Electrochemical Adsorption on Pt Nanoparticles in Alkaline Solution Observed Using In Situ High Energy Resolution X-ray Absorption Spectroscopy



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experiment
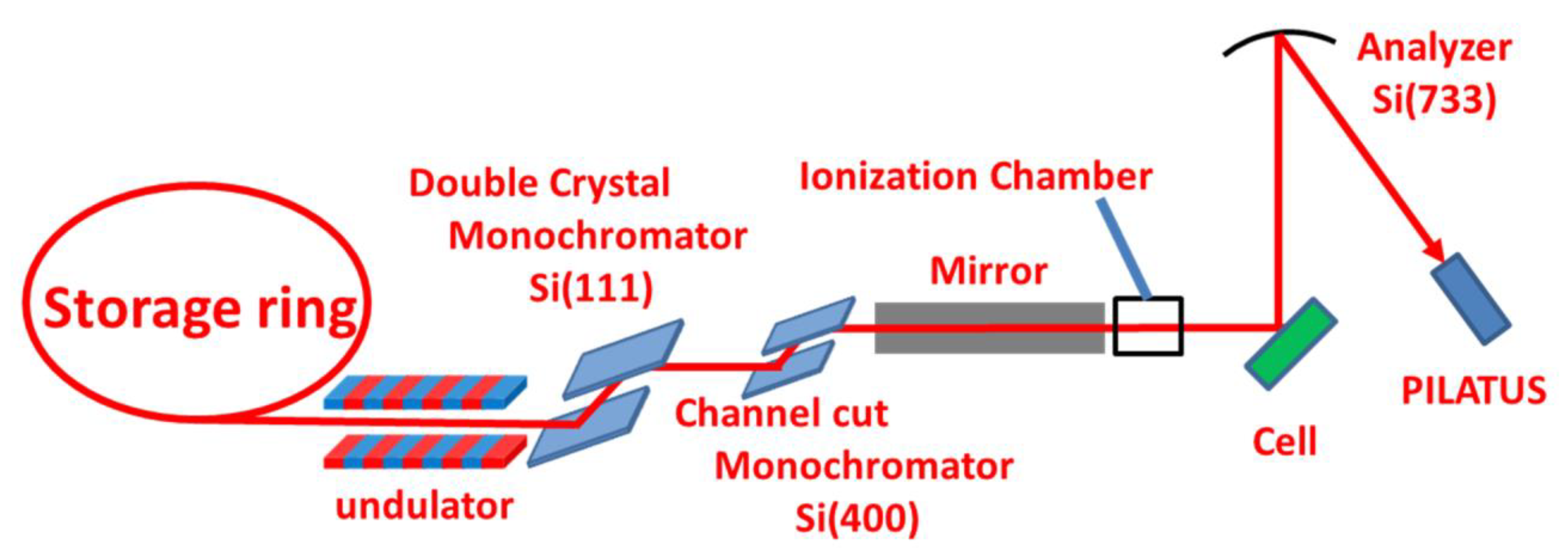
2.1. Electrochemical Cell
2.2. HERFD-XAS
2.3. Data Analysis
3. Results and Discussion
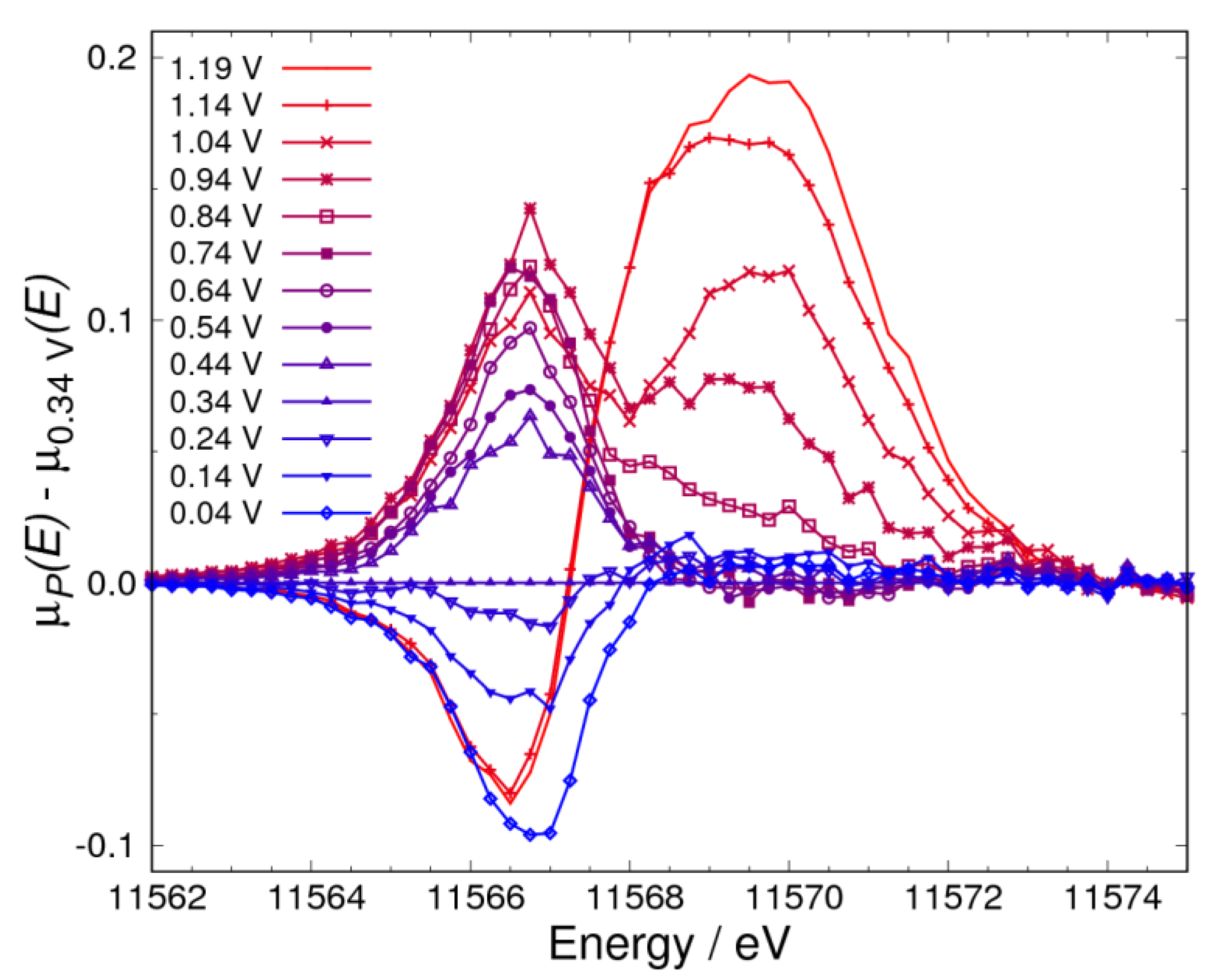
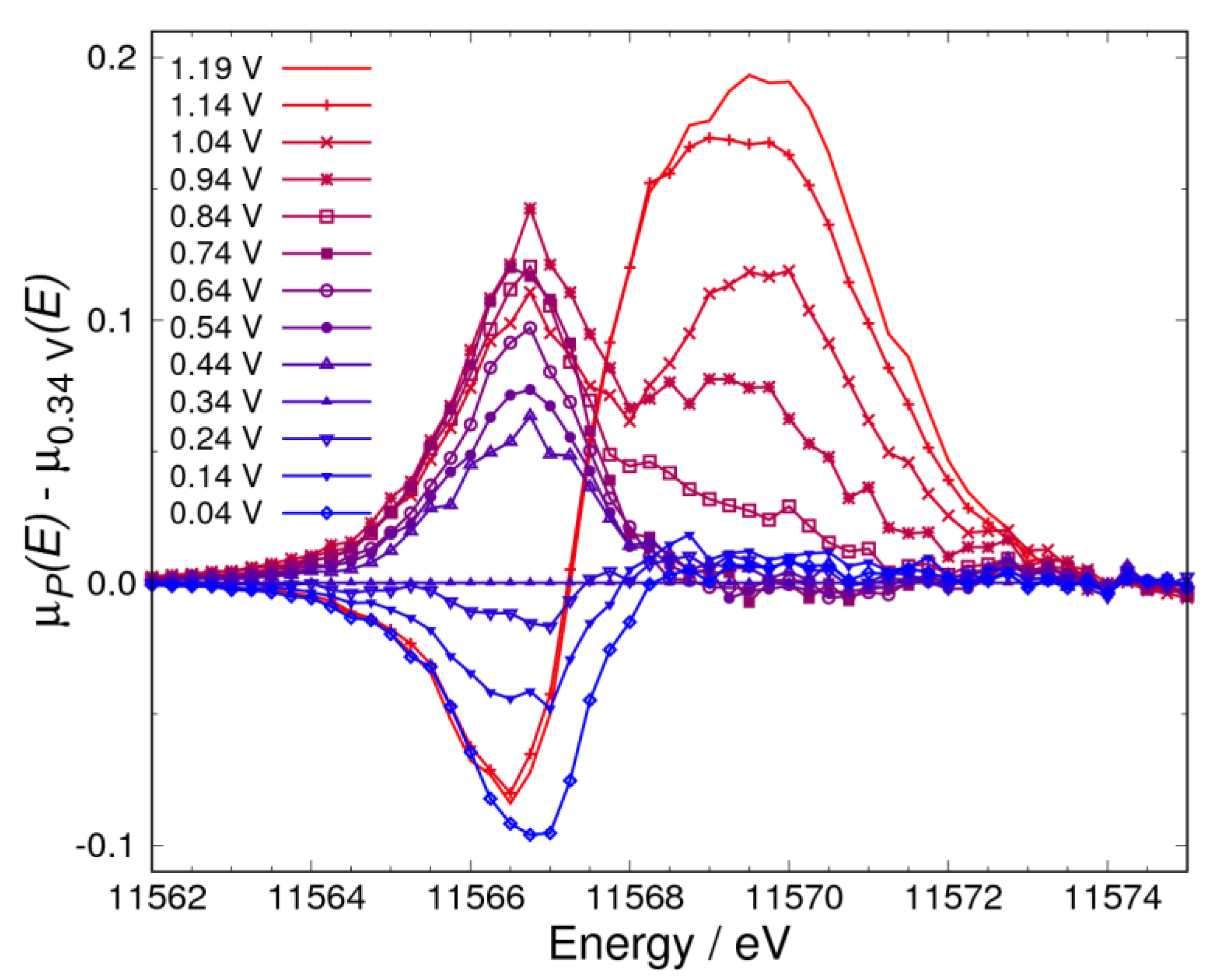
3.1. Δμ Analysis
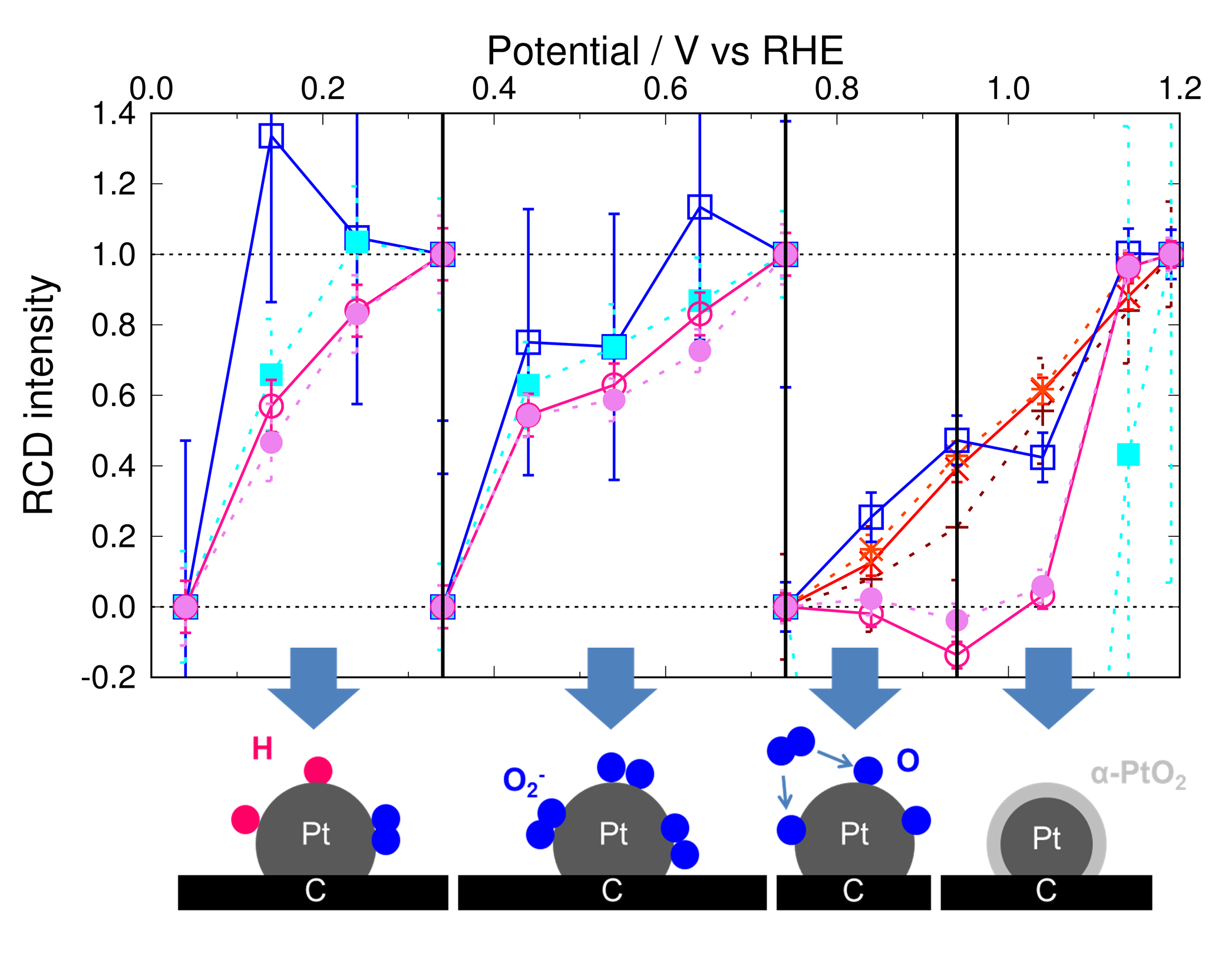
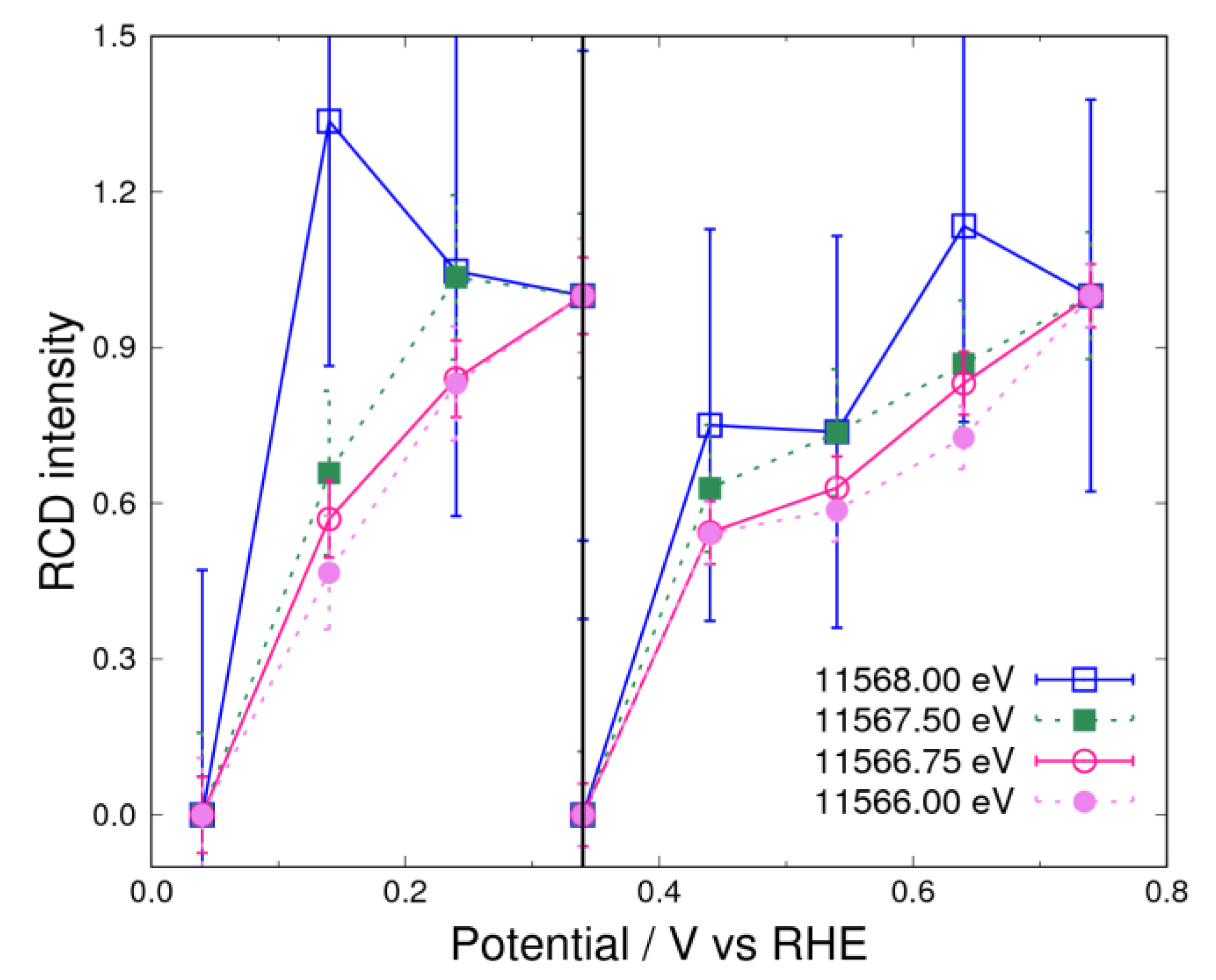
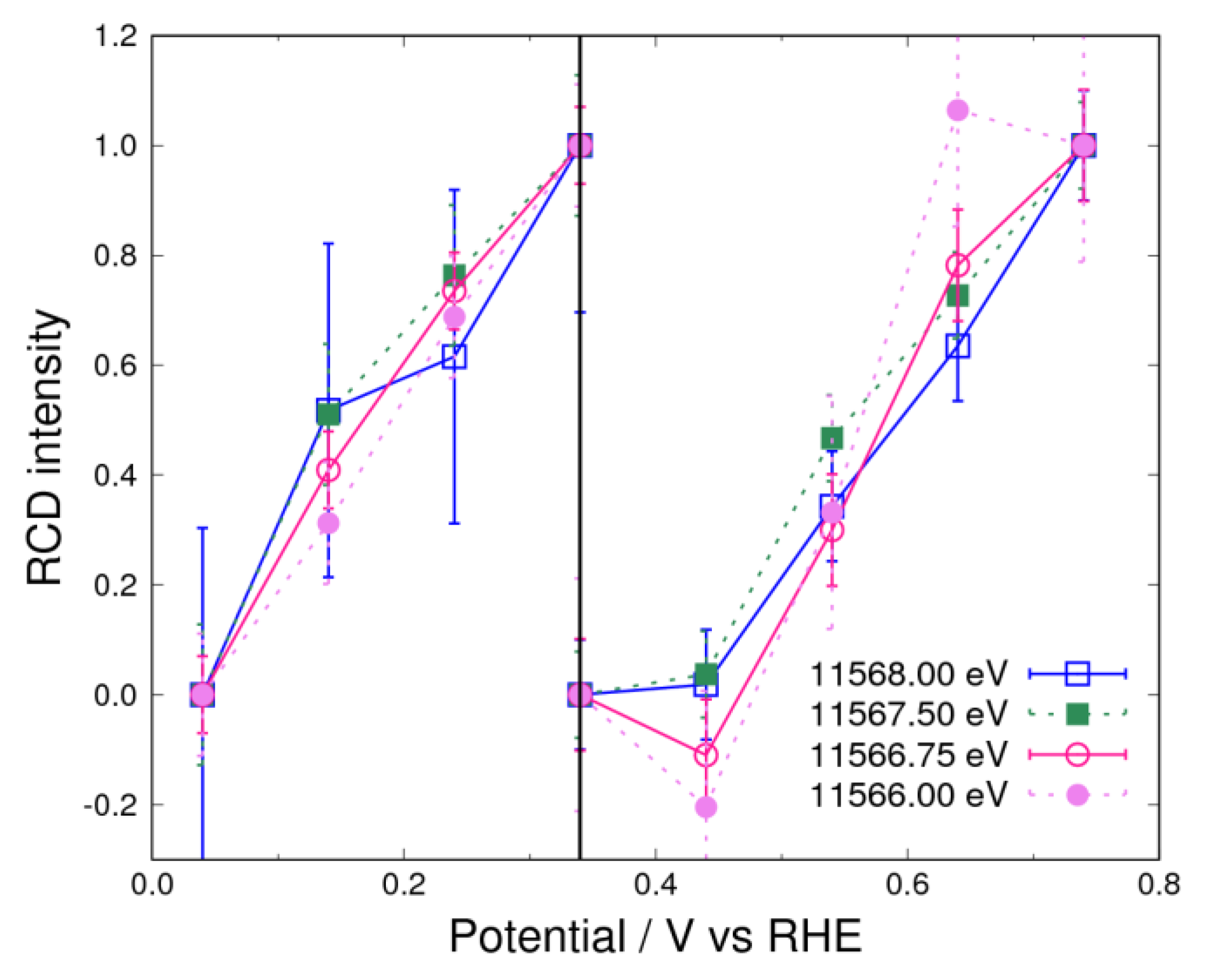
3.2. Adsorbates on Pt Surface at Low Potential (from 0.04 V to 0.74 V) in N2 Atmosphere
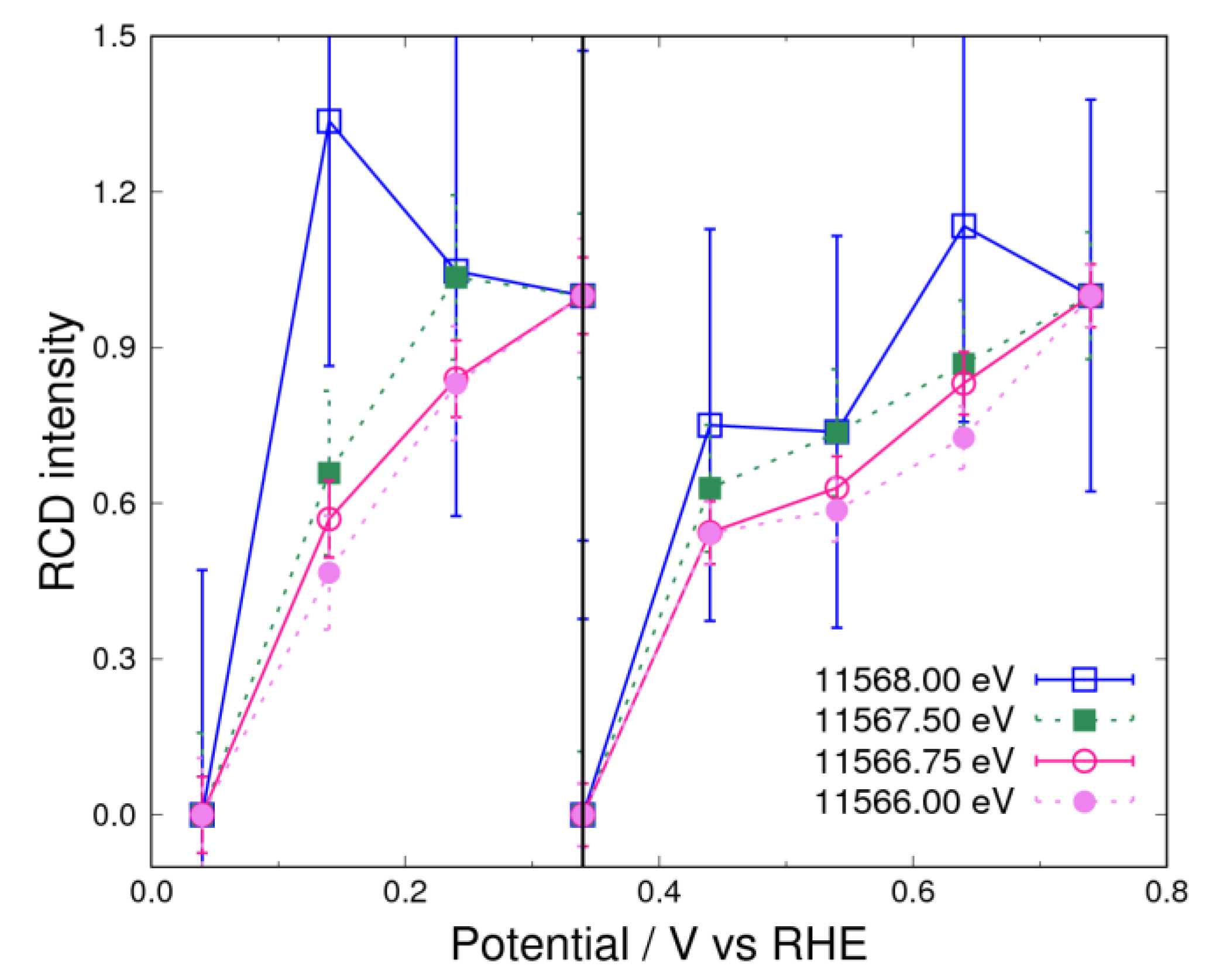
3.3. Adsorbates on Pt Surface at Low Potential (from 0.04 V to 0.74 V) in O2 Atmosphere
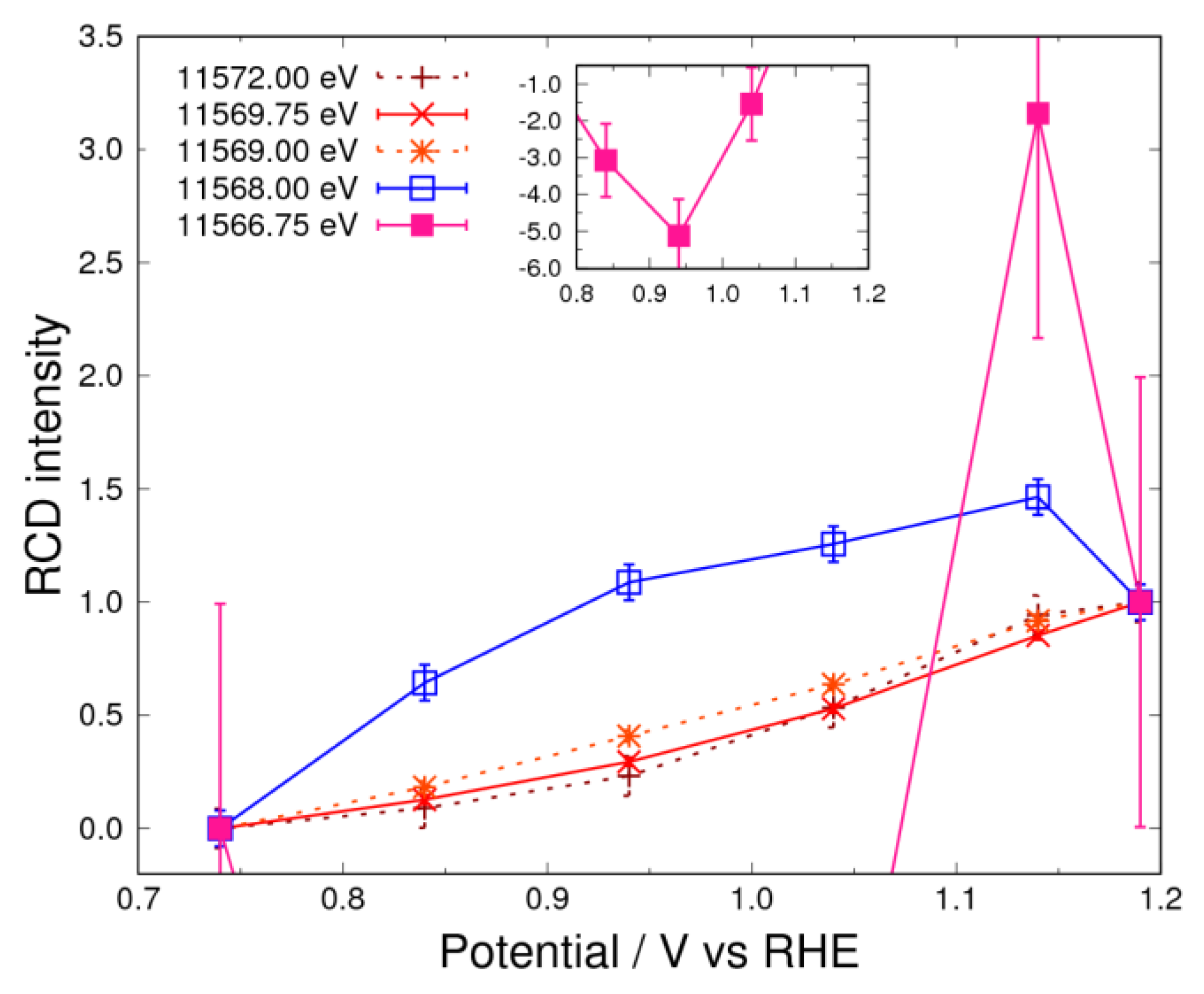
3.4. Pt Oxidation at High Potential (above 0.74 V) in N2 Atmosphere
3.5. Pt Oxidation at High Potential (above 0.74 V) in the O2 Atmosphere
3.6. ORR Mechanism in Alkaline Solution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Debe, M.K. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 2012, 486, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Suntivich, J.; Gasteiger, H.A.; Yabuuchi, N.; Nakanishi, H.; Goodenough, J.B.; Shao-Horn, Y. Design principles for oxygen-reduction activity on perovskite oxide catalysts for fuel cells and metal–air batteries. Nat. Chem. 2011, 3, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Marković, N.M.; Ross, P.N., Jr. Surface science studies of model fuel cell electrocatalysts. Surf. Sci. Rep. 2002, 45, 117–229. [Google Scholar] [CrossRef]
- Liu, S.; White, M.G.; Liu, P. Mechanism of oxygen reduction reaction on Pt(111) in alkaline solution: Importance of chemisorbed water on surface. J. Phys. Chem. C 2016, 120, 15288–15298. [Google Scholar] [CrossRef]
- Jin, W.; Du, H.; Zheng, S.; Xu, H.; Zheng, Y. Comparison of the oxygen reduction reaction between NaOH and KOH solutions on a Pt electrode: The electrolyte-dependent effect. J. Phys. Chem. B 2010, 114, 6542–6548. [Google Scholar] [CrossRef]
- Ramaswamy, N.; Mukerjee, S. Fundamental mechanistic understanding of electrocatalysis of oxygen reduction on Pt and non-Pt surfaces: Acid versus alkaline media. Adv. Phys. Chem. 2012, 2012, 491604. [Google Scholar] [CrossRef]
- Merte, L.R.; Behafarid, F.; Miller, D.J.; Friebel, D.; Cho, S.; Mbuga, F.; Sokaras, D.; Alonso-Mori, R.; Weng, T.-C.; Nordlund, D.; et al. Electrochemical oxidation of size-selected Pt nanoparticles studied using in situ high-energy-resolution x-ray absorption spectroscopy. ACS Catal. 2012, 2, 2371–2376. [Google Scholar] [CrossRef]
- Ramaker, D.E.; Korovina, A.; Croze, V.; Melke, J.; Roth, C. Following ORR intermediates adsorbed on a Pt cathode catalyst during break-in of a PEM fuel cell by in operando X-ray absorption spectroscopy. Phys. Chem. Chem. Phys. 2014, 16, 13645–13653. [Google Scholar] [CrossRef]
- Coleman, E.J.; Chowdhury, M.H.; Co, A.C. Insights into the oxygen reduction reaction activity of Pt/C and PtCu/C catalysts. ACS Catal. 2015, 5, 1245–1253. [Google Scholar] [CrossRef]
- Cui, Y.-T.; Harada, Y.; Niwa, H.; Hatanaka, T.; Nakamura, N.; Ando, M.; Yoshida, T.; Ishii, K.; Matsumura, D.; Oji, H.; et al. Wetting induced oxidation of Pt-based nano catalysts revealed by in situ high energy resolution x-ray absorption spectroscopy. Sci. Rep. 2017, 7, 1482. [Google Scholar] [CrossRef]
- Nagamatsu, S.; Arai, T.; Yamamoto, M.; Ohkura, T.; Oyanagi, H.; Ishizaka, T.; Kawanami, H.; Uruga, T.; Tada, M.; Iwasawa, Y. Potential-dependent restructuring and hysteresis in the structural and electronic transformations of Pt/C, Au(Core)-Pt(Shell)/C, and Pd(Core)-Pt(Shell)/C cathode catalysts in polymer electrolyte fuel cells characterized by in situ x-ray absorption fine structure. J. Phys. Chem. C 2013, 117, 13094–13107. [Google Scholar]
- Sasaki, K.; Marinkovic, N.; Isaacs, H.S.; Adzic, R.R. Synchrotron-based in situ characterization of carbon-supported platinum and platinum monolayer electrocatalysts. ACS Catal. 2016, 6, 69–76. [Google Scholar] [CrossRef]
- Tripković, A.V.; Popović, K.D.; Grgur, B.N.; Blizanac, B.; Ross, P.N.; Marković, N.M. Methanol electrooxidation on supported Pt and PtRu catalysts in acid and alkaline solutions. Electrochim. Acta 2002, 47, 3707–3714. [Google Scholar] [CrossRef]
- Prabhuram, J.; Manoharan, R. Investigation of methanol oxidation on unsupported platinum electrodes in strong alkali and strong acid. J. Power Source 1998, 74, 54–61. [Google Scholar] [CrossRef]
- Asazawa, K.; Yamada, K.; Tanaka, H.; Oka, A.; Taniguchi, M.; Kobayashi, T. A Platinum-free zero-carbon-emission easy fuelling direct hydrazine fuel cell for vehicles. Angew. Chem. Int. Ed. 2007, 46, 8024–8027. [Google Scholar] [CrossRef]
- Varcoe, J.R.; Slade, R.C.T. Prospects for alkaline anion-exchange membranes in low temperature fuel cells. Fuel Cells 2005, 5, 187–200. [Google Scholar] [CrossRef]
- Adams, L.A.; Poynton, S.D.; Tamain, C.; Slade, R.C.T.; Varcoe, J.R. A carbon dioxide tolerant aqueous-electrolyte-free anion-exchange membrane alkaline fuel cell. ChemSusChem Chem. Sustain. Energy Mater. 2008, 1, 79–81. [Google Scholar] [CrossRef] [PubMed]
- Varcoe, J.R.; Slade, R.C.T.; Wright, G.L.; Chen, Y. Steady-state dc and impedance investigations of H2/O2 alkaline membrane fuel cells with commercial Pt/C, Ag/C, and Au/C cathodes. J. Phys. Chem. B 2006, 110, 21041–21049. [Google Scholar] [CrossRef]
- Varcoe, J.R.; Slade, R.C.T.; Yee, E.L.H.; Poynton, S.D.; Driscoll, D.J.; Apperley, D.C. Poly(ethylene-co-tetrafluoroethylene)-derived radiation-grafted anion-exchange membrane with properties specifically tailored for application in metal-cation-free alkaline polymer electrolyte fuel cells. Chem. Mater. 2007, 19, 2686–2693. [Google Scholar] [CrossRef]
- Sakamoto, T.; Masuda, T.; Yoshimoto, K.; Kishi, H.; Yamaguchi, S.; Matsumura, D.; Tamura, K.; Hori, A.; Horiuchi, Y.; Serov, A.; et al. NiO/Nb2O5/C hydrazine electrooxidation catalysts for anion exchange membrane fuel cells. J. Electrochem. Soc. 2017, 163, F229–CF234. [Google Scholar] [CrossRef]
- Huang, W.; Ahlfield, J.M.; Zhang, X.; Kohl, P.A. Platinum supported on functionalized carbon nanotubes for oxygen reduction reaction in PEM/AEM hybrid fuel cells. J. Electrochem. Soc. 2017, 164, F217–F223. [Google Scholar] [CrossRef]
- Ahlfield, J.M.; Liu, L.; Kohl, P.A. PEM/AEM junction design for bipolar membrane fuel cells. J. Electrochem. Soc. 2017, 164, F1165–CF1171. [Google Scholar] [CrossRef]
- Shao, M.; Liu, P.; Adzic, R.R. Superoxide anion is the intermediate in the oxygen reduction reaction on platinum electrodes. J. Am. Chem. Soc. 2006, 128, 7408–7409. [Google Scholar] [CrossRef]
- Quaino, P.; Luque, N.B.; Nazmutdinov, R.; Santos, E.; Schmickler, W. Why is gold such a good catalyst for oxygen reduction in alkaline media? Angew. Chem. Int. Ed. 2012, 51, 12997–13000. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.M.C.; Quaino, P.; Arce, M.D.; Santos, E.; Schmickler, W. Electrochemical adsorption of OH on Pt(111) in alkaline solutions: Combining DFT and molecular dynamics. Chem. Phys. Chem. 2014, 15, 2003–2009. [Google Scholar] [CrossRef]
- Glatzel, P.; Singh, J.; Kvashnina, K.O.; van Bokhoven, J.A. In situ characterization of the 5d density of states of Pt nanoparticles upon adsorption of CO. J. Am. Chem. Soc. 2010, 132, 2555–2557. [Google Scholar] [CrossRef] [PubMed]
- Imai, H.; Izumi, K.; Matsumoto, M.; Kubo, Y.; Kato, K.; Imai, Y. In situ and real-time monitoring of oxide growth in a few monolayers at surfaces of platinum nanoparticles in aqueous media. J. Am. Chem. Soc. 2009, 131, 6293–6300. [Google Scholar] [CrossRef]
- Miller, D.J.; Öberg, H.; Kaya, S.; Sanchez Casalongue, H.; Friebel, D.; Anniyev, T.; Ogasawara, H.; Bluhm, H.; Pettersson, L.G.M.; Nilsson, A. Oxidation of Pt(111) under near-ambient conditions. Phys. Rev. Lett. 2011, 107, 195502. [Google Scholar] [CrossRef]
- Kityakarn, S.; Saida, T.; Sode, A.; Ishiguro, N.; Sekizawa, O.; Uruga, T.; Nagasawa, K.; Yamamoto, T.; Yokoyama, T.; Tada, M. In situ time-resolved XAFS of transitional states of Pt/C cathode electrocatalyst in an MEA during PEFC loading with transient voltages. Top. Catal. 2014, 57, 903–910. [Google Scholar] [CrossRef]
- Kusano, S.; Matsumura, D.; Asazawa, K.; Kishi, H.; Sakamoto, T.; Yamaguchi, S.; Tanaka, H.; Mizuki, J. Study of catalytic reaction at electrode–electrolyte interfaces by a CV-XAFS method. J. Electr. Mater. 2017, 46, 3634–3638. [Google Scholar] [CrossRef]
- Friebel, D.; Miller, D.J.; O’Grady, C.P.; Anniyev, T.; Bargar, J.; Bergmann, U.; Ogasawara, H.; Wikfeldt, K.T.; Pettersson, L.G.M.; Nilsson, A. In situ x-ray probing reveals fingerprints of surface platinum oxide. Phys. Chem. Chem. Phys. 2011, 13, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Imaoka, T.; Chun, W.-J.; Enoki, O.; Katoh, H.; Takenaga, M.; Sonoi, A. Size-specific catalytic activity of platinum clusters enhances oxygen reduction reactions. Nat. Chem. 2009, 1, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Nesselberger, M.; Roefzaad, M.; Hamou, R.F.; Biedermann, P.U.; Schweinberger, F.F.; Kunz, S.; Schloegl, K.; Wiberg, G.K.H.; Ashton, S.; Heiz, U.; et al. The effect of particle proximity on the oxygen reduction rate of size-selected platinum clusters. Nat. Mater. 2013, 12, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Fan, F.-R.F.; Bard, A.J. Electrochemistry of oxygen in concentrated NaOH solutions: Solubility, diffusion coefficients, and superoxide formation. J. Am. Chem. Soc. 2009, 131, 177–181. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kusano, S.; Matsumura, D.; Ishii, K.; Tanaka, H.; Mizuki, J. Electrochemical Adsorption on Pt Nanoparticles in Alkaline Solution Observed Using In Situ High Energy Resolution X-ray Absorption Spectroscopy. Nanomaterials 2019, 9, 642. https://doi.org/10.3390/nano9040642
Kusano S, Matsumura D, Ishii K, Tanaka H, Mizuki J. Electrochemical Adsorption on Pt Nanoparticles in Alkaline Solution Observed Using In Situ High Energy Resolution X-ray Absorption Spectroscopy. Nanomaterials. 2019; 9(4):642. https://doi.org/10.3390/nano9040642
Chicago/Turabian StyleKusano, Shogo, Daiju Matsumura, Kenji Ishii, Hirohisa Tanaka, and Jun’ichiro Mizuki. 2019. "Electrochemical Adsorption on Pt Nanoparticles in Alkaline Solution Observed Using In Situ High Energy Resolution X-ray Absorption Spectroscopy" Nanomaterials 9, no. 4: 642. https://doi.org/10.3390/nano9040642