Tangeretin-Assisted Platinum Nanoparticles Enhance the Apoptotic Properties of Doxorubicin: Combination Therapy for Osteosarcoma Treatment


Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Agents
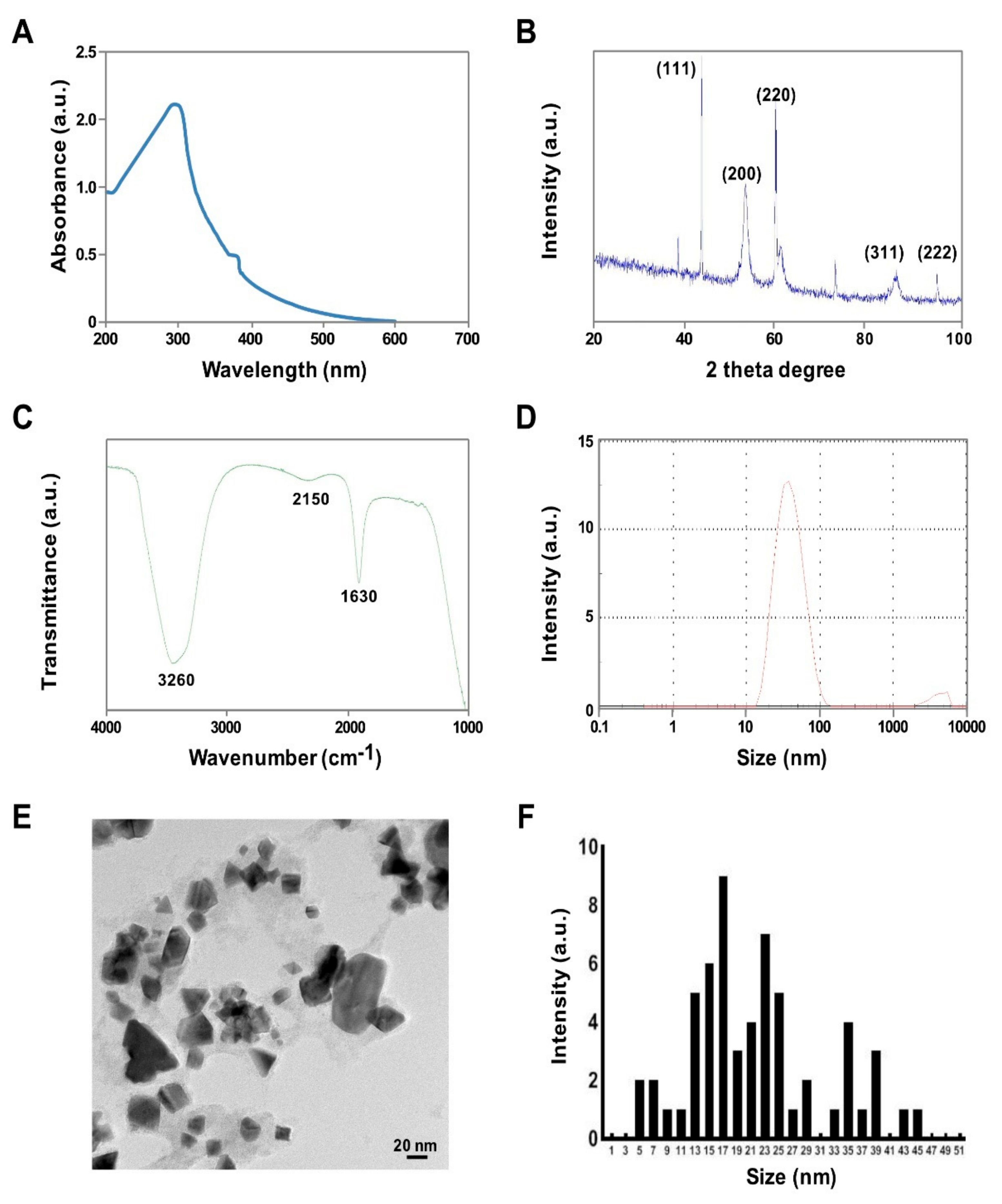
2.2. PtNPs Synthesis and Characterization
2.3. Cell Culture Conditions
2.4. Cell Viability Assay
2.5. BrdU Cell Proliferation Assay
2.6. Determination of PtNPs Concentration Using Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
2.7. Assessment of Membrane Integrity
2.8. Cell Mortality Assay
2.9. Cell Morphology
2.10. Determination of ROS, MDA, Nitric Oxide (NO), and Carbonylated Protein Levels
2.11. Measurement of Antioxidative Marker Levels
2.12. Measurement of 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dG) and 8-oxo-G Levels
2.13. Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
2.14. Statistical Analysis
3. Results and Discussion
3.1. Synthesis and Ccharacterization of PtNPs Using Tangeretin
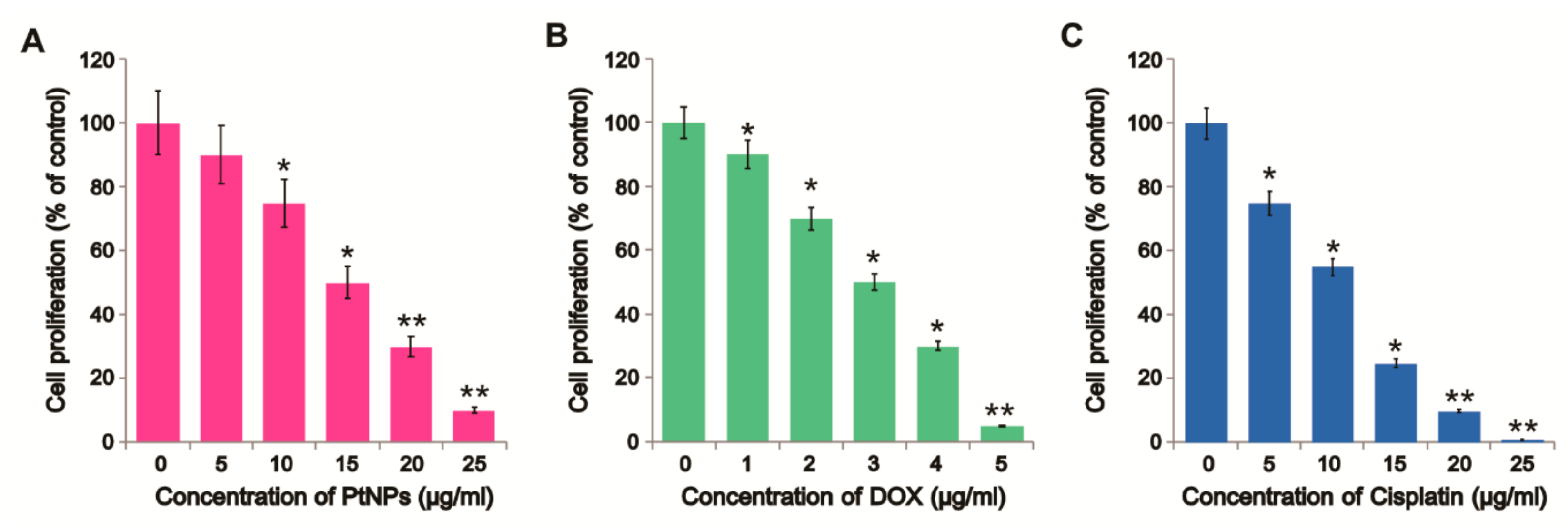
3.2. Dose-Dependent Effect of PtNPs, DOX, and CIS on U2OS Cell Viability
3.3. Dose-Dependent Effect of PtNPs, DOX, and CIS on U2OS Cell Proliferation
3.4. Cellular Quantification of PtNPs Using ICP-MS
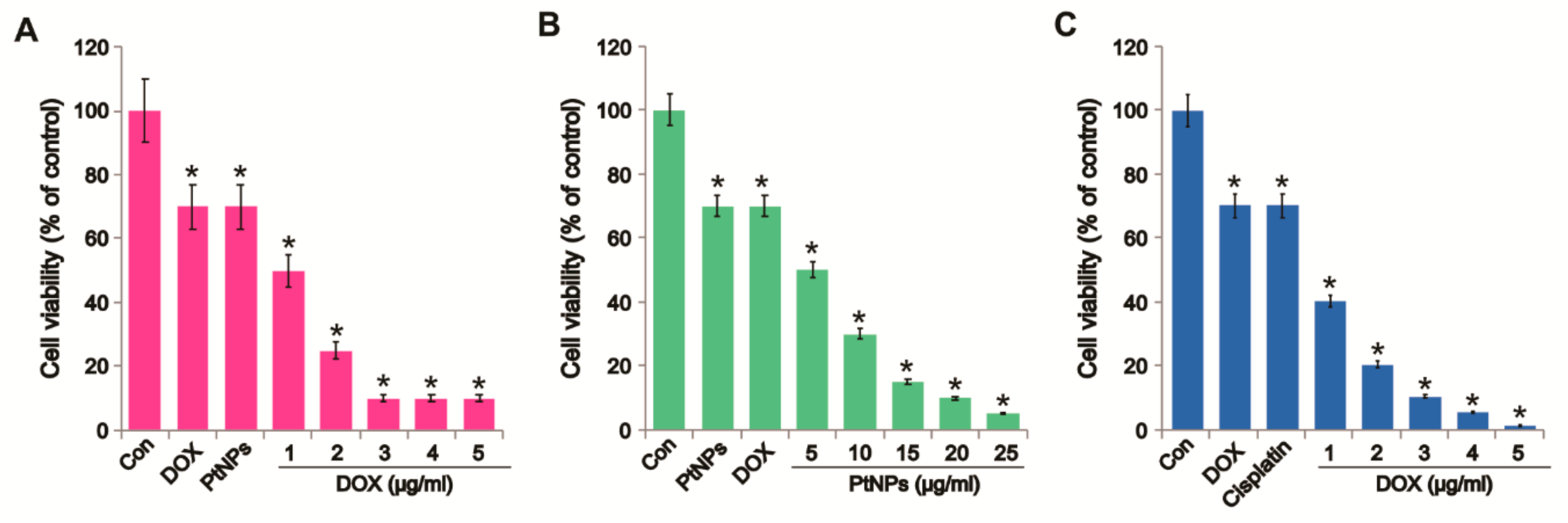
3.5. Combined Effects of PtNPs and DOX
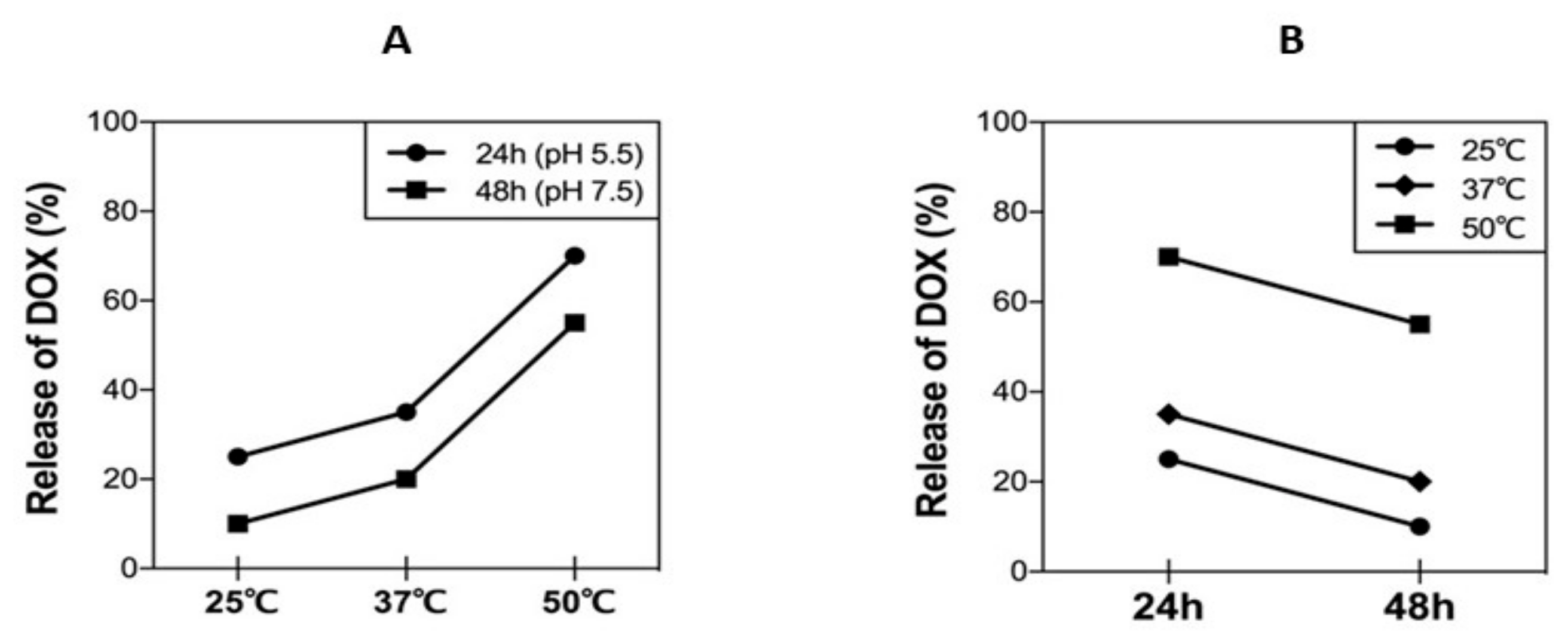
3.6. DOX Release Kinetics under Different Physiological Conditions
3.7. Determination of Combination Index of PtNPs and DOX
3.8. PtNPs and DOX Enhance LDH Leakage and Cell Death
3.9. PtNPs and DOX Alter Cell Morphology
3.10. PtNPs and DOX Induce ROS Generation and Lipid Peroxidation in U2OS Cells
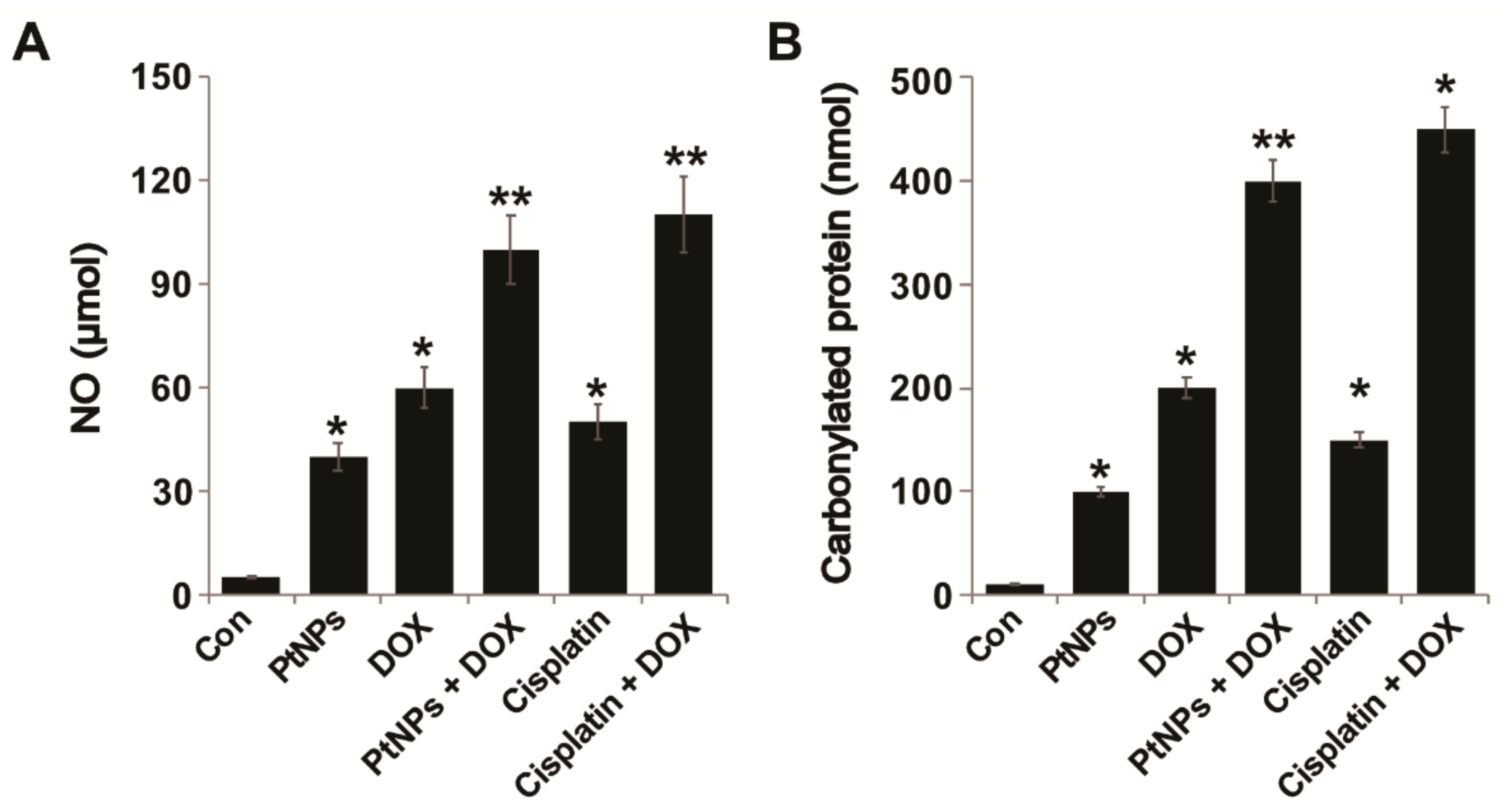
3.11. PtNPs and DOX Increase the Levels of NO and Carbonylated Protein
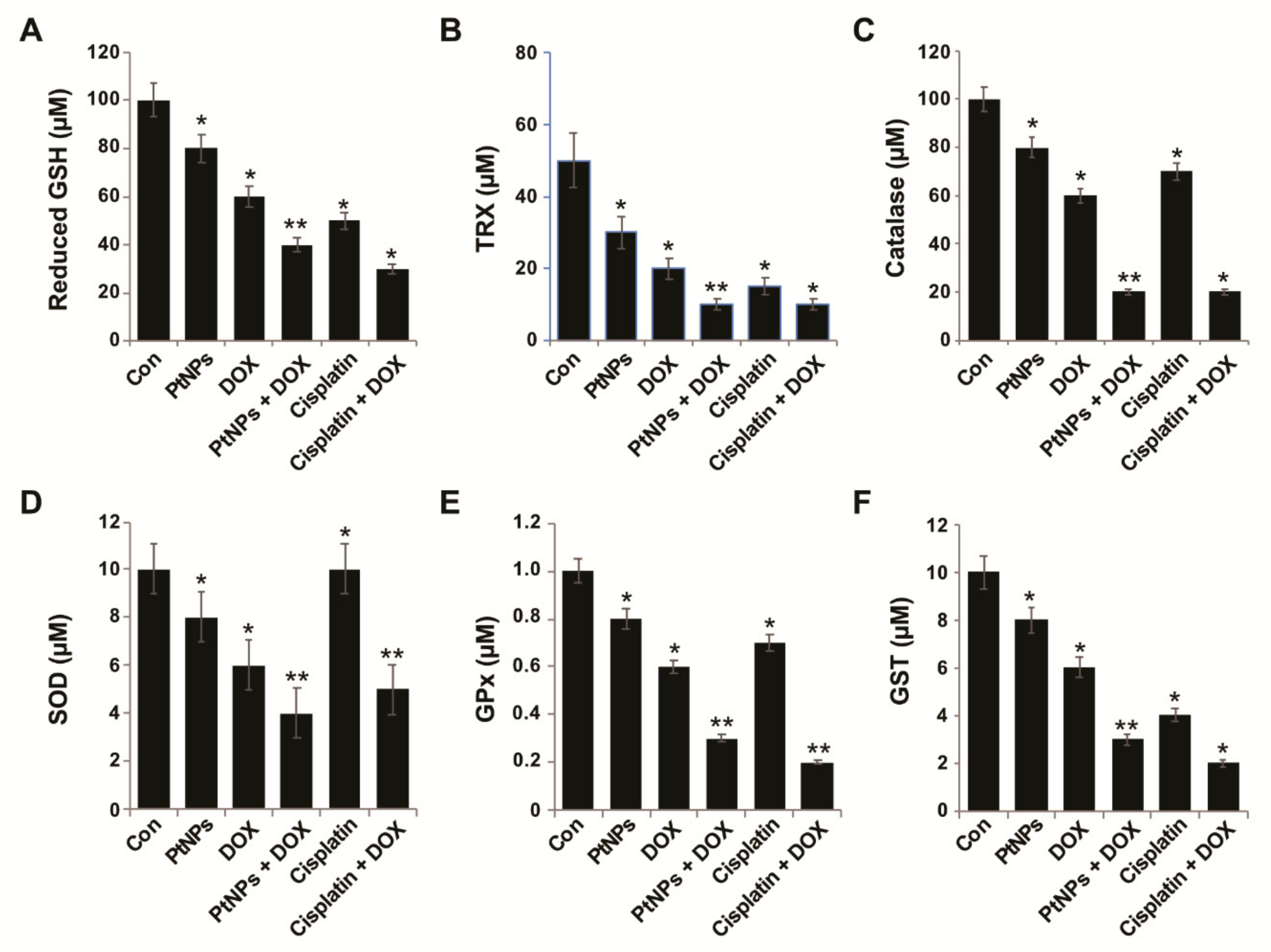
3.12. Effect of PtNPs and DOX on Antioxidants
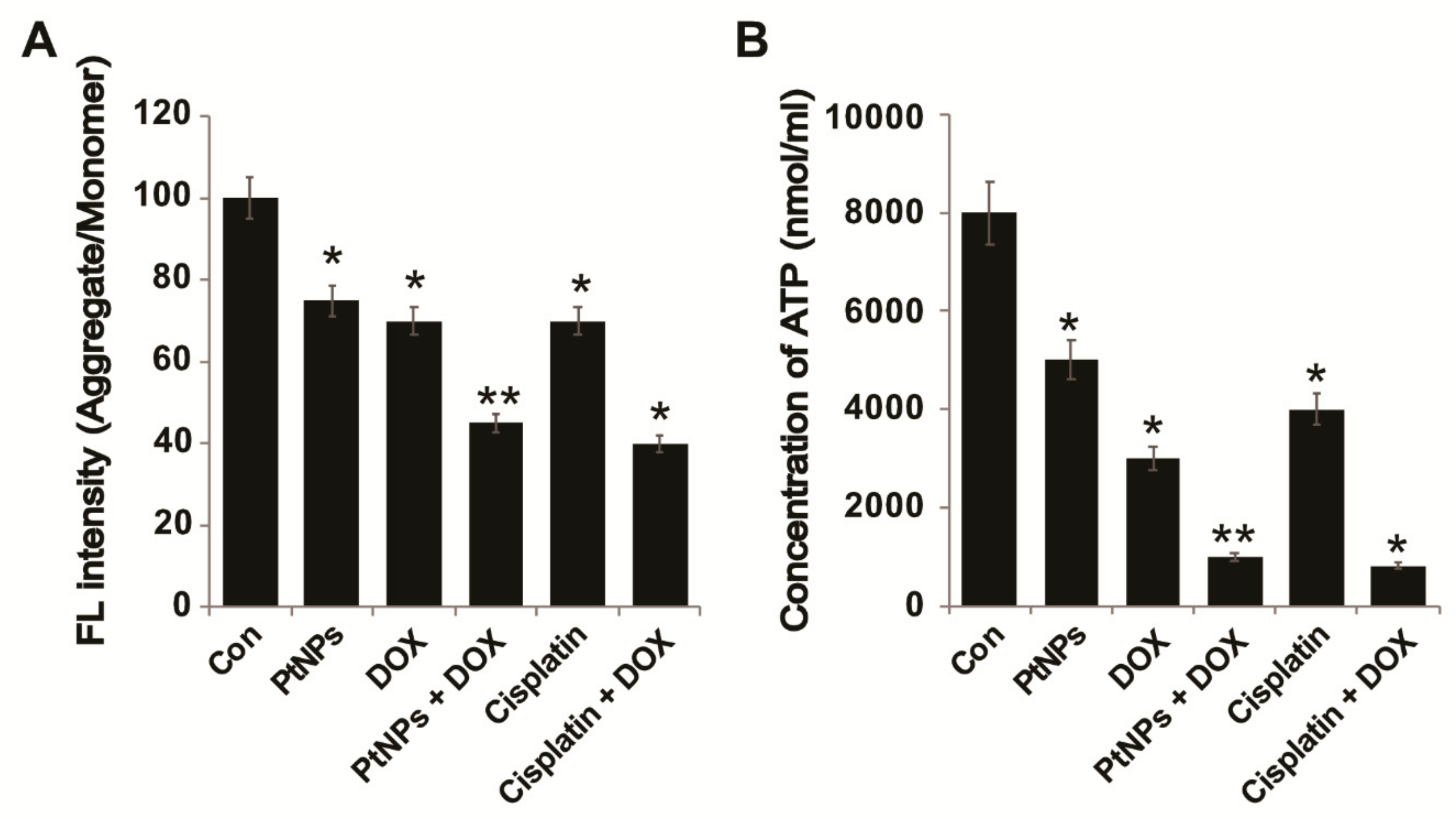
3.13. PtNPs and DOX Reduce the Mitochondrial Membrane Potential (MMP) and ATP Levels
3.14. PtNPs and DOX Enhance Proapoptotic and Downregulate Antiapoptotic Gene Expression
3.15. PtNPs and DOX Increase 8-oxo-dG and oxo-G Levels
3.16. PtNPs and DOX Alter DNA Repair Gene Expression
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Longhi, A.; Errani, C.; De Paolis, M.; Mercuri, M.; Bacci, G. Primary bone osteosarcoma in the pediatric age: State of the art. Cancer Treat. Rev. 2006, 32, 423–436. [Google Scholar] [CrossRef]
- Nthumba, P.M. Osteosarcoma of the jaws: A review of literature and a case report on synchronous multicentric osteosarcomas. World J. Surg. Oncol. 2012, 10, 240. [Google Scholar] [CrossRef]
- Anninga, J.K.; Gelderblom, H.; Fiocco, M.; Kroep, J.R.; Taminiau, A.H.; Hogendoorn, P.C.; Egeler, R.M. Chemotherapeutic adjuvant treatment for osteosarcoma: Where do we stand? Eur. J. Cancer 2011, 47, 2431–2445. [Google Scholar] [CrossRef]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef]
- Schwartz, C.L.; Gorlick, R.; Teot, L.; Krailo, M.; Chen, Z.; Goorin, A.; Grier, H.E.; Bernstein, M.L.; Meyers, P. Multiple drug resistance in osteogenic sarcoma: INT0133 from the Children’s Oncology Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 2057–2062. [Google Scholar] [CrossRef]
- Woodcock, J.; Griffin, J.P.; Behrman, R.E. Development of novel combination therapies. N. Engl. J. Med. 2011, 364, 985–987. [Google Scholar] [CrossRef]
- Cho, K.; Wang, X.; Nie, S.; Chen, Z.; Shin, D.M. Therapeutic Nanoparticles for Drug Delivery in Cancer. Clin. Cancer Res. 2008, 14, 1310–1316. [Google Scholar] [CrossRef]
- Sun, T.-M.; Du, J.-Z.; Yao, Y.-D.; Mao, C.-Q.; Dou, S.; Huang, S.-Y.; Zhang, P.-Z.; Leong, K.W.; Song, E.-W.; Wang, J. Simultaneous Delivery of siRNA and Paclitaxel via a “Two-in-One” Micelleplex Promotes Synergistic Tumor Suppression. ACS Nano 2011, 5, 1483–1494. [Google Scholar] [CrossRef]
- Wang, H.; Lee, D.-K.; Chen, K.-Y.; Chen, J.-Y.; Zhang, K.; Silva, A.; Ho, C.-M.; Ho, D. Mechanism-Independent Optimization of Combinatorial Nanodiamond and Unmodified Drug Delivery Using a Phenotypically Driven Platform Technology. ACS Nano 2015, 9, 3332–3344. [Google Scholar] [CrossRef]
- Song, X.R.; Cai, Z.; Zheng, Y.; He, G.; Cui, F.Y.; Gong, D.Q.; Hou, S.X.; Xiong, S.J.; Lei, X.J.; Wei, Y.Q. Reversion of multidrug resistance by co-encapsulation of vincristine and verapamil in PLGA nanoparticles. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2009, 37, 300–305. [Google Scholar] [CrossRef]
- Zhao, F.; Yin, H.; Li, J. Supramolecular self-assembly forming a multifunctional synergistic system for targeted co-delivery of gene and drug. Biomaterials 2014, 35, 1050–1062. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, E.; Cui, Y.; Huang, Y. Nanotechnology-based combination therapy for overcoming multidrug-resistant cancer. Cancer Biol. Med. 2017, 14, 212–227. [Google Scholar] [CrossRef]
- Patil, Y.; Sadhukha, T.; Ma, L.; Panyam, J. Nanoparticle-mediated simultaneous and targeted delivery of paclitaxel and tariquidar overcomes tumor drug resistance. J. Control. Release Off. J. Control. Release Soc. 2009, 136, 21–29. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, D.; Zhang, Q.; Chen, Y.; Zheng, D.; Hao, L.; Duan, C.; Jia, L.; Liu, G.; Liu, Y. Synergistic effect of folate-mediated targeting and verapamil-mediated P-gp inhibition with paclitaxel -polymer micelles to overcome multi-drug resistance. Biomaterials 2011, 32, 9444–9456. [Google Scholar] [CrossRef]
- Mitrus, I.; Sochanik, A.; Cichon, T.; Szala, S. Combination of combretastatin A4 phosphate and doxorubicin-containing liposomes affects growth of B16-F10 tumors. Acta Biochim. Pol. 2009, 56, 161–165. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, F.; Pan, Y.; Li, Z.; Xue, X.; Okeke, C.I.; Wang, Y.; Li, C.; Peng, L.; Wang, P.C.; et al. Nanodrug Formed by Coassembly of Dual Anticancer Drugs to Inhibit Cancer Cell Drug Resistance. ACS Appl. Mater. Interfaces 2015, 7, 19295–19305. [Google Scholar] [CrossRef]
- Mignani, S.; Bryszewska, M.; Klajnert-Maculewicz, B.; Zablocka, M.; Majoral, J.P. Advances in combination therapies based on nanoparticles for efficacious cancer treatment: An analytical report. Biomacromolecules 2015, 16, 1–27. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.H.; Qasim, M.; Kim, J.H. Nanoparticle-Mediated Combination Therapy: Two-in-One Approach for Cancer. Int. J. Mol. Sci. 2018, 19, 3264. [Google Scholar] [CrossRef]
- Frederick, C.A.; Williams, L.D.; Ughetto, G.; Van der Marel, G.A.; Van Boom, J.H.; Rich, A.; Wang, A.H. Structural comparison of anticancer drug-DNA complexes: Adriamycin and daunomycin. Biochemistry 1990, 29, 2538–2549. [Google Scholar] [CrossRef]
- Zhang, X.F.; Gurunathan, S. Combination of salinomycin and silver nanoparticles enhances apoptosis and autophagy in human ovarian cancer cells: An effective anticancer therapy. Int. J. Nanomed. 2016, 11, 3655–3675. [Google Scholar] [CrossRef]
- Yuan, Y.G.; Gurunathan, S. Combination of graphene oxide-silver nanoparticle nanocomposites and cisplatin enhances apoptosis and autophagy in human cervical cancer cells. Int. J. Nanomed. 2017, 12, 6537–6558. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.H.; Kim, J.H. Combination Effect of Silver Nanoparticles and Histone Deacetylases Inhibitor in Human Alveolar Basal Epithelial Cells. Molecules 2018, 23, 2046. [Google Scholar] [CrossRef]
- Gurunathan, S.; Jeyaraj, M.; Kang, M.H.; Kim, J.H. The Effects of Apigenin-Biosynthesized Ultra-Small Platinum Nanoparticles on the Human Monocytic THP-1 Cell Line. Cells 2019, 8, 444. [Google Scholar] [CrossRef]
- Sliwinska, A.; Kwiatkowski, D.; Czarny, P.; Toma, M.; Wigner, P.; Drzewoski, J.; Fabianowska-Majewska, K.; Szemraj, J.; Maes, M.; Galecki, P. The levels of 7, 8-dihydrodeoxyguanosine (8-oxoG) and 8-oxoguanine DNA glycosylase 1 (OGG1)–A potential diagnostic biomarkers of Alzheimer’s disease. J. Neurol. Sci. 2016, 368, 155–159. [Google Scholar] [CrossRef]
- Gurunathan, S.; Qasim, M.; Park, C.; Yoo, H.; Choi, D.Y.; Song, H.; Park, C.; Kim, J.H.; Hong, K. Cytotoxicity and Transcriptomic Analysis of Silver Nanoparticles in Mouse Embryonic Fibroblast Cells. Int. J. Mol. Sci. 2018, 19, 3618. [Google Scholar] [CrossRef]
- Teranishi, T.; Hosoe, M.; Tanaka, T.; Miyake, M. Size Control of Monodispersed Pt Nanoparticles and Their 2D Organization by Electrophoretic Deposition. J. Phys. Chem. B 1999, 103, 3818–3827. [Google Scholar] [CrossRef]
- Long, N.V.; Chien, N.D.; Uchida, M.; Matsubara, T.; Randy, J.; Masayuki, N. Directed and random self-assembly of Pt–Au nanoparticles. Mater. Chem. Phys. 2010, 124, 1193–1197. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kalishwaralal, K.; Vaidyanathan, R.; Venkataraman, D.; Pandian, S.R.; Muniyandi, J.; Hariharan, N.; Eom, S.H. Biosynthesis, purification and characterization of silver nanoparticles using Escherichia coli. Colloids Surf. B Biointerfaces 2009, 74, 328–335. [Google Scholar] [CrossRef]
- Sheny, D.S.; Philip, D.; Mathew, J. Synthesis of platinum nanoparticles using dried Anacardium occidentale leaf and its catalytic and thermal applications. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 114, 267–271. [Google Scholar] [CrossRef]
- Al-Radadi, N.S. Green synthesis of platinum nanoparticles using Saudi’s Dates extract and their usage on the cancer cell treatment. Arab. J. Chem. 2019, 12, 330–349. [Google Scholar] [CrossRef]
- Castro, L.; Blázquez, M.L.; González, F.; Muñoz, J.Á.; Ballester, A. Biosynthesis of silver and platinum nanoparticles using orange peel extract: Characterisation and applications. IET Nanobiotechnol. 2015, 9, 252–258. [Google Scholar] [CrossRef]
- Huang, J.; He, C.; Liu, X.; Xiao, Y.; Mya, K.Y.; Chai, J. Formation and characterization of water-soluble platinum nanoparticles using a unique approach based on the hydrosilylation reaction. Langmuir 2004, 20, 5145–5148. [Google Scholar] [CrossRef]
- Nadaroglu, H.; Gungor, A.A.; Ince, S.; Babagil, A. Green synthesis and characterisation of platinum nanoparticles using quail egg yolk. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 172, 43–47. [Google Scholar] [CrossRef]
- Michel, J.A.; Morris Iii, W.H.; Lukehart, C.M. Synthesis of shaped Pt nanoparticles using common anions or small molecules as shape-directing agents: Observation of a strong halide or pseudo-halide effect. J. Mater. Chem. A 2015, 3, 2012–2018. [Google Scholar] [CrossRef]
- Narayanan, R.; El-Sayed, M.A. Shape-Dependent Catalytic Activity of Platinum Nanoparticles in Colloidal Solution. Nano Lett. 2004, 4, 1343–1348. [Google Scholar] [CrossRef]
- Shah, M.A. Growth of uniform nanoparticles of platinum by an economical approach at relatively low temperature. Sci. Iran. 2012, 19, 964–966. [Google Scholar] [CrossRef]
- Dobrucka, R.; Kaczmarek, M.; Łagiedo, M.; Kielan, A.; Dlugaszewska, J. Evaluation of biologically synthesized Au-CuO and CuO-ZnO nanoparticles against glioma cells and microorganisms. Saudi Pharm. J. 2019, 27, 373–383. [Google Scholar] [CrossRef]
- Pal, J.; Deb, M.K.; Deshmukh, D.K.; Sen, B.K. Microwave-assisted synthesis of platinum nanoparticles and their catalytic degradation of methyl violet in aqueous solution. Appl. Nanosci. 2014, 4, 61–65. [Google Scholar] [CrossRef]
- Dauthal, P.; Mukhopadhyay, M. Biosynthesis of palladium nanoparticles using Delonix regia leaf extract and its catalytic activity for nitro-aromatics hydrogenation. Ind. Eng. Chem. Res. 2013, 52, 18131–18139. [Google Scholar] [CrossRef]
- Sahin, B.; Aygun, A.; Gunduz, H.; Sahin, K.; Demir, E.; Akocak, S.; Sen, F. Cytotoxic effects of platinum nanoparticles obtained from pomegranate extract by the green synthesis method on the MCF-7 cell line. Colloids Surf. B Biointerfaces 2018, 163, 119–124. [Google Scholar] [CrossRef]
- Sack, M.; Alili, L.; Karaman, E.; Das, S.; Gupta, A.; Seal, S.; Brenneisen, P. Combination of Conventional Chemotherapeutics with Redox-Active Cerium Oxide Nanoparticles—A Novel Aspect in Cancer Therapy. Mol. Cancer Ther. 2014, 13, 1740–1749. [Google Scholar] [CrossRef]
- Ji, G.-R.; Yu, N.-C.; Xue, X.; Li, Z.-G. 4-Hydroxy-2-nonenal Induces Apoptosis by Inhibiting AKT Signaling in Human Osteosarcoma Cells. Sci. World J. 2014, 2014, 7. [Google Scholar] [CrossRef]
- Pilco-Ferreto, N.; Calaf, G.M. Influence of doxorubicin on apoptosis and oxidative stress in breast cancer cell lines. Int. J. Oncol. 2016, 49, 753–762. [Google Scholar] [CrossRef]
- Alshatwi, A.A.; Athinarayanan, J.; Vaiyapuri Subbarayan, P. Green synthesis of platinum nanoparticles that induce cell death and G2/M-phase cell cycle arrest in human cervical cancer cells. J. Mater. Sci. Mater. Med. 2015, 26, 5330. [Google Scholar] [CrossRef]
- Kumari, M.; Purohit, M.P.; Patnaik, S.; Shukla, Y.; Kumar, P.; Gupta, K.C. Curcumin loaded selenium nanoparticles synergize the anticancer potential of doxorubicin contained in self-assembled, cell receptor targeted nanoparticles. Eur. J. Pharm. Biopharm. Off. J. Arb. Fur Pharm. Verfahr. E.V. 2018, 130, 185–199. [Google Scholar] [CrossRef]
- Gurunathan, S.; Jeyaraj, M.; Kang, M.H.; Kim, J.H. Graphene Oxide(-)Platinum Nanoparticle Nanocomposites: A Suitable Biocompatible Therapeutic Agent for Prostate Cancer. Polymers 2019, 11, 733. [Google Scholar] [CrossRef]
- Tanaka, M.; Okinaga, T.; Iwanaga, K.; Matsuo, K.; Toyono, T.; Sasaguri, M.; Ariyoshi, W.; Tominaga, K.; Enomoto, Y.; Matsumura, Y. Anticancer effect of novel platinum nanocomposite beads on oral squamous cell carcinoma cells. J. Biomed. Mater. Res. Part B Appl. Biomater. 2019. [Google Scholar] [CrossRef]
- Kutwin, M.; Sawosz, E.; Jaworski, S.; Wierzbicki, M.; Strojny, B.; Grodzik, M.; Ewa Sosnowska, M.; Trzaskowski, M.; Chwalibog, A. Nanocomplexes of Graphene Oxide and Platinum Nanoparticles against Colorectal Cancer Colo205, HT-29, HTC-116, SW480, Liver Cancer HepG2, Human Breast Cancer MCF-7, and Adenocarcinoma LNCaP and Human Cervical Hela B Cell Lines. Materials 2019, 12, 909. [Google Scholar] [CrossRef]
- Wang, L.; Wang, W.; Rui, Z.; Zhou, D. The effective combination therapy against human osteosarcoma: Doxorubicin plus curcumin co-encapsulated lipid-coated polymeric nanoparticulate drug delivery system. Drug Deliv. 2016, 23, 3200–3208. [Google Scholar] [CrossRef]
- Schott, C.R.; Ludwig, L.; Mutsaers, A.J.; Foster, R.A.; Wood, G.A. The autophagy inhibitor spautin-1, either alone or combined with doxorubicin, decreases cell survival and colony formation in canine appendicular osteosarcoma cells. PLoS ONE 2018, 13, e0206427. [Google Scholar] [CrossRef]
- Chen, L.; Ye, H.L.; Zhang, G.; Yao, W.M.; Chen, X.Z.; Zhang, F.C.; Liang, G. Autophagy inhibition contributes to the synergistic interaction between EGCG and doxorubicin to kill the hepatoma Hep3B cells. PLoS ONE 2014, 9, e85771. [Google Scholar] [CrossRef]
- Plichta, Z.; Kozak, Y.; Panchuk, R.; Sokolova, V.; Epple, M.; Kobylinska, L.; Jendelova, P.; Horak, D. Cytotoxicity of doxorubicin-conjugated poly[N-(2-hydroxypropyl)methacrylamide]-modified gamma-Fe2O3 nanoparticles towards human tumor cells. Beilstein J. Nanotechnol. 2018, 9, 2533–2545. [Google Scholar] [CrossRef]
- Al-Shakarchi, W.; Alsuraifi, A.; Abed, M.; Abdullah, M.; Richardson, A.; Curtis, A.; Hoskins, C. Combined Effect of Anticancer Agents and Cytochrome C Decorated Hybrid Nanoparticles for Liver Cancer Therapy. Pharmaceutics 2018, 10, 48. [Google Scholar] [CrossRef]
- Ortiz-Lazareno, P.C.; Bravo-Cuellar, A.; Lerma-Díaz, J.M.; Jave-Suárez, L.F.; Aguilar-Lemarroy, A.; Domínguez-Rodríguez, J.R.; González-Ramella, O.; De Célis, R.; Gómez-Lomelí, P.; Hernández-Flores, G. Sensitization of U937 leukemia cells to doxorubicin by the MG132 proteasome inhibitor induces an increase in apoptosis by suppressing NF-kappa B and mitochondrial membrane potential loss. Cancer Cell Int. 2014, 14, 13. [Google Scholar] [CrossRef]
- Al-Sheddi, E.S.; Farshori, N.N.; Al-Oqail, M.M.; Al-Massarani, S.M.; Saquib, Q.; Wahab, R.; Musarrat, J.; Al-Khedhairy, A.A.; Siddiqui, M.A. Anticancer Potential of Green Synthesized Silver Nanoparticles Using Extract of Nepeta deflersiana against Human Cervical Cancer Cells (HeLA). Bioinorg. Chem. Appl. 2018, 2018, 12. [Google Scholar] [CrossRef]
- Nel, A.; Xia, T.; Madler, L.; Li, N. Toxic potential of materials at the nanolevel. Science (New York) 2006, 311, 622–627. [Google Scholar] [CrossRef]
- Gurunathan, S.; Han, J.W.; Eppakayala, V.; Jeyaraj, M.; Kim, J.-H. Cytotoxicity of Biologically Synthesized Silver Nanoparticles in MDA-MB-231 Human Breast Cancer Cells. BioMed Res. Int. 2013, 2013, 10. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxidative Med. Cell. Longev. 2017, 2017, 11. [Google Scholar] [CrossRef]
- Zhou, T.; Prather, E.R.; Garrison, D.E.; Zuo, L. Interplay between ROS and Antioxidants during Ischemia-Reperfusion Injuries in Cardiac and Skeletal Muscle. Int. J. Mol. Sci. 2018, 19, 417. [Google Scholar] [CrossRef]
- Dai, F.; Wang, Q.; Fan, G.-J.; Du, Y.-T.; Zhou, B. ROS-driven and preferential killing of HepG2 over L-02 cells by a short-term cooperation of Cu(II) and a catechol-type resveratrol analog. Food Chem. 2018, 250, 213–220. [Google Scholar] [CrossRef]
- Han, J.W.; Gurunathan, S.; Jeong, J.K.; Choi, Y.J.; Kwon, D.N.; Park, J.K.; Kim, J.H. Oxidative stress mediated cytotoxicity of biologically synthesized silver nanoparticles in human lung epithelial adenocarcinoma cell line. Nanoscale Res. Lett. 2014, 9, 459. [Google Scholar] [CrossRef]
- Gurunathan, S.; Han, J.W.; Park, J.H.; Kim, E.; Choi, Y.J.; Kwon, D.N.; Kim, J.H. Reduced graphene oxide-silver nanoparticle nanocomposite: A potential anticancer nanotherapy. Int. J. Nanomed. 2015, 10, 6257–6276. [Google Scholar] [CrossRef]
- Yuan, Y.G.; Peng, Q.L.; Gurunathan, S. Silver nanoparticles enhance the apoptotic potential of gemcitabine in human ovarian cancer cells: Combination therapy for effective cancer treatment. Int. J. Nanomed. 2017, 12, 6487–6502. [Google Scholar] [CrossRef]
- Yuan, Y.G.; Peng, Q.L.; Gurunathan, S. Combination of palladium nanoparticles and tubastatin-A potentiates apoptosis in human breast cancer cells: A novel therapeutic approach for cancer. Int. J. Nanomed. 2017, 12, 6503–6520. [Google Scholar] [CrossRef]
- Zhang, X.F.; Huang, F.H.; Zhang, G.L.; Bai, D.P.; Massimo, D.F.; Huang, Y.F.; Gurunathan, S. Novel biomolecule lycopene-reduced graphene oxide-silver nanoparticle enhances apoptotic potential of trichostatin A in human ovarian cancer cells (SKOV3). Int. J. Nanomed. 2017, 12, 7551–7575. [Google Scholar] [CrossRef]
- Zhang, X.F.; Yan, Q.; Shen, W.; Gurunathan, S. Trichostatin A Enhances the Apoptotic Potential of Palladium Nanoparticles in Human Cervical Cancer Cells. Int. J. Mol. Sci. 2016, 17, 1354. [Google Scholar] [CrossRef]
- Almeer, R.S.; Ali, D.; Alarifi, S.; Alkahtani, S.; Almansour, M. Green Platinum Nanoparticles Interaction With HEK293 Cells: Cellular Toxicity, Apoptosis, and Genetic Damage. Dose-Response 2018, 16, 1559325818807382. [Google Scholar] [CrossRef]
- Sawyer, D.B.; Fukazawa, R.; Arstall, M.A.; Kelly, R.A. Daunorubicin-induced apoptosis in rat cardiac myocytes is inhibited by dexrazoxane. Circ. Res. 1999, 84, 257–265. [Google Scholar] [CrossRef]
- Huang, C.; Lu, X.; Tong, L.; Wang, J.; Zhang, W.; Jiang, B.; Yang, R. Requirement for endogenous heat shock factor 1 in inducible nitric oxide synthase induction in murine microglia. J. Neuroinflamm. 2015, 12, 189. [Google Scholar] [CrossRef]
- Cao, G.-J.; Fisher, C.M.; Jiang, X.; Chong, Y.; Zhang, H.; Guo, H.; Zhang, Q.; Zheng, J.; Knolhoff, A.M.; Croley, T.R.; et al. Platinum nanoparticles: An avenue for enhancing the release of nitric oxide from S-nitroso-N-acetylpenicillamine and S-nitrosoglutathione. Nanoscale 2018, 10, 11176–11185. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Schaur, R.J. Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Mol. Asp. Med. 2003, 24, 149–159. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef]
- Szokalska, A.; Makowski, M.; Nowis, D.; Wilczynski, G.M.; Kujawa, M.; Wojcik, C.; Mlynarczuk-Bialy, I.; Salwa, P.; Bil, J.; Janowska, S.; et al. Proteasome inhibition potentiates antitumor effects of photodynamic therapy in mice through induction of endoplasmic reticulum stress and unfolded protein response. Cancer Res. 2009, 69, 4235–4243. [Google Scholar] [CrossRef]
- Ortega, A.L.; Mena, S.; Estrela, J.M. Glutathione in cancer cell death. Cancers 2011, 3, 1285–1310. [Google Scholar] [CrossRef]
- Kretz-Remy, C.; Arrigo, A.P. Gene expression and thiol redox state. Methods Enzymol. 2002, 348, 200–215. [Google Scholar]
- Franco, R.; Sánchez-Olea, R.; Reyes-Reyes, E.M.; Panayiotidis, M.I. Environmental toxicity, oxidative stress and apoptosis: Ménage à Trois. Mutat. Res. Genet. Toxicol. Environ. Mutagenesis 2009, 674, 3–22. [Google Scholar] [CrossRef]
- Song, J.J.; Lee, Y.J. Differential role of glutaredoxin and thioredoxin in metabolic oxidative stress-induced activation of apoptosis signal-regulating kinase 1. Biochem. J. 2003, 373, 845–853. [Google Scholar] [CrossRef]
- Hlavacova, M.; Gumulec, J.; Stracina, T.; Fojtu, M.; Raudenska, M.; Masarik, M.; Novakova, M.; Paulova, H. Different doxorubicin formulations affect plasma 4-hydroxy-2-nonenal and gene expression of aldehyde dehydrogenase 3A1 and thioredoxin reductase 2 in rat. Physiol. Res. 2015, 64 (Suppl. S5), S653–S660. [Google Scholar]
- Arner, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef]
- Lee, J.H.; Cha, K.E.; Kim, M.S.; Hong, H.W.; Chung, D.J.; Ryu, G.; Myung, H. Nanosized polyamidoamine (PAMAM) dendrimer-induced apoptosis mediated by mitochondrial dysfunction. Toxicol. Lett. 2009, 190, 202–207. [Google Scholar] [CrossRef]
- Jeong, J.-K.; Gurunathan, S.; Kang, M.-H.; Han, J.W.; Das, J.; Choi, Y.-J.; Kwon, D.-N.; Cho, S.-G.; Park, C.; Seo, H.G.; et al. Hypoxia-mediated autophagic flux inhibits silver nanoparticle-triggered apoptosis in human lung cancer cells. Sci. Rep. 2016, 6, 21688. [Google Scholar] [CrossRef]
- Mordente, A.; Meucci, E.; Silvestrini, A.; Martorana, G.E.; Giardina, B. Anthracyclines and mitochondria. Adv. Exp. Med. Biol. 2012, 942, 385–419. [Google Scholar] [CrossRef]
- Gharanei, M.; Hussain, A.; Janneh, O.; Maddock, H.L. Doxorubicin induced myocardial injury is exacerbated following ischaemic stress via opening of the mitochondrial permeability transition pore. Toxicol. Appl. Pharmacol. 2013, 268, 149–156. [Google Scholar] [CrossRef]
- Carvalho, F.S.; Burgeiro, A.; Garcia, R.; Moreno, A.J.; Carvalho, R.A.; Oliveira, P.J. Doxorubicin-induced cardiotoxicity: From bioenergetic failure and cell death to cardiomyopathy. Med. Res. Rev. 2014, 34, 106–135. [Google Scholar] [CrossRef]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef]
- Govender, R.; Phulukdaree, A.; Gengan, R.M.; Anand, K.; Chuturgoon, A.A. Silver nanoparticles of Albizia adianthifolia: The induction of apoptosis in human lung carcinoma cell line. J. Nanobiotechnol. 2013, 11, 5. [Google Scholar] [CrossRef]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Mohan, G.M.; Shailender Naik, G.; Malla, R. Synergistic effects of coralyne and paclitaxel on cell migration and proliferation of breast cancer cells lines. Biomed. Pharmacother. 2017, 91, 436–445. [Google Scholar] [CrossRef]
- Lu, Y.; Yagi, T. Apoptosis of Human Tumor Cells by Chemotherapeutic Anthracyclines is Enhanced by Bax Overexpression. J. Radiat. Res. 1999, 40, 263–272. [Google Scholar] [CrossRef][Green Version]
- Jiang, X.; Wang, X. Cytochrome C-mediated apoptosis. Annu. Rev. Biochem. 2004, 73, 87–106. [Google Scholar] [CrossRef]
- Mieyal, J.J.; Gallogly, M.M.; Qanungo, S.; Sabens, E.A.; Shelton, M.D. Molecular Mechanisms and Clinical Implications of Reversible Protein S-Glutathionylation. Antioxid. Redox Signal. 2008, 10, 1941–1988. [Google Scholar] [CrossRef]
- Radak, Z.; Boldogh, I. 8-Oxo-7,8-dihydroguanine: Links to gene expression, aging, and defense against oxidative stress. Free Radic. Biol. Med. 2010, 49, 587–596. [Google Scholar] [CrossRef]
- Eustaquio, T.; Leary, J.F. Single-cell nanotoxicity assays of superparamagnetic iron oxide nanoparticles. Methods Mol. Biol. (Clifton N.J.) 2012, 926, 69–85. [Google Scholar] [CrossRef]
- Guo, C.; Ding, P.; Xie, C.; Ye, C.; Ye, M.; Pan, C.; Cao, X.; Zhang, S.; Zheng, S. Potential application of the oxidative nucleic acid damage biomarkers in detection of diseases. Oncotarget 2017, 8, 75767. [Google Scholar] [CrossRef]
- Fleming, A.M.; Zhou, J.; Wallace, S.S.; Burrows, C.J. A Role for the Fifth G-Track in G-Quadruplex Forming Oncogene Promoter Sequences during Oxidative Stress: Do These “Spare Tires” Have an Evolved Function? ACS Cent. Sci. 2015, 1, 226–233. [Google Scholar] [CrossRef]
- Guo, C.; Li, X.; Wang, R.; Yu, J.; Ye, M.; Mao, L.; Zhang, S.; Zheng, S. Association between Oxidative DNA Damage and Risk of Colorectal Cancer: Sensitive Determination of Urinary 8-Hydroxy-2′-deoxyguanosine by UPLC-MS/MS Analysis. Sci. Rep. 2016, 6, 32581. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.H.; Jeyaraj, M.; Kim, J.H. Differential Immunomodulatory Effect of Graphene Oxide and Vanillin-Functionalized Graphene Oxide Nanoparticles in Human Acute Monocytic Leukemia Cell Line (THP-1). Int. J. Mol. Sci. 2019, 20, 247. [Google Scholar] [CrossRef]
- Tsoli, M.; Kuhn, H.; Brandau, W.; Esche, H.; Schmid, G. Cellular Uptake and Toxicity of Au55 Clusters. Small 2005, 1, 841–844. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, D.; Yang, S.; Wang, Y.; Tang, Z.; Fu, X. Co-administration of genistein with doxorubicin-loaded polypeptide nanoparticles weakens the metastasis of malignant prostate cancer by amplifying oxidative damage. Biomater. Sci. 2018, 6, 827–835. [Google Scholar] [CrossRef]
- Hirano, T. Repair system of 7, 8-dihydro-8-oxoguanine as a defense line against carcinogenesis. J. Radiat. Res. 2008, 49, 329–340. [Google Scholar] [CrossRef]
- Sampath, H. Oxidative DNA damage in disease—Insights gained from base excision repair glycosylase-deficient mouse models. Environ. Mol. Mutagenesis 2014, 55, 689–703. [Google Scholar] [CrossRef]
- Van Berlo, D.; Hullmann, M.; Wessels, A.; Scherbart, A.M.; Cassee, F.R.; Gerlofs-Nijland, M.E.; Albrecht, C.; Schins, R.P.F. Investigation of the effects of short-term inhalation of carbon nanoparticles on brains and lungs of c57bl/6j and p47(phox-/-) mice. Neurotoxicology 2014, 43, 65–72. [Google Scholar] [CrossRef]
- El-Said, K.S.; Ali, E.M.; Kanehira, K.; Taniguchi, A. Molecular mechanism of DNA damage induced by titanium dioxide nanoparticles in toll-like receptor 3 or 4 expressing human hepatocarcinoma cell lines. J. Nanobiotechnol. 2014, 12, 48. [Google Scholar] [CrossRef]
- Nallanthighal, S.; Chan, C.; Murray, T.M.; Mosier, A.P.; Cady, N.C.; Reliene, R. Differential effects of silver nanoparticles on DNA damage and DNA repair gene expression in Ogg1-deficient and wild type mice. Nanotoxicology 2017, 11, 996–1011. [Google Scholar] [CrossRef]
- Singh, N.; Nelson, B.C.; Scanlan, L.D.; Coskun, E.; Jaruga, P.; Doak, S.H. Exposure to Engineered Nanomaterials: Impact on DNA Repair Pathways. Int. J. Mol. Sci. 2017, 18, 1515. [Google Scholar] [CrossRef]
- Biola-Clier, M.; Beal, D.; Caillat, S.; Libert, S.; Armand, L.; Herlin-Boime, N.; Sauvaigo, S.; Douki, T.; Carriere, M. Comparison of the DNA damage response in BEAS-2B and A549 cells exposed to titanium dioxide nanoparticles. Mutagenesis 2017, 32, 161–172. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | List of Primers |
|---|---|
| APEX1 | F:ATTGGCTGGAGGGCAGATCT |
| R:CCACTGGGTGAGGTTTTCTGA | |
| OGG1 | F:TCCTCCCTAGGTTTCCTCTC |
| R:TGAGACTAGTGACAGTGTTGG | |
| P53 | F:AGAGACCGTACAGAAGA |
| R:CTGTAGCATGGGATCCTTT | |
| P21 | F:GTTGCTGTCCGGACTACCG |
| R:AAAAACAATGCCACCACTCC | |
| Caspase-3 | F:AGGGGTCATTTATGGGACA |
| R:TACACGGGATCTGTTTCTTTG | |
| Cyt C | F: GCGTGTCCTTGGACTTAGAG |
| R: GGCGGCTGTGTAAGAGTATC | |
| Bax | F:CGAGCTGATCAGAACCATCA |
| R:GAAAAATGCCTTTCCCCTTC | |
| POLB | F:GTTTCAGAAGAGGTGCAGAG |
| R:AGTGAAATAGAGAACACCACAG | |
| Bcl-2 | F:TAAGCTGTCACAGAGGGGCT |
| R:TGAAGAGTTCCTCCACCACC | |
| CREB1 | F:CAGTTCAGTCTTCCTGTAAGGACT |
| R:CGTTTGTCATGGTTAGTGTC | |
| UNG | F:CTCTGCTTTAGTGTTCAAAGG |
| R:GAGTTCTGATTTAGCCAGGA | |
| R:CCTTTGTACCGTTGCATCCT | |
| GAPDH | F:AGGTCGGTGTGAACGGATTTG |
| R:TGTAGACCATGTAGTTGAGGTCA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurunathan, S.; Jeyaraj, M.; Kang, M.-H.; Kim, J.-H. Tangeretin-Assisted Platinum Nanoparticles Enhance the Apoptotic Properties of Doxorubicin: Combination Therapy for Osteosarcoma Treatment. Nanomaterials 2019, 9, 1089. https://doi.org/10.3390/nano9081089
Gurunathan S, Jeyaraj M, Kang M-H, Kim J-H. Tangeretin-Assisted Platinum Nanoparticles Enhance the Apoptotic Properties of Doxorubicin: Combination Therapy for Osteosarcoma Treatment. Nanomaterials. 2019; 9(8):1089. https://doi.org/10.3390/nano9081089
Chicago/Turabian StyleGurunathan, Sangiliyandi, Muniyandi Jeyaraj, Min-Hee Kang, and Jin-Hoi Kim. 2019. "Tangeretin-Assisted Platinum Nanoparticles Enhance the Apoptotic Properties of Doxorubicin: Combination Therapy for Osteosarcoma Treatment" Nanomaterials 9, no. 8: 1089. https://doi.org/10.3390/nano9081089
APA StyleGurunathan, S., Jeyaraj, M., Kang, M.-H., & Kim, J.-H. (2019). Tangeretin-Assisted Platinum Nanoparticles Enhance the Apoptotic Properties of Doxorubicin: Combination Therapy for Osteosarcoma Treatment. Nanomaterials, 9(8), 1089. https://doi.org/10.3390/nano9081089