage is 0.75 eV, which is lower than the previous result of 1T- (0.85 eV) [41]. Based on the exiting learning, it can be learned that the lower energy barrier may facilitate the formation of the H-H bond.
To understand the kinetic mechanism of HER at 18.75% H coverage, the activation energies of the Tafel reaction were calculated, in which two adjacent pre-bonded H atoms at top site of Mo2 and P1 combine to form an molecule. Figure S3 shows the kinetic energy barrier profile of HER on the (111) surface at 18.75% H coverage. Clearly, generation needs to experience two transition states. The first step needs to overcome a relatively high activation energy barrier of 0.75 eV. However, it drops to 0.18 eV for the second step. These low activation energy barriers revel that generation through Tafel reaction is favored on the (111) surface at 18.75% H coverage.
3.3. Doping on the (111) Surface
Doping has been extensively used to tune the physical and chemical properties of materials. It has been proven that doping on transition-mental is an effective way, which influence the chemical activity of catalysts [55,56]. The hydrogen generation of catalytic activity of 2H can be promoted by the introduction of metal dopants such as Fe, Ni and Co [57,58,59]. Experiments found that N, S doping of MoP can enhance the HER activity [60,61]. To systematically recognize HER activity of the doping effects on HER activity for the (111) facet of have been done from a theoretical perspective.
Figure 7a illustrates the substitutional doing of the Mo site with a Co, Cu, Mn, Ni, Fe or Cr atom, and doping of the P site with an S or N atom was evaluated. The Hubbard correction was considered for the metal dopant of Fe. Geometry optimizations of the doping system show that doping of the Mo site by using a Co, Fe, Mn, Ni or Cr atom, as well as doping of the P site by using a N or S atom could not induce visible structural change to the (111) facet. On the other hand, doping with a Cu atom induces slightly larger lattice distortion, which is 0.8% tensile strain, compared with the clear (111) facet. The calculated of the doped system was summarized in Figure 7b. Clearly, doping of the P site with an N or S atom and doping of the Mo site with an Fe atom give a value of closer to zero, which promote HER catalytic activity for the (111) facet of MoP2.
4. Conclusions
In this work, HER catalytic activities for the (111) facet of were systematically investigated from theoretical perspective using periodic DFT calculations. The electronic structure, Gibbs free energy of hydrogen adsorption and ab initio thermodynamics of hydrogen coverage were calculated to reveal the HER characteristics of .
From the electronic structures of six low Miller-index facets, we found the thermodynamic stability of the clean facets follows the order of (111) > (110) > (101) > (011) > (100) > (001). As the most stable facet, the (111) facet has the metallic behavior with none having zero DOS at the Fermi level. The values of indicated that the (111) facet can favor HER over a large hydrogen coverage range. The zigzagged patterns before 75% hydrogen coverage suggest a synergy among Mo1, P1 and Mo2 sites, which are attributed to repeat occupancy sites of H atoms. From charge density differences and Bader analysis, we found that H adsorption will lead to a rearrangement of the electron and change the bond length of Mo-P on the (111) facet. Meanwhile, H adsorption at one site will facilitate hydrogen adsorption at the other site. From the ab initio atomistic thermodynamics analysis, we obtained that the most stable phase at 1 atm and 298 K is 18.75% H coverage, in which the active sites for HER is Mo2 site. Finally, we found doping of the P site with an N or S atom and doping the Mo site with an Fe atom can promote HER catalytic activity for the (111) facet of .
Supplementary Materials
The following are available online at https://www.mdpi.com/2079-4991/9/9/1270/s1, Figure S1: All possible adsorption sites on (111) facet, Figure S2. Top view of six clean MoP2 surfaces, including (a) (100), (b) (111), (c) (110), (d) (001), (e) (101) and (f) (011) facets. Mo: dark green; P: purple. Table S1. Adsorption energy of the optimized hydrogen adsorption sites for (111) facet.
Author Contributions
R.H.S. designed the research. G.Y.Y. did computational studies. L.H.Y., W.J.Y., and M.J.Y. discussed. R.H.S and G.Y.Y. wrote the paper. All authors edited the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Project No. 21573152 and No. 91841301).
Acknowledgments
The authors gratefully acknowledge the financial support for this study from the National Natural Science Foundation of China.
Conflicts of Interest
There are no conflicts to declare.
References
- Cheng, X.; Shi, Z.; Glass, N.; Zhang, L.; Zhang, J.; Song, D.; Liu, Z.-S.; Wang, H.; Shen, J. A review of PEM hydrogen fuel cell contamination: Impacts, mechanisms, and mitigation. J. Power Sources 2007, 165, 739–756. [Google Scholar] [CrossRef]
- Cao, X.H.; Tan, C.L.; Sindoro, M.; Zhang, H. Hybrid micro-/nano-structures derived from metal-organic frameworks: Preparation and application in energy storage and convension. Chem. Soc. Rev. 2017, 46, 2660–2677. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Cui, W.; Liu, Q.; Xing, Z.C. Recent Progress in Cobalt-Based Heterogeneous Catalysts for Electrochemical Water Splitting. Adv. Mater. 2016, 28, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Li, P.K.; Zhu, J.G.; Handoko, A.D.; Zhang, R.F.; Wang, H.T.; Legut, D.; Wen, X.D.; Fu, Z.; Shen, Z.; Zhang, Q. High-throughput theoretical optimization of hydrogen evolution reaction on MXens by transition mental modification. J. Mater. Chem. A 2018, 6, 4271–4278. [Google Scholar] [CrossRef]
- Kronberg, R.; Hakala, M.; Holmberg, N.; Laasonen, K. Hydrogen adsorption on MoS2 -surfaces: A DFT study on preferential sites and the effect of sulfur and hydrogen coverage. Phys. Chem. Chem. Phys. 2017, 19, 16231–16241. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.G.; Warren, E.L.; McKone, J.R.; Boettcher, S.W.; Mi, Q.; Santori, E.A.; Lewis, N.S. Solar water splitting cells. Chem. Rev. 2010, 110, 6446–6473. [Google Scholar] [CrossRef]
- Chen, L.; Wang, M.; Han, K.; Zhang, P.; Gloaguen, F.; Sun, L. A super-efficient cobalt catalyst for electrochemical hydrogen production from neutral water with 80 mV overpotential. Energy Environ. Sci. 2014, 7, 329–334. [Google Scholar] [CrossRef]
- Abbas, M.A.; Bang, J.H. Rising Again: Opportunities and Challenges for Platinum-Free Electrocatalysts. Chem. Mater. 2015, 27, 7218–7235. [Google Scholar] [CrossRef]
- Islam, M.M.; Calatayud, M.; Pacchioni, G. Hydrogen Adsorption and Diffusion on the Anatase TiO2(101) Surface: A First-Principles Investigation. J. Phys. Chem. C 2011, 115, 6809–6814. [Google Scholar] [CrossRef]
- González-Navarrete, P.; Monica, C. On the reductive hydrogenation process of gas-phase metal dioxides: H2 activation or reduction of the metal center, what is more important? Theor. Chem. Acc. 2019, 138, 98. [Google Scholar] [CrossRef]
- Chen, W.-F.; Muckerman, J.T.; Fujita, E. ChemInform Abstract: Recent Developments in Transition Metal Carbides and Nitrides as Hydrogen Evolution Electrocatalysts. Chemin- 2013, 44, 8896–8909. [Google Scholar] [CrossRef]
- Kong, D.; Wang, H.; Lu, Z.; Cui, Y. CoSe2 Nanoparticles Grown on Carbon Fiber Paper: An Efficient and Stable Electrocatalyst for Hydrogen Evolution Reaction. J. Am. Chem. Soc. 2014, 136, 4897–4900. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, T.F.; Jørgensen, K.P.; Bonde, J.; Nielsen, J.H.; Horch, S.; Chorkendroff, I. Identification of active edge sites for electrochemical H2 evolution from MoS2 nanocatalysts. Science 2017, 317, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.; Alam Sk, M.; Thia, L.; Ge, X.; Lim, R.J.; Wang, J.-Y.; Lim, K.H.; Wang, X. Molybdenum phosphide as an efficient electrocatalyst for the hydrogen evolution reaction. Energy Environ. Sci. 2014, 7, 2624–2629. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.F.; Sasaki, K.; Ma, C.; Frenkel, A.I.; Marinkovic, N.; Muckerman, J.T.; Zhu, Y.; Adzic, R.R. Hydrogen-Evolution Catalysts Based on Non-Noble Metal Nickel-Molybdenum Nitride Nanosheets. Angew. Chem. 2012, 124, 6235–6239. [Google Scholar] [CrossRef]
- Xiao, P.; Chen, W.; Wang, X. A Review of Phosphide-Based Materials for Electrocatalytic Hydrogen Evolution. Adv. Energy Mater. 2015, 5, 1500985. [Google Scholar] [CrossRef]
- Oyama, S.T.; Gott, T.; Zhou, H.Y.; Lee, Y.K. Transition metal phosphide hydroprocessing catalysts: A review. Calay. Today 2003, 143, 94–107. [Google Scholar] [CrossRef]
- McEnaney, J.M.; Crompton, J.C.; Callejas, J.F.; Popczun, E.J.; Read, C.G.; Lewis, N.S.; Schaak, R.E. Electrocatalytic hydrogen evolution using amorphous tungsten phosphide nanoparticles. Chem. Commun. 2014, 50, 11026–11028. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Pu, Z.; Tu, Z.; Amiinu, I.S.; Liu, S.; Wang, P.; Mu, S. Integrated design and construction of WP/W nanorod array electrodes toward efficient hydrogen evolution reaction. Chem. Eng. J. 2017, 327, 705–712. [Google Scholar] [CrossRef]
- Jiang, P.; Liu, Q.; Liang, Y.; Tian, J.; Asiri, A.M.; Sun, X. A Cost-Effective 3D Hydrogen Evolution Cathode with High Catalytic Activity: FeP Nanowire Array as the Active Phase. Angew. Chem. 2014, 126, 13069–13073. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, Q.; Asiri, A.M.; Sun, X.; Luo, Y. Self-Supported FeP Nanorod Arrays: A Cost-Effective 3D Hydrogen Evolution Cathode with High Catalytic Activity. ACS Catal. 2014, 4, 4065–4069. [Google Scholar] [CrossRef]
- Zhou, Z.; Wei, L.; Wang, Y.; Karahan, H.E.; Chen, Z.; Lei, Y.; Chen, X.; Zhai, S.; Liao, X.; Chen, Y. Hydrogen evolution reaction activity of nickel phosphide is highly sensitive to electrolyte pH. J. Mater. Chem. A 2017, 5, 20390–20397. [Google Scholar] [CrossRef]
- Chen, X.; Wang, D.; Wang, Z.; Zhou, P.; Wu, Z.; Jiang, F. Molybdenum phosphide: A new highly efficient catalyst for the electrochemical hydrogen evolution reaction. Chem. Commun. 2014, 50, 11683–11685. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.N.; Li, S.H.; Tan, H.Q.; Khan, S.U.; Ma, Y.Y.; Zang, H.Y.; Wang, Y.H.; Li, Y.G. MoP/Mo2C@C: A New Combination of Electrocatalysts for Highly Efficient Hydrogen Evolution over the Entire pH Range. ACS Appl. Mater. Interfaces 2017, 9, 16270–16279. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.Y.; Wu, C.X.; Feng, X.J.; Tan, H.Q.; Yan, L.K.; Liu, Y.; Kang, Z.H.; Wang, E.B.; Li, Y.G. Highly efficient hydrogen evolution from seawater by a low-cost and stable CoMoPC electrocatalyst superior to Pt/C. Energy Environ. Sci. 2017, 10, 788–798. [Google Scholar] [CrossRef]
- Li, D.; Baydoun, H.; Verani, C.N.; Brock, S.L. Efficient Water Oxidation Using CoMnP Nanoparticles. J. Am. Chem. Soc. 2016, 138, 4006–4009. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Liu, Q.; Asiri, A.M.; Sun, X. Closely Interconnected Network of Molybdenum Phosphide Nanoparticles: A Highly Efficient Electrocatalyst for Generating Hydrogen from Water. Adv. Mater. 2014, 26, 5702–5707. [Google Scholar] [CrossRef]
- Wu, T.L.; Pi, M.Y.; Zhang, D.K.; Chen, S.J. Three-dimensional porous structural MoP2 nanoparticles as a novel and superior catalyst for electrochemical hydrogen evolution. J. Power Sources 2016, 328, 551–557. [Google Scholar] [CrossRef]
- Sheng, M.; Yano, J.; You, B.; Jiang, N.; Gul, S.; Sun, Y. High-Performance Overall Water Splitting Electrocatalysts Derived from Cobalt-Based Metal–Organic Frameworks. Chem. Mater. 2015, 27, 7636–7642. [Google Scholar]
- Du, H.; Gu, S.; Liu, R.; Li, C.M. Tungsten diphosphide nanorods as an efficient catalyst for electrochemical hydrogen evolution. J. Power Sources 2015, 278, 540–545. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, R.; Zhang, J.; Shi, Y.; Zhang, B. Anion-exchange synthesis of nanoporous FeP nanosheets as electrocatalysts for hydrogen evolution reaction. Chem. Commun. 2013, 49, 6656–6658. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.L.; Chen, S.J.; Zhang, D.K.; Hou, J.K. Facile preparation of semimetallic MoP2 as a novel visible light driven photocatalyst with high photocatalytic activity. J. Mater. Chem. A 2015, 3, 0360–10367. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, B. Recent advances in transition metal phosphide nanomaterials: Synthesis and applications in hydrogen evolution reaction. Chem. Soc. Rev. 2016, 45, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Sun, Y.; Xu, N.; Manna, K.; Yao, M.; Süss, V.; Leermakers, I.; Young, O.; Forster, T.; Schmidt, M.; et al. Extremely high magnetoresistance and conductivity in the type-II Weyl semimetals WP2 and MoP2. Nat. Commun. 2017, 8, 1642. [Google Scholar] [CrossRef] [PubMed]
- Pu, Z.; Saana, A.I.; Wang, M.; Yang, Y.; Mu, S. Semimetallic MoP2: An active and stable hydrogen evolution electrocatalyst over the whole pH range. Nanoscale 2016, 8, 8500–8504. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhu, W.; Tang, C.; Wang, J.; Asiri, A.M.; Sun, X. A self-standing nanoporous MoP 2 nanosheet array: An advanced pH-universal catalytic electrode for the hydrogen evolution reaction. J. Mater. Chem. A 2016, 4, 7169–7173. [Google Scholar]
- Gao, Y.; Zhang, M.; Ding, J.; Hong, S.; Masa, J.; Liu, S.; Sun, Z. Simple synthesis of two-dimensional MoP2 nanosheets for efficient electrocatalytic hydrogen evolution. Electrochem. Commun. 2018, 97, 27–31. [Google Scholar] [CrossRef]
- Owens-Baird, B.; Kolen’ko, Y.V.; Kovnir, K. Structure-Activity Relationships for Pt-Free Metal Phosphide Hydrogen Evolution Electrocatalysts. Chem. Eur. J. 2018, 24, 7298–7311. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2015, 152, J23–J26. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef] [PubMed]
- Mo, L.B.; Wang, Y.; Bai, Y.; Xiang, Q.Y.; Li, Q.; Yao, W.Q.; Wang, J.O.; Ibrahim, K.; Wang, H.H.; Wan, C.H.; et al. Hydrogen Impurity Defects in Rutile TiO2. Sci. Rep. 2015, 5, 17634. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Jiang, D.E. Mechanism of Hydrogen Evolution Reaction on 1T-MoS2 from First Principles. ACS Catal. 2016, 6, 4953–4961. [Google Scholar] [CrossRef]
- An, Y.R.; Fan, X.L.; Liu, H.J.; Luo, Z.F. Improved catalytic performance of monolayer nano-triangles WS2 and MoS2 on HER by 3d metals doping. Comp. Mater. Sci. 2019, 159, 333–340. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Perdew, burke, and ernzerhof reply. Phys. Rev. Lett. 1998, 80, 891. [Google Scholar] [CrossRef]
- Hafner, J.; Kresse, G. The Vienna AB-Initio Simulation Program VASP: An Efficient and Versatile Tool for Studying the Structural, Dynamic, and Electronic Properties of Materials. In Properties of Complex Inorganic Solids; Springer: Boston, MA, USA, 1997; pp. 69–82. [Google Scholar]
- Huber, K.P.; Herzberg, G. Molecular spectra and molecular structure: IV constants of diatomic molecules; Van Rostrand-Reinhold: New York, NY, USA, 1997. [Google Scholar]
- Rogal, J.; Reuter, K. Ab Initio Atomistic Thermodynamics for Surfaces: A Primer; Max-planck-gesellschaft zur foerderung der wissenschaften ev berlin (germany fr) fritz-haber-inst: Berlin, Germany, 2006. [Google Scholar]
- Rundqvist, S.; Lundström, T. X-ray Studies of Molybdenum and Tungsten Phosphides. Acta Chem. Scand. 1963, 17, 37–46. [Google Scholar] [CrossRef]
- Winkler, B.; Knorr, K.; Hytha, M.; Milman, V.; Soto, V.; Avalos, M.; Avalos-Borja, M. Crystal chemistry of molybdenum phosphides from density functional theory calculations. J. Phys. Chem. Solids 2003, 64, 405–411. [Google Scholar] [CrossRef]
- Mou, J.; Gao, Y.; Wang, J.; Ma, J.; Ren, H. Hydrogen evolution reaction activity related to the facet-dependent electrocatalytic performance of NiCoP from first principles. RSC Adv. 2019, 9, 11755–11761. [Google Scholar] [CrossRef] [Green Version]
- Scaranto, J.; Idriss, H. DFT studies of bulk and surfaces of the electrocatalyst cobalt phosphide CoP2. Chem. Phys. Lett. X 2019, 2, 100008. [Google Scholar] [CrossRef]
- Liang, Z.; Zhong, X.L.; Li, T.Q.; Chen, M.; Feng, G. DFT Study on the Hydrogen Evolution Reaction for Different Facets of Co2P. Chem. Electro. Chem. 2019, 6, 260–267. [Google Scholar]
- George, P.P.; Genish, I.; Maklouf, S.B.; Koltypin, Y.; Gedanken, A. A New Approach to the Synthesis of Transition Metal Phosphide Nanocrystallites (MoP, MoP2, Cu3P and CuP2) by Using Reaction under Autogenic Pressure at Elevated Temperatures (RAPET) Technique. Int. J. Nanosci. 2017, 16, 1650030. [Google Scholar] [CrossRef]
- Wang, H.; Tsai, C.; Kong, D.; Chan, K.; Abild-Pedersen, F.; Nørskov, J.K.; Cui, Y. Transition-metal doped edge sites in vertically aligned MoS2 catalysts for enhanced hydrogen evolution. Nano Res. 2015, 8, 566–575. [Google Scholar] [CrossRef]
- Hong, X.; Chan, K.; Tsai, C.; Norskov, J.K. How doped MoS2 breaks transition-metal scaling relations for CO2 electrochemical reduction. ACS Catal. 2016, 6, 4428–4437. [Google Scholar] [CrossRef]
- Dai, X.; Du, K.; Li, Z.; Liu, M.; Ma, Y.; Sun, H.; Zhang, X.; Yang, Y. Co-Doped MoS2 Nanosheets with the Dominant CoMoS Phase Coated on Carbon as an Excellent Electrocatalyst for Hydrogen Evolution. ACS Appl. Mater. Interfaces 2015, 7, 27242–27253. [Google Scholar] [CrossRef] [PubMed]
- Tedstone, A.A.; Lewis, D.J.; O’Brien, P. Synthesis, Properties, and Applications of Transition Metal-Doped Layered Transition Metal Dichalcogenides. Chem. Mater. 2016, 28, 1965–1974. [Google Scholar] [CrossRef]
- Merki, D.; Vrubel, H.; Fierro, S.; Hu, X.; Rovelli, L. Fe, Co, and Ni ions promote the catalytic activity of amorphous molybdenum sulfide films for hydrogen evolution. Chem. Sci. 2012, 3, 2515–2525. [Google Scholar] [CrossRef] [Green Version]
- Kibsgaard, J.; Jaramillo, T.F. Molybdenum Phosphosulfide: An Active, Acid-Stable, Earth-Abundant Catalyst for the Hydrogen Evolution Reaction. Angew. Chem. 2014, 126, 14661–14665. [Google Scholar] [CrossRef]
- Anjum, M.A.R.; Lee, J.S. Sulfur and Nitrogen Dual-Doped Molybdenum Phosphide Nanocrystallites as an Active and Stable Hydrogen Evolution Reaction Electrocatalyst in Acidic and Alkaline Media. ACS Catal. 2017, 7, 3030–3038. [Google Scholar] [CrossRef]
Figure 1.
Bulk . Mo: dark green; P: purple.

Figure 2.
Project and total density of states of the (111) facet.

Figure 3.
Optimized structures of the (111) facet with the H coverages: (a) 18.75%, (b) 37.5%, (c) 56.25%, (d) 75% and (e) 100%.
Figure 3.
Optimized structures of the (111) facet with the H coverages: (a) 18.75%, (b) 37.5%, (c) 56.25%, (d) 75% and (e) 100%.

Figure 4.
Adsorption energy () and adsorption free energy ( ) as functions of hydrogen coverage on .
Figure 4.
Adsorption energy () and adsorption free energy ( ) as functions of hydrogen coverage on .

Figure 5.
The charge density differences for the adsorbed H atoms with H coverage of (a) 1/16, (b) 2/16 and (c) 3/16. Top and bottom panels represent overlooking and sidelooking, respectively. Color codes for Mo, P and H are dark green, purple and brown, respectively. Charge accumulation and depletion are plotted by the yellow and light blue regions with the isosurface value of 1.5×10−3 e/bohr3.
Figure 5.
The charge density differences for the adsorbed H atoms with H coverage of (a) 1/16, (b) 2/16 and (c) 3/16. Top and bottom panels represent overlooking and sidelooking, respectively. Color codes for Mo, P and H are dark green, purple and brown, respectively. Charge accumulation and depletion are plotted by the yellow and light blue regions with the isosurface value of 1.5×10−3 e/bohr3.

Figure 6.
at 298K with pressure and ranging from: (a) −1.4 to 0 eV and (b) −0.3 to −0.1 eV. Note that the dot line is used to clearly reflect the values of at 1 atm and 298 K. The intersection points show the values of for different coverages at 1 atm and 298K.
Figure 6.
at 298K with pressure and ranging from: (a) −1.4 to 0 eV and (b) −0.3 to −0.1 eV. Note that the dot line is used to clearly reflect the values of at 1 atm and 298 K. The intersection points show the values of for different coverages at 1 atm and 298K.

Figure 7.
(a) Top view of the (111) facet with two doping atom sites. (b) Gibbs free energies of adsorption for the (111) facet and its doped facets.
Figure 7.
(a) Top view of the (111) facet with two doping atom sites. (b) Gibbs free energies of adsorption for the (111) facet and its doped facets.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The lattice parameters of bulk .
| a (Å) | b (Å) | c (Å) | |
|---|---|---|---|
| Present | 3.147 | 11.242 | 5.009 |
| Calculation a | 3.142 | 11.132 | 4.949 |
| Experiment b | 3.145 a | 11.184 | 4.984 |
Table 2.
Calculated surface energies and surface packing density (SPD) of low Miller-index facets for .
Table 2.
Calculated surface energies and surface packing density (SPD) of low Miller-index facets for .
| (111) | (110) | (101) | (011) | (100) | (001) | |
|---|---|---|---|---|---|---|
| Surface energy(meVÅ−2) | 104.78 | 134.34 | 134.85 | 146.73 | 147.35 | 173.52 |
| SPD (atom nm−2) | 18 | 17 | 15 | 13 | 11 | 11 |
Table 3.
Bond lengths and partial atomic charges from Bader analysis with H coverage of 1/16, 2/16 and 3/16.
Table 3.
Bond lengths and partial atomic charges from Bader analysis with H coverage of 1/16, 2/16 and 3/16.
| Clean facet | 1/16 H atom | 2/16 H atoms | 3/16 H atoms | |
|---|---|---|---|---|
| (Å) | 2.313 | 2.330 | 2.380 | 2.394 |
| (Å) | 2.382 | 2.366 | 2.435 | 2.411 |
| (e) | −0.360 | −0.354 | 0.146 | 0.200 |
| (e) | 0.659 | 0.707 | 0.722 | 0.736 |
| (e) | 0.696 | 0.670 | 0.681 | 0.698 |
Table 4.
Adsorption energies at the Mo1, P1 and Mo2 sites. Here, 1 means there is an adsorbed H atom, while 0 stands for 0 adsorbed H atoms.
Table 4.
Adsorption energies at the Mo1, P1 and Mo2 sites. Here, 1 means there is an adsorbed H atom, while 0 stands for 0 adsorbed H atoms.
| Adsorption Energy | Mo1 site | P1 site | Mo2 site |
|---|---|---|---|
| −0.106 | 0 | 0 | |
| 0 | −0.038 | 0 | |
| 0 | 0 | 0.083 | |
| 1 | −0.194 | 0 | |
| 1 | 1 | −0.286 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gao, Y.; Li, H.; Wang, J.; Ma, J.; Ren, H. New Insight on Hydrogen Evolution Reaction Activity of MoP2 from Theoretical Perspective. Nanomaterials 2019, 9, 1270. https://doi.org/10.3390/nano9091270
AMA Style
Gao Y, Li H, Wang J, Ma J, Ren H. New Insight on Hydrogen Evolution Reaction Activity of MoP2 from Theoretical Perspective. Nanomaterials. 2019; 9(9):1270. https://doi.org/10.3390/nano9091270
Chicago/Turabian StyleGao, Yuyue, Hongyan Li, Jingyu Wang, Jianyi Ma, and Haisheng Ren. 2019. "New Insight on Hydrogen Evolution Reaction Activity of MoP2 from Theoretical Perspective" Nanomaterials 9, no. 9: 1270. https://doi.org/10.3390/nano9091270
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.