Spray-Dried, Nanoencapsulated, Multi-Drug Anti-Tuberculosis Therapy Aimed at Once Weekly Administration for the Duration of Treatment

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Nanoparticle Preparation and Characterisation
2.3. In Vivo Assays
2.3.1. Mice
2.3.2. Mycobacteria and Infection
2.3.3. Chemotherapy Preparation and Treatment
2.4. Statistical Analysis
3. Results
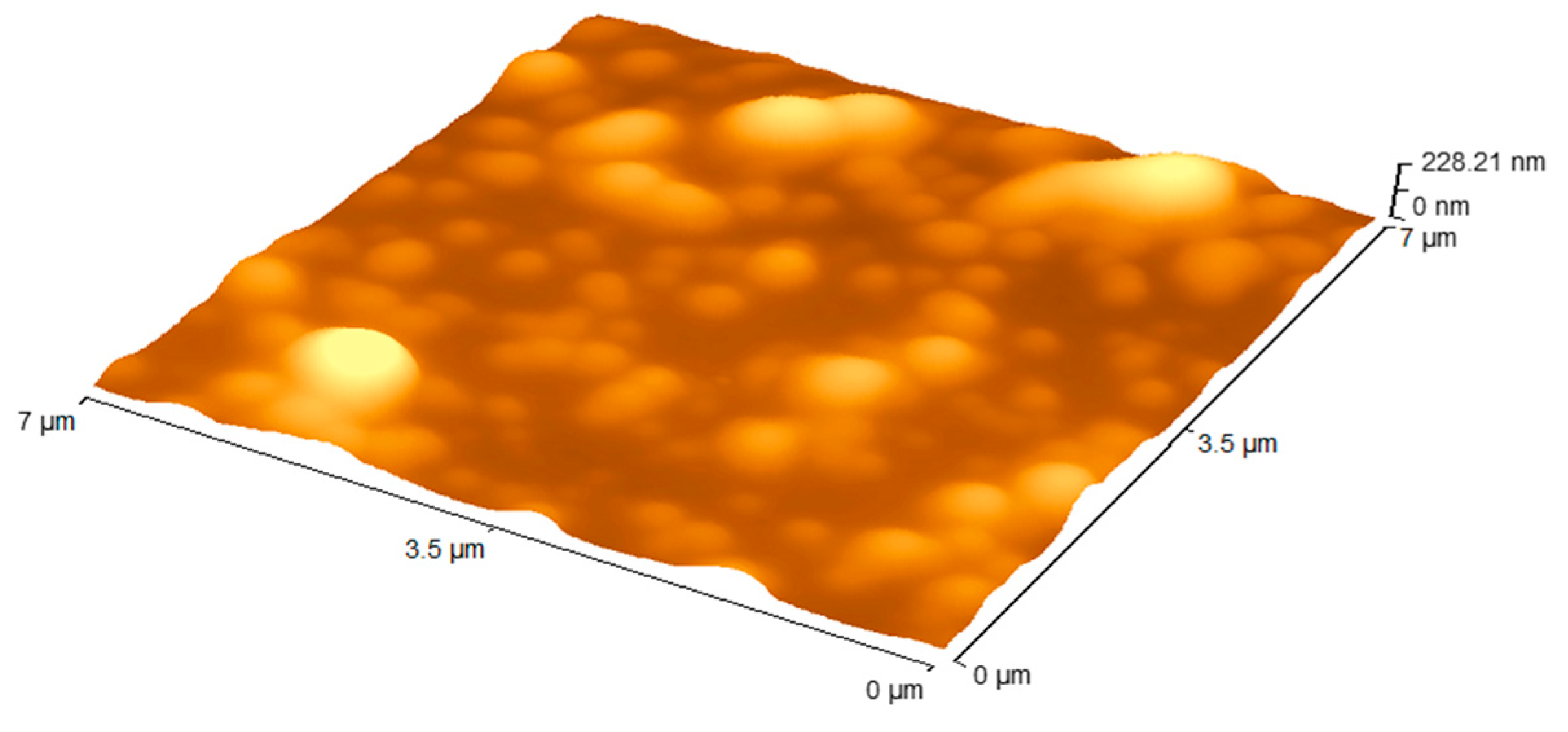
3.1. Nanoparticle Formulations
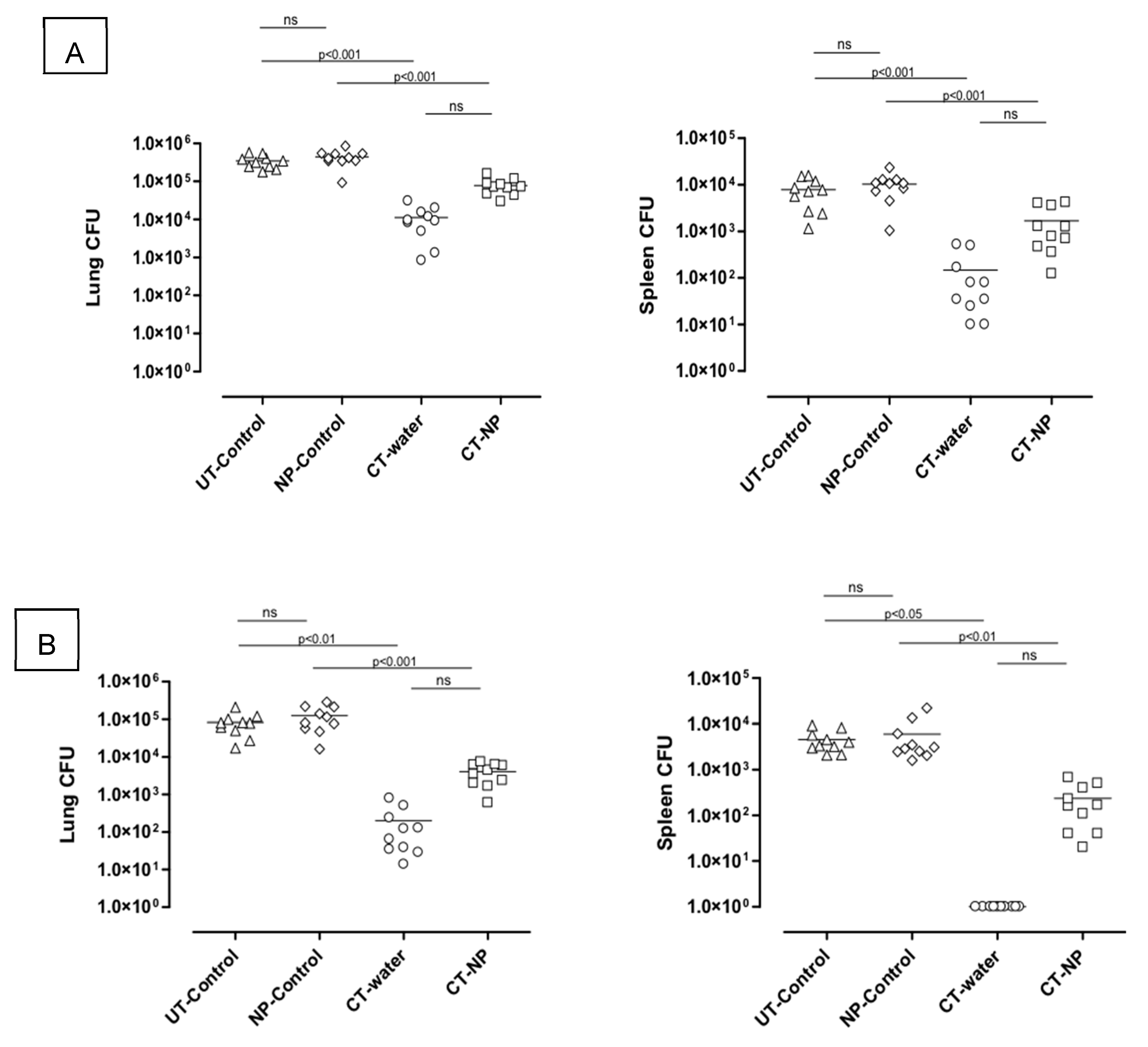
3.2. In Vivo Study
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

References
- Hershkovitz, I.; Donoghue, H.D.; Minnikin, D.E.; May, H.; OY-C, L.; Feldman, M.; Galili, E.; Spigelman, M.; Rothschild, B.M.; Bar-Gal, G.K. Tuberculosis origin: The Neolithic scenario. Tuberculosis 2015, 95, S122–S126. [Google Scholar] [CrossRef] [PubMed]
- Global Tuberculosis Report 2018; World Health Organization Geneva: Geneva, Switzerland, 2018.
- Costa, A.; Pinheiro, M.; Magalhães, J.; Ribeiro, R.; Seabra, V.; Reis, S.; Sarmento, B. The formulation of nanomedicines for treating tuberculosis. Adv. Drug Deliv. Rev. 2016, 102, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Dartois, V. The path of anti-tuberculosis drugs: From blood to lesions to mycobacterial cells. Nat. Rev. Microbiol. 2014, 12, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Sharma, A.; Pandey, R.; Khuller, G.K. Chemotherapeutic efficacy of poly (dl-lactide-co-glycolide) nanoparticle encapsulated antitubercular drugs at sub-therapeutic dose against experimental tuberculosis. Int. J. Antimicrob. Ag. 2004, 24, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Zahoor, A.; Sharma, S.; Khuller, G.K. Nanoparticle encapsulated antitubercular drugs as a potential oral drug delivery system against murine tuberculosis. Tuberculosis 2003, 83, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.M.; Pandey, R.; Sharma, S.; Khuller, G.K.; Basaraba, R.J.; Orme, I.M.; Lenaerts, A.J. Oral Therapy Using Nanoparticle-Encapsulated Antituberculosis Drugs in Guinea Pigs Infected with Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2005, 49, 4335–4338. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miranda, M.S.; Rodrigues, M.T.; Domingues, R.M.A.; Torrado, E.; Reis, R.L.; Pedrosa, J.; Gomes, M.E. Exploring inhalable polymeric dry powders for anti-tuberculosis drug delivery. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 93, 1090–1103. [Google Scholar] [CrossRef] [PubMed]
- Pham, D.; Fattal, E.; Tsapis, N. Pulmonary drug delivery systems for tuberculosis treatment. Int. J. Pharm. 2015, 478, 517–529. [Google Scholar] [CrossRef]
- Turner, P.V.; Brabb, T.; Pekow, C.; Vasbinder, M.A. Administration of substances to laboratory animals: Routes of administration and factors to consider. JAALAS 2011, 50, 600. [Google Scholar]
- Date, A.A.; Hanes, J.; Ensign, L.M. Nanoparticles for oral delivery: Design, evaluation and state-of-the-art. J. Control Release 2016, 240, 504–526. [Google Scholar] [CrossRef]
- Horváti, K.; Bacsa, B.; Szabó, N.; Fodor, K.; Balka, G.; Rusvai, M.; Szabó, E.; Hudecz, F.; Bősze, S. Antimycobacterial activity of peptide conjugate of pyridopyrimidine derivative against Mycobacterium tuberculosis in a series of in vitro and in vivo models. Tuberculosis 2015, 95, 207. [Google Scholar] [CrossRef] [PubMed]
- de Faria, T.J.; Roman, M.; de Souza, N.M.; De Vecchi, R.; Vitor de Assis, J.; Gomes dos Santos, A.L.; Bechtold, I.H.; Winter, N.; José Soares, M.; Silva, L.P.; et al. An Isoniazid Analogue Promotes Mycobacterium tuberculosis Nanoparticle Interactions and Enhances Bacterial Killing by Macrophages. Antimicrob. Agents Chemother. 2012, 56, 2259–2267. [Google Scholar] [CrossRef] [PubMed]
- Moretton, M.A.; Hocht, C.; Taira, C.; Sosnik, A. Rifampicin-loaded ‘flower-like’ polymeric micelles for enhanced oral bioavailability in an extemporaneous liquid fixed-dose combination with isoniazid. Nanomedicine 2014, 9, 1635–1650. [Google Scholar] [CrossRef] [PubMed]
- Semete, B.; Booysen, L.; Lemmer, Y.; Kalombo, L.; Katata, L.; Verschoor, J.; Swai, H.S. In vivo evaluation of the biodistribution and safety of PLGA nanoparticles as drug delivery systems. Nanomedicine 2010, 6, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Semete, B.; Booysen, L.I.J.; Kalombo, L.; Venter, J.D.; Katata, L.; Ramalapa, B.; Verschoor, J.; Swai, H.S. In vivo uptake and acute immune response to orally administered chitosan and PEG coated PLGA nanoparticles. Toxicol. Appl. Pharmacol. 2010, 249, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Semete, B.; Booysen, L.; Kalombo, L.; Ramalapa, B.; Hayeshi, R.; Swai, H.S. Effects of protein binding on the biodistribution of PEGylated PLGA nanoparticles post oral administration. Int. J Pharm. 2012, 424, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Booysen, L.L.I.J.; Kalombo, L.; Brooks, E.; Hansen, R.; Gilliland, J.; Gruppo, V.; Lungenhofer, P.; Semete-Makokotlela, B.; Swai, H.S.; Kotze, A.F.; et al. In vivo/in vitro pharmacokinetic and pharmacodynamic study of spray-dried poly-(dl-lactic-co-glycolic) acid nanoparticles encapsulating rifampicin and isoniazid. Int. J. Pharm. 2013, 444, 10–17. [Google Scholar] [CrossRef]
- Ziaee, A.; Albadarin, A.B.; Padrela, L.; Femmer, T.; O’Reilly, E.; Walker, G. Spray drying of pharmaceuticals and biopharmaceuticals: Critical parameters and experimental process optimization approaches. Eur. J. Pharm. Sci. 2019, 127, 300–318. [Google Scholar] [CrossRef]
- Dobry, D.E.; Settell, D.M.; Baumann, J.M.; Ray, R.J.; Graham, L.J.; Beyerinck, R.A. A Model-Based Methodology for Spray-Drying Process Development. J. Pharm. Innov. 2009, 4, 133–142. [Google Scholar] [CrossRef]
- Calver, A.D.; Falmer, A.A.; Murray, M.; Strauss, O.J.; Streicher, E.M.; Hanekom, M.; Liversage, T.; Masibi, M.; van Helden, P.D.; Warren, R.M.; et al. Emergence of increased resistance and extensively drug-resistant tuberculosis despite treatment adherence, South Africa. Emerg. Infect. Dis. 2010, 16, 264. [Google Scholar] [CrossRef]
- Lenaerts, A.J.; Gruppo, V.; Marietta, K.S.; Johnson, C.M.; Driscoll, D.K.; Tompkins, N.M.; Rose, J.D.; Reynolds, R.C.; Orme, I.M. Preclinical Testing of the Nitroimidazopyran PA-824 for Activity against Mycobacterium tuberculosis in a Series of In Vitro and In Vivo Models. Antimicrob. Antimicrob. Agents Chemother. 2005, 49, 2294–2301. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Contreras, L.; Sung, J.C.; Muttil, P.; Padilla, D.; Telko, M.; VerBerkmoes, J.L.; Elbert, K.J.J.; Hickey, A.J.; Edwards, D.A. Dry Powder PA-824 Aerosols for Treatment of Tuberculosis in Guinea Pigs. Antimicrob. Agents Chemother. 2010, 54, 1436–1442. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mandiwana, V.; Kalombo, L.; Venter, K.; Sathekge, M.; Grobler, A.; Zeevaart, J. Samarium oxide as a radiotracer to evaluate the in vivo biodistribution of PLGA nanoparticles. J. Nanopart. Res. 2015, 17, 1–11. [Google Scholar] [CrossRef][Green Version]
- Dutt, M.; Khuller, G.K. Liposomes and PLG microparticles as sustained release antitubercular drug carriers—an in vitro–in vivo study. Int. J. Antimicrob. Ag. 2001, 18, 245–252. [Google Scholar] [CrossRef]
- Pandey, R.; Sharma, S.; Khuller, G.K. Oral poly(lactide-co-glycolide) nanoparticle based antituberculosis drug delivery: Toxicological and chemotherapeutic implications. Indian J. Exp. Biol. 2006, 44, 459–467. [Google Scholar] [PubMed]
- Ahmad, Z.; Pandey, R.; Sharma, S.; Khuller, G.K. Novel chemotherapy for tuberculosis: Chemotherapeutic potential of econazole- and moxifloxacin-loaded PLG nanoparticles. Int. J. Antimicrob. Ag. 2008, 31, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Gelperina, S.; Kisich, K.; Iseman, M.D.; Heifets, L. The Potential Advantages of Nanoparticle Drug Delivery Systems in Chemotherapy of Tuberculosis. Am. J. Respir. Crit. Care Med. 2005, 172, 1487–1490. [Google Scholar] [CrossRef]
- Semete, B.; Kalombo, L.; Katata, L.; Chelule, P.; Booysen, L.; Lemmer, Y.; Naidoo, S.; Ramalapa, B.; Hayeshi, R.; Swai, H.S. Potential of Improving the Treatment of Tuberculosis Through Nanomedicine. Mol. Cryst. Liq. Cryst. 2012, 556, 317. [Google Scholar] [CrossRef]
- Du Toit, L.C.; Pillay, V.; Danckwerts, M.P. Tuberculosis chemotherapy: Current drug delivery approaches. Respir. Res. 2006, 7, 118. [Google Scholar] [CrossRef]
- Singh, H.; Bhandari, R.; Kaur, I.P. Encapsulation of Rifampicin in a solid lipid nanoparticulate system to limit its degradation and interaction with Isoniazid at acidic pH. Int. J. Pharm. 2013, 446, 106–111. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Size (nm) | PDI * | Zeta Potential (mv) | EE ** (%) | Drug Loading (%) |
|---|---|---|---|---|---|
| PLGA-INH | 328.7 ± 32.9 | 0.2 ± 0.01 | 17.7 ± 1.6 | 62.4 | 24.1 |
| PLGA-PZA | 348.3 ± 44.2 | 0.3 ± 0.02 | 19.4 ± 1.4 | 75.2 | 19.5 |
| PLGA-RIF | 252.2 ± 17.7 | 0.2 ± 0.01 | 17.9 ± 1.1 | 82.2 | 9.0 |
| PLGA-DRUG FREE | 259.6 ± 2.6 | 0.1 ± 0.01 | 11.4 ± 2.1 | N/A | N/A |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalombo, L.; Lemmer, Y.; Semete-Makokotlela, B.; Ramalapa, B.; Nkuna, P.; Booysen, L.L.L.I.J.; Naidoo, S.; Hayeshi, R.; Verschoor, J.A.; Swai, H.S. Spray-Dried, Nanoencapsulated, Multi-Drug Anti-Tuberculosis Therapy Aimed at Once Weekly Administration for the Duration of Treatment. Nanomaterials 2019, 9, 1167. https://doi.org/10.3390/nano9081167
Kalombo L, Lemmer Y, Semete-Makokotlela B, Ramalapa B, Nkuna P, Booysen LLLIJ, Naidoo S, Hayeshi R, Verschoor JA, Swai HS. Spray-Dried, Nanoencapsulated, Multi-Drug Anti-Tuberculosis Therapy Aimed at Once Weekly Administration for the Duration of Treatment. Nanomaterials. 2019; 9(8):1167. https://doi.org/10.3390/nano9081167
Chicago/Turabian StyleKalombo, Lonji, Yolandy Lemmer, Boitumelo Semete-Makokotlela, Bathabile Ramalapa, Patric Nkuna, Laetitia L.L.I.J. Booysen, Saloshnee Naidoo, Rose Hayeshi, Jan A. Verschoor, and Hulda S. Swai. 2019. "Spray-Dried, Nanoencapsulated, Multi-Drug Anti-Tuberculosis Therapy Aimed at Once Weekly Administration for the Duration of Treatment" Nanomaterials 9, no. 8: 1167. https://doi.org/10.3390/nano9081167
APA StyleKalombo, L., Lemmer, Y., Semete-Makokotlela, B., Ramalapa, B., Nkuna, P., Booysen, L. L. L. I. J., Naidoo, S., Hayeshi, R., Verschoor, J. A., & Swai, H. S. (2019). Spray-Dried, Nanoencapsulated, Multi-Drug Anti-Tuberculosis Therapy Aimed at Once Weekly Administration for the Duration of Treatment. Nanomaterials, 9(8), 1167. https://doi.org/10.3390/nano9081167