First-Principles Study of Thermo-Physical Properties of Pu-Containing Gd2Zr2O7
by
 and
and
Pengcheng Li
1, 
Fengai Zhao
2,
Haiyan Xiao
1,*,
Haibin Zhang
3,*,
Hengfeng Gong
4,
Sa Zhang
1,
Zijiang Liu
5 and
Xiaotao Zu
1,2 1
School of Physics, University of Electronic Science and Technology of China, Chengdu 610054, China
2
Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 610054, China
3
Institute of Nuclear Physics and Chemistry, Chinese Academy of Engineering Physics, Mianyang 621900, China
4
Department of ATF R&D, China Nuclear Power Technology Research Institute Co., Ltd., Shenzhen 518000, China
5
Department of Physics, Lanzhou City University, Lanzhou 730070, China
*
Authors to whom correspondence should be addressed.
Nanomaterials 2019, 9(2), 196; https://doi.org/10.3390/nano9020196
Submission received: 2 January 2019
/
Revised: 30 January 2019
/
Accepted: 31 January 2019
/
Published: 3 February 2019
Abstract
:A density functional theory plus Hubbard U method is used to investigate how the incorporation of Pu waste into Gd2Zr2O7 pyrochlore influences its thermo-physical properties. It is found that immobilization of Pu at Gd-site of Gd2Zr2O7 has minor effects on the mechanical and thermal properties, whereas substitution of Pu for Zr-site results in remarkable influences on the structural parameters, elastic moduli, elastic isotropy, Debye temperature and electronic structure. The discrepancy in thermo-physical properties between Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7 may be a result of their different structural and electronic structures. This study provides a direct insight into the thermo-physical properties of Pu-containing Gd2Zr2O7, which will be important for further investigation of nuclear waste immobilization by pyrochlores.
1. Introduction
As the nuclear industry develops fast, ways to treat spent fuel and nuclear waste safely, such as plutonium and minor actinides (Np, Am, Cm), has become an important environmental conservation issue [1,2,3]. It is acceptable to store spent fuel and separated waste in stainless steel vessels in the short term, but in the long term it is hoped that this material will be transformed into more secure and manageable solids [1,4,5]. One method proposed for the treatment of plutonium is immobilization in zirconate pyrochlores, particularly Gd2Zr2O7, which has high thermal stability, high chemical durability, and high radiation tolerance [6,7,8,9]. Besides, Gd is an effective neutron absorber [6].
Experimentally, Pu is often substituted by nonradioactive cerium (Ce), since they share the same crystallographic structure, and thermo-physical and chemical properties [10,11]. Zhao et al. synthesized (Gd1−xCex)2Zr2O7+x (0) solid solutions, indicating that Ce3+ ions can be incorporated into the Gd3+ sites. They proposed that the content of immobilized at Gd-site was less than 40 mol% [12]. On the other hand, Gd2(Zr1−xCex)2O7 (0) solid solutions have been synthesized by Reid et al. [13] and Patwe et al. [14]. They found that Ce can be immobilized at Zr sites entirely as Ce4+, which leads to structural transformation from pyrochlore to fluorite phase, and its composition ranges from Gd2Ce0.2Zr1.8O7 to Gd2Ce1.7Zr0.3O7. Similar solution behavior and structural properties for Pu incorporation in Gd2Zr2O7 have been obtained by first-principles calculations [15,16,17]. However, different electronic structures can be obtained for Ce and Pu immobilization at the Gd-site of Gd2Zr2O7, i.e., the band gap increases and reduces when Ce and Pu substitutes for Gd site, respectively [15,18]. These differences mainly result from the different <Ce-O> and <Pu-O> interactions at band edges. The different electronic structure may result in varying mechanical properties. For Young’s modulus, which is described by with the Madelung constant and r0 the interionic spacing [19], it is very sensitive to r0. For ionic crystals, the r0 is affected by bond interactions. According to these analyses, large discrepancies in electronic structures between Ce and Pu incorporation in Gd2Zr2O7 may lead to different Young’s modulus. This indicates that for the mechanical properties of pyrochlores, Ce may not be a good substitute for Pu, despite the two having several similar thermo-physical properties. Thus far, there are no reports on the mechanical properties of Pu immobilization in Gd2Zr2O7. It is necessary to explore how Pu doping influences the mechanical properties of Gd2Zr2O7, because the knowledge of thermo-mechanical characteristics, for example, elastic moduli and Debye temperature is important for safe fuel disposal [20]. It provides new perspectives into the behavior of actinide incorporation in pyrochlores for their applications in harsh environments.
In this work, the structural, mechanical and electronic properties of Pu incorporation in Gd2Zr2O7 are investigated by the density functional theory plus the Hubbard U method (DFT+U). The remaining part of the paper is structured as follows: Section 2 lists computational details; Section 3 contains our results and discussions, involving the structural stability, elastic constants with elastic moduli of Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7, as well as the ductility, elastic anisotropy, Debye temperature and electronic structures of Pu-doped Gd2Zr2O7. In Section 4, we summarize our conclusions.
2. Computational Details
The density functional theory method within the Vienna Ab-initio Simulation Package (VASP) [21,22] are employed in all the computations. The Hubbard U correction [23] is considered to take into account the strong correlation interaction between 5f electrons of Pu and the effective U values are taken to be 4 eV. The projector augment wave (PAW) method [24] is used to describe the interaction between electrons and ions. As for the exchange-correlation functional, a number of generalized gradient approximation (GGA) functionals have been reported in the literature [25,26,27] and the functional parametrized by Perdew, Burke and Ernzerhof [28] is employed in this work. In the calculations, a 2 × 2 × 2 k-point sampling in reciprocal space is employed, with a cutoff energy of 600 eV for the plane wave basis sets. Figure 1 illustrates the schematic view of geometrical structure for the considered compounds, i.e., Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7 (y = 0, 0.5, 1.0, 1.5, 2.0). The special quasi-random structure method is used to build the structural models for Pu immobilization at Gd-site and Zr-site [29,30,31,32].
3. Results and Discussion
3.1. Structural Stability of Pu Incorporation into Gd2Zr2O7
As Pu substitutes for Gd3+ and Zr4+ in Gd2Zr2O7, the corresponding valence states for Pu are Pu3+ and Pu4+, respectively. Because in both PuO2 and Pu2O3 the Pu 5f electrons are strongly correlated, Hubbard U correction is thus necessary. In the revised manuscript, we present the density of state distribution for both PuO2 and Pu2O3 at Ueff = 0 eV and Ueff = 4 eV in Figure 2. It is shown that without Hubbard U correction, i.e., at Ueff = 0 eV, the Pu 5f electrons are itinerant and delocalized over the Fermi lever, resulting in metallic states. At Ueff = 4 eV, the Pu 5f electrons are localized and the system becomes insulating, which is consistent with the experimental finding [32]. The calculated lattice constant of 5.46 Å for PuO2 and 11.18 Å for Pu2O3 obtained at Ueff = 4 eV are comparable to the experimental values of 5.39 Å [33] and 10.98 Å [34], respectively. The calculated band gap for Pu2O3 at Ueff = 4 eV is 1.757 eV, which corresponds to the experimental value of 2 eV [35]. Thus, we use Ueff = 4 eV in our subsequent calculations for Pu immobilization in Gd2Zr2O7. On the other hand, the 4 eV for Ueff is also consistent with the value of 4–5 eV that are reported in the literature [36,37].
A structural optimization is first performed for both Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7. The calculated lattice constants, oxygen positional parameter and bond distances for Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7 are listed in Table 1 and Table 2, respectively. The changes of lattice constant and for Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7 with Pu concentrations are shown in Figure 3. The calculated lattice constant of 10.666 Å for Gd2Zr2O7 is slightly larger than the experimental value of 10.54 Å [38], while consistent with other calculations of 10.66 Å [18]. The calculated a0 of 10.802 Å for Pu2Zr2O7 is comparable with the experimental value of 10.70 Å [39]. As the Pu content increases, the lattice constant gradually increases for both Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7, and it changes more significantly for Gd2Zr2−yPuyO7 than that for Gd2−yPuyZr2O7. This is caused by the fact that the effective ionic radius of 1.053 Å [40] for Gd3 is in good agreement with the value of +~1.1 Å [40] for Pu3+, but the effective ionic radius of 0.72 Å [40] for Zr4+ is much smaller than the value of 0.96 Å [40] for Pu4+. With regard to oxygen positional parameter , the calculated value of 0.339 for Gd2Zr2O7 is smaller than the experimental value of 0.345 [41], and is comparable to the calculated value of 0.339 reported by Wang et al. [18]. For Gd2−yPuyZr2O7, the changes slightly as the Pu content increases, which indicates that the Gd2−yPuyZr2O7 remains the pyrochlore structure. Wang et al. [18] has observed similar phenomenon for Gd2−yCeyZr2O7. For Gd2Zr2−yPuyO7, we find that the increases a lot, varying from 0.339 to 0.350, suggesting that the Gd2Zr2−yPuyO7 tends to be a defect fluorite structure as the Pu content increases [15]. Comparing the lattice constant and oxygen positional parameter for Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7, we find that the formation of Gd2Zr2O7-Pu2Zr2O7 solid solution is more preferable than that of Gd2Zr2O7 and Gd2Pu2O7. As for bond distances, the calculated <Gd-O48f> distance of 2.553 Å in Gd2Zr2O7 is a little larger than the experimental value of 2.483 Å [38], and is comparable to other calculated value of 2.548 Å [18]. Meanwhile, the calculated value of 2.109 Å for <Zr-O48f> distance is consistent with the experimental value [38] and other calculated value [18] of 2.110 Å. For Gd2−yPuyZr2O7, the <Gd-O48f> and <Pu-O48f> distances increase slightly and the <Gd-O8b> and <Pu-O8b> distances decrease slightly as the Pu content increases. Comparing the <Gd-O> and <Pu-O> bonds, we find that the <Pu-O48f> and <Pu-O8b> distances are slightly larger than <Gd-O48f> and <Gd-O8b> distances, respectively, i.e., Pu substitution for Gd-site leads to small increase in the bonding distance. For Gd2Zr2−yPuyO7, the situation is different. The <Pu-O48f> bond is about 0.12–0.19 Å larger than the <Zr-O48f> bond. Simultaneously, the <Gd-O8b> bond increases a little as the Pu content increases. Consequently, there is a remarkable increase in the lattice constant of Gd2−yPuyZr2O7.
3.2. Elastic Constants and Elastic Moduli of Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7
Elastic constants are response functions to the external forces and are very important in the materials’ properties [42]. Table 3 lists the calculated elastic constants along with available experimental and theoretical values. For Gd2Zr2O7, the calculated C11, C12 and C44 are 285.1, 102.5 and 82.1 GPa, respectively, showing good agreement with other calculations [43]. For Pu2Zr2O7, the calculated C11 = 270.6 GPa, C12 = 107.3 GPa and C44 = 81.2 GPa differ from reference [2], in which different calculational parameters are employed. It is noted that the elastic stability criteria are satisfied for all the investigated systems, i.e., C11 > |C12|, C44 > 0, and (C11 + 2C12) > 0 [44], implying that they are all mechanically stable.
Figure 4 presents the changes of elastic constants with Pu content for both Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7. For Gd2−yPuyZr2O7, as the Pu content increases, the elastic constants are affected minorly. As the Pu concentration increases, the C11 and C12 decreases and increases slightly, respectively, and the change in C44, is nearly negligible. As for Gd2Zr2−yPuyO7, the variation of elastic constants with Pu content is more considerable. As the y value changes, the C11 and C12 first decreases, then increases, and finally decreases again. As for C44, it first decreases to y = 1.5, and then increases. Generally speaking, as the Pu content increases, there are more significant changes on Zr-site than Gd-site, meaning that Pu immobilization at Zr-site leads to remarkable variations in the mechanical properties of Gd2Zr2O7. Zhao et al. [47] also reported that Nd substitution of Zr-site of Gd2Zr2O7 greatly affects the mechanical properties.
From the calculated elastic constants, the elastic moduli, including the bulk modulus (B), shear modulus (G) and Young’s modulus (E), can be deduced [48,49,50,51], i.e.,
Here, the Voigt and Reuss evaluations for B and G are represented by V and R, respectively. Table 3 lists the calculated B, G, E, and others’ theoretical and experimental values for both Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7. For Gd2Zr2O7, the calculated B = 163.4 GPa, G = 85.7 GPa, E = 218.8 GPa are comparable with experimental [20,45,46] and other calculated [43] results. Figure 5 shows the variation of elastic moduli for both Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7. For Gd2−yPuyZr2O7, the elastic moduli change very slightly as the Pu content increases. When Pu is immobilized at Zr-site, there are remarkable changes. The bulk modulus first decreases, reaching a minimum of 134.3 GPa at y = 1.0, then rises up to 142.3 GPa at y = 1.5 and finally decreases to 136.9 GPa at y = 2.0. The shear modulus decreases to 58.9 GPa at y = 1.0 and increases slightly to 63.6 GPa at y = 2.0. The Young’s modulus decreases sharply from 218.8 GPa to 154.2 GPa as the y varies from 0 to 1.0, but increases again to 165.7 GPa at y = 1.5, finally changing slightly. The <Zr-O> bonds determine the total stiffness of A2Zr2O7 pyrochlore, because the corner-sharing ZrO6 octahedra constitutes its backbone, and the A3+ fills the interstices [19,52]. Therefore, the substitution of Zr4+ by Pu4+ causes the change of <Zr-O> bonds to <Pu-O> bonds and influences the Young’s modulus, especially for these ionic bonds [19,52]. The Young’s modulus E is described by for ionic bonds, in which Ma represents the Madelung constant and ro represents the interionic distance [19]. The <Zr-O> bonds in Gd2Zr2O7 are affected little by Pu substitution for Gd-site, leading to slight effects on the Young’s modulus. For Gd2Zr2−yPuyO7, the <Zr-O48f> bond length of 2.11 is smaller than the value of 2.26 for <Pu-O48f>, resulting in remarkable effects on the Young’s modulus. The bulk modulus, shear modulus and Young’s modulus for Nd doping of Gd2Zr2O7 have been calculated by Zhao et al. [47], who also reported that Nd immobilization at Zr-site has more remarkable influences on the elastic moduli than that at Gd-site.
For each ion in Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7, we analyze the Bader charge to explore the origin of the discrepancy in the elastic moduli between Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7. The Bader charge values are listed in Table 4. As Pu is immobilized at Gd-site and Zr-site, the average Bader charge for Pu are 2.10 |e| and 2.33 |e|, respectively, corresponding to the nominal +3 |e| and +4 |e| in oversimplified classical model [2]. For Gd2−yPuyZr2O7, the Bader charge for Gd and Pu ions are similar to each other, i.e., 2.15 and 2.10|e|, respectively. Wang et al. [18] calculated the Bader charge of Gd2−yCeyZr2O7 and reported very similar results. Additionally, the bonding distance for <Gd-O48f> and <Gd-O8b> are determined to be 2.57 Å and 2.30 Å, which are comparable to the values of 2.60 Å for <Pu-O48f> and 2.36 Å for <Pu-O8b>, respectively. Obviously, the <Gd-O> and <Pu-O> bonding interaction are very similar to each other, which explains why the mechanical properties of Gd2Zr2O7 are affected slightly by Pu immobilization at Gd-site. For Gd2Zr2−yPuyO7, the situation is much different. The average Bader charge are 2.33 |e| for Pu ions and 2.26 |e| for Zr ions. Considering that the <Pu-O48f> distance of 2.26 is larger than the <Zr-O48f> distance of 2.11 and the ionic radius of 0.96 for Pu ions is larger than that of 0.72 for Zr ions [40], it is suggested that the <Zr-O> bonds exhibit weaker ionicity than <Pu-O> bonds in Gd2Zr2−yPuyO7. Because of the brittleness of the ionic bonds, the immobilization of Pu at Zr sites will thus increase the ionicity and decrease the elastic moduli.
3.3. Ductility, Elastic Anisotropy, Debye Temperature and Electronic Structures of Pu-Doped Gd2Zr2O7
Pugh’s indicator () is used to reflect the ductility of materials. If , the material shows ductility; or, it is brittle [54]. Table 5 presents the calculated Pugh’s indicators. For Gd2Zr2O7, our value of 1.907 is comparable with the experimental values of 1.913 [45,46], 1.891 [20] and other calculated value of 2.004 [53]. For both Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7, our calculations show that the Pugh’s indicators are all larger than 1.75, implying that all the considered composites are ductile. Poisson’s indicator ( can be employed to evaluate the relative ductility of materials. When is around 0.1, the material shows brittle covalent properties. When is bigger than 0.25, it exhibits ductile ionic properties [55]. Table 5 lists the calculated, experimental and others’ calculated Poisson’s ratio for both Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7. For Gd2Zr2O7, the calculated Poisson’s ratio of 0.277 could be comparable to the experimental values of 0.276 [45,46], 0.274 [20] and other calculated results of 0.286 [53] and 0.273 [43]. The Poisson’s ratios for Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7 are all larger than 0.25, as presented in Table 5, i.e., the Pu-substituted Gd2Zr2O7 exhibit good ductility.
Elastic anisotropy is an important parameter for phase transformations, dislocation dynamics and geophysical applications [56]. Ranganathan and co-workers [57,58] proposed the universal elastic anisotropic index to indicate the elastic anisotropy of cubic crystals. The index is investigated for all the considered compositions., where describes an isotropic crystal [57,58]. The calculated for both Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7 are shown in Table 5. For Gd2Zr2O7, our calculated value of 0.01355 is comparable with other calculated value of 0.00420 [53]. As Pu is immobilized at Gd-site, we find that the values for all the compositions are nearly zero, indicating that the Gd2−yPuyZr2O7 compounds are isotropic elastically. As for the immobilization of Pu at Zr-site, the values are 0.21353 for Gd2Zr1.0Pu1.0O7 and 0.10062 for Gd2Zr0.5Pu1.5O7, indicative of elastic anisotropy.
The thermal properties of materials can be analyzed by the Debye temperature [4]. Table 5 lists the calculated and available experimental Debye temperature. The calculated value of 580.2 K for Gd2Zr2O7 is larger than the experimental result of 513.3 K [20], but shows better agreement with experimental value than another calculated value of 612.9 K [53]. As the Pu concentration increases, the value decreases for both Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7. In particular, the values for Gd2Zr2−yPuyO7 are smaller than those for Gd2−yPuyZr2O7. These results imply that the Gd2Zr2−yPuyO7 compositions have a lower melting point and weaker interatomic binding force than the Gd2-yPuyZr2O7 compositions. Zhao et al. [59] calculated the Debye temperature for Th immobilization at Gd-site and Zr-site of Gd2Zr2O7 and observed similar phenomena, i.e., the Th-substituted Gd2Zr2O7 have a lower Debye temperature and especially smaller Debye temperature can be obtained by the Th immobilization at Zr-site than that at Gd-site.
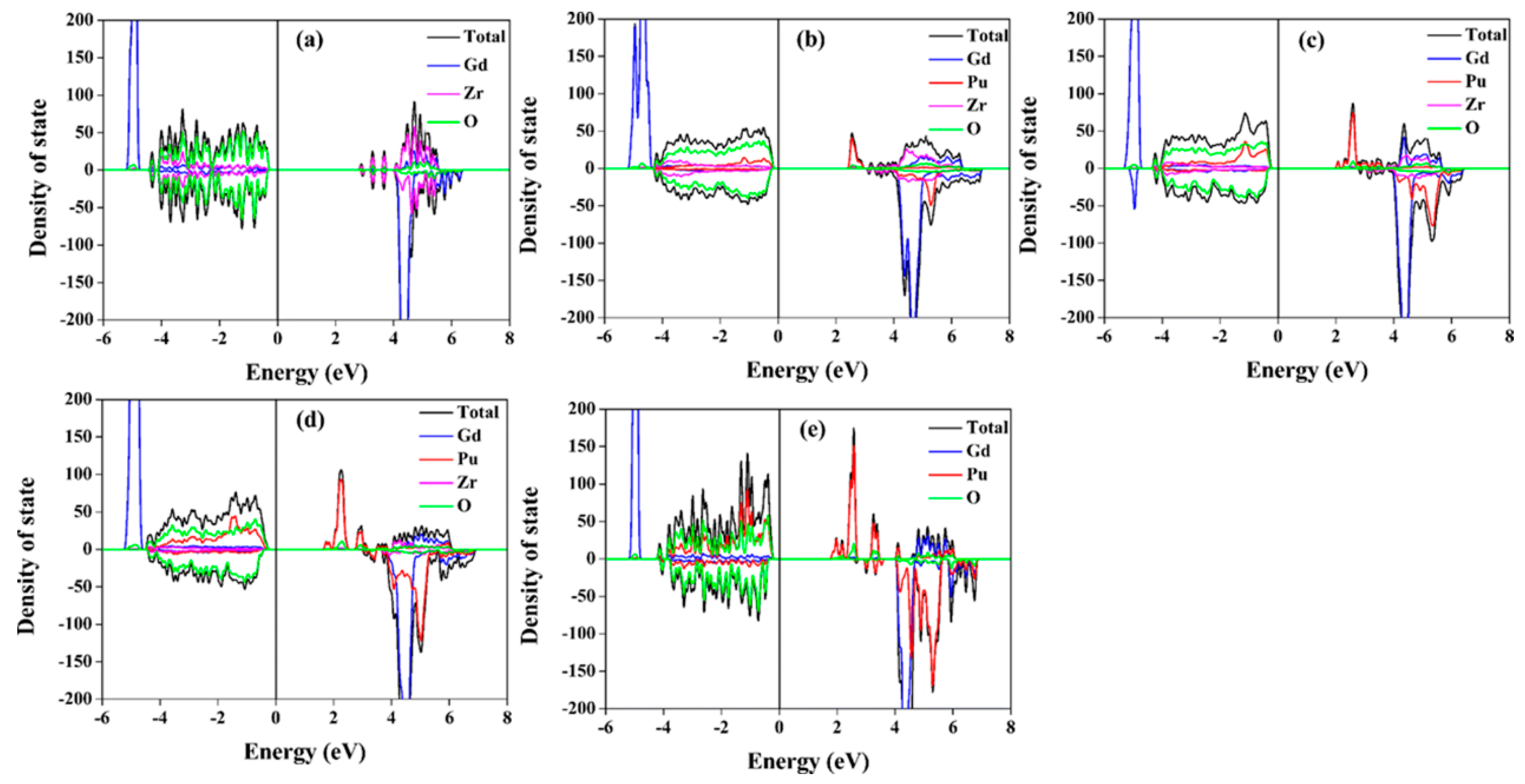
The total and projected density of state (DOS) distributions for Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7 are illustrated in Figure 6 and Figure 7, respectively. Table 1 and Table 2 list the band gap values. It is shown that all the compositions have large band gap values. For Gd2Zr2O7, the calculated band gap is 2.86 eV. For Pu incorporation into Gd-site, the band gap values of 2.27–2.37 eV are smaller than that of Gd2Zr2O7 and nearly independent of the Pu content. For Gd2Zr2−yPuyO7, the changes in the band gap is more significant, ranging from 2.33 to 1.68 eV. It is indicated that Pu immobilization at Gd-site and Zr-site has different influences on the electronic structure of Gd2Zr2O7. The same conclusion can be drawn from the density of state distribution. For Gd2−yPuyZr2O7, O 2p orbital dominates and hybridizes with very few Gd 5d, Pu 5f and Zr 4d orbitals in the energy range of −5–1 eV, and the Pu 5f orbitals hybridize with the O 2p orbitals at the bottom of valence band. For Gd2Zr2−yPuyO7, the O 2p orbital dominates and hybridizes with Pu 5f orbitals and very few Zr 4d and Gd 5d orbitals at the valence band. Obviously, in Gd2Zr2O7, different electronic structures can be obtained from the immobilization of Pu at Gd and Zr sites. These different electronic structures may result in discrepancies in the thermo-physical properties of Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7.
4. Conclusions
The mechanical and electronic properties of Pu-containing Gd2Zr2O7 are studied by a DFT+U method. For Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7, the elastic stability criteria are satisfied for all the calculated elastic constants, i.e., all the compounds are mechanically stable. As Pu immobilizes at Gd-site in Gd2Zr2O7, because the bonding distance and covalency of <Gd-O> and <Pu-O> bonds are comparable to each other, the elastic constants, elastic moduli, elastic isotropy and Debye temperature of Gd2Zr2O7 are all affected a little. As for Gd2Zr2−yPuyO7, the elastic constants and elastic moduli change remarkably as compared with Gd2Zr2O7. The substitution of Pu for Zr sites increases the ionicity and decreases the elastic moduli, because the <Zr-O> bonds exhibit weaker ionicity than <Pu-O> bonds. In addition, the Debye temperature is decreased and the band gap is greatly reduced. Our calculations suggest that the Gd2Zr2O7 is a promising material for immobilizing nuclear waste such as Pu, while the thermo-physical of Gd2Zr2O7 may be influenced significantly after nuclear waste incorporation.
Author Contributions
Conceptualization, H.X.; Methodology, H.X. and P.L.; Validation, H.X. and F.Z.; Investigation, P.L.; Data Curation, P.L., F.Z. and S.Z.; Writing—Original Draft Preparation, P.L.; Writing—Review & Editing, H.Z., H.G., Z.L. and X.Z.; Supervision, H.X.
Acknowledgments
H.X. was supported by NSAF Joint Foundation of China (Grant No. U1530129). Z.L. was supported by National Natural Science Foundation of China (Grant No. 11464025), the New Century Excellent Talents in University under Grant No. NECT-11-0906 and the Key Talent Foundation of Gansu Province. The theoretical calculations were performed using the supercomputer resources at TianHe-1 located at National Supercomputer Center in Tianjin.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ewing, R.C. Nuclear waste forms for actinides. Proc. Natl. Acad. Sci. USA 1999, 96, 3432–3439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.Y.; Jiang, M.; Zhao, F.A.; Liu, Z.J.; Zu, X.T. Thermal and mechanical stability, electronic structure and energetic properties of Pu-containing pyrochlores: La2−yPuyZr2O7 and La2Zr2−yPuyO7 (0 ≤ y ≤ 2). J. Nucl. Mater. 2015, 466, 162–171. [Google Scholar] [CrossRef]
- Ewing, R.C.; Weber, W.J.; Lian, J. Nuclear waste disposal—Pyrochlore (A2B2O7): Nuclear waste form for the immobilization of plutonium and “minor” actinides. J. Appl. Phys. 2004, 95, 5949–5971. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 6th ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1986; pp. 106–107. [Google Scholar]
- Chroneos, A.; Rushton, M.J.D.; Jiang, C.; Tsoukalas, L.H. Nuclear wasteform materials: Atomistic simulation case studies. J. Nucl. Mater. 2013, 441, 29–39. [Google Scholar] [CrossRef]
- Wang, S.X.; Begg, B.D.; Wang, L.M.; Ewing, R.C.; Weber, W.J.; Kutty, K.V.G. Radiation stability of gadolinium zirconate: A waste form for plutonium disposition. J. Mater. Res. 1999, 14, 4470–4473. [Google Scholar] [CrossRef]
- Sickafus, K.E.; Minervini, L.; Grimes, R.W.; Valdez, J.A.; Ishimaru, M.; Li, F.; McClellan, K.J.; Hartmann, T. Radiation tolerance of complex oxides. Science 2000, 289, 748–751. [Google Scholar] [CrossRef] [PubMed]
- Weber, W.J. Plutonium immobilization and radiation effects. Science 2000, 289, 2051–2052. [Google Scholar] [CrossRef]
- Devanathan, R.; Weber, W.J.; Gale, J.D. Radiation tolerance of ceramics—Insights from atomistic simulation of damage accumulation in pyrochlores. Energy Environ. Sci. 2010, 3, 1551–1559. [Google Scholar] [CrossRef]
- Patwe, S.J.; Tyagi, A.K. Solubility of Ce4+ and Sr2+ in the pyrochlore lattice of Gd2Zr2O7 for simulation of Pu and alkaline earth metal. Ceram. Int. 2006, 32, 545–548. [Google Scholar] [CrossRef]
- Mandal, B.P.; Tyagi, A.K. Pyrochlores: Potential multifunctional materials. BARC Newslett. 2010, 313, 6–13. [Google Scholar]
- Zhao, P.Z.; Li, L.Y.; Xu, S.M.; Zhang, Q. Synthesis and structural transformations of (Gd1−xCex)2Zr2O7+x: An analogue for Pu immobilization. Acta Phys. Chim. Sin. 2013, 29, 1168–1172. [Google Scholar]
- Reid, D.P.; Stennett, M.C.; Hyatt, N.C. The fluorite related modulated structures of the Gd2(Zr2−xCex)O7 solid solution: An analogue for Pu disposition. J. Solid State Chem. 2012, 191, 2–9. [Google Scholar] [CrossRef]
- Patwe, S.J.; Ambekar, B.R.; Tyagi, A.K. Synthesis, characterization and lattice thermal expansion of some compounds in the system Gd2CexZr2−xO7. J. Alloys Compd. 2005, 389, 243–246. [Google Scholar] [CrossRef]
- Zhao, F.A.; Xiao, H.Y.; Jiang, M.; Liu, Z.J.; Zu, X.T. A DFT+U study of Pu immobilization in Gd2Zr2O7. J. Nucl. Mater. 2015, 467, 937–948. [Google Scholar] [CrossRef]
- Williford, R.E. Computer simulation of Pu3+ and Pu4+ substitutions in gadolinium zirconate. J. Nucl. Mater. 2001, 299, 140–147. [Google Scholar] [CrossRef]
- Cleave, A.; Grimes, R.W.; Sickafus, K. Plutonium and uranium accommodation in pyrochlore oxides. Philos. Mag. 2005, 85, 967–980. [Google Scholar] [CrossRef]
- Wang, X.J.; Xiao, H.Y.; Zu, X.T.; Weber, W.J. Study of cerium solubility in Gd2Zr2O7 by DFT + U calculations. J. Nucl. Mater. 2011, 419, 105–111. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, F.; Xiao, P. Role and determining factor of substitutional defects on thermal conductivity: A study of La2(Zr1−xBx)2O7 (B = Hf, Ce, 0 ≤ x ≤ 0.5) pyrochlore solid solutions. Acta Mater. 2014, 68, 106–115. [Google Scholar] [CrossRef]
- Shimamura, K.; Arima, T.; Idemitsu, K.; Inagaki, Y. Thermophysical properties of rare-earth-stabilized zirconia and zirconate pyrochlores as surrogates for actinide-doped zirconia. Int. J. Thermophys. 2007, 28, 1074–1084. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmiiller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Cohen, R.E. More accurate generalized gradient approximation for solids. Phys. Rev. B 2006, 73, 235116. [Google Scholar] [CrossRef]
- Constantin, L.A.; Terentjevs, A.; Della Sala, F.; Fabiano, E. Gradient-dependent upper bound for the exchange-correlation energy and application to density functional theory. Phys. Rev. B 2015, 91, 041120(R). [Google Scholar] [CrossRef]
- Constantin, L.A.; Terentjevs, A.; Della Sala, F.; Cortona, P.; Fabiano, E. Semiclassical atom theory applied to solid-state physics. Phys. Rev. B 2016, 93, 045126. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Wei, S.H.; Ferreira, L.G.; Bernard, J.E.; Zunger, A. Electronic properties of random alloys: Special quasirandom structures. Phys. Rev. B 1990, 42, 9622–9649. [Google Scholar] [CrossRef]
- Zunger, A.; Wei, S.; Ferreira, L.G.; Bernard, J.E. Special quasirandom structures. Phys. Rev. Lett. 1990, 65, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Wolverton, C.; Sofo, J.; Chen, L.Q.; Liu, Z.K. First-principles study of binary bcc alloys using special quasirandom structures. Phys. Rev. B 2004, 69, 214202. [Google Scholar] [CrossRef]
- Jiang, C.; Stanek, C.R.; Sickafus, K.E.; Uberuaga, B.P. First-principles prediction of disordering tendencies in pyrochlore oxides. Phys. Rev. B 2009, 79, 104203. [Google Scholar] [CrossRef]
- McCleskey, T.M.; Bauer, E.; Jia, Q.; Burrell, A.K.; Scott, B.L.; Conradson, S.D.; Mueller, A.; Roy, L.; Wen, X.; Scuseria, G.E.; et al. Optical band gap of NpO2 and PuO2 from optical absorbance of epitaxial films. J. Appl. Phys. 2013, 113. [Google Scholar] [CrossRef]
- Chikalla, T.D.; McNeilly, C.E.; Skavdahl, R.E. The plutonium-oxygen system. J. Nucl. Mater. 1964, 12, 131–141. [Google Scholar] [CrossRef]
- Jomard, G.; Amadon, B.; Bottin, F.; Torrent, M. Structural, thermodynamic, and electronic properties of plutonium oxides from first principles. Phys. Rev. B 2008, 78, 075125. [Google Scholar] [CrossRef]
- Sun, B.; Zhang, P.; Zhao, X.-G. First-principles local density approximation plus U and generalized gradient approximation plus U study of plutonium oxides. J. Chem. Phys. 2008, 128, 084705. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.-D.; Martin, R.L.; Henderson, T.M.; Scuseria, G.E. Density Functional Theory Studies of the Electronic Structure of Solid State Actinide Oxides. Chem. Rev. 2013, 113, 1063–1096. [Google Scholar] [CrossRef] [PubMed]
- Mandal, B.P.; Banerji, A.; Sathe, V.; Deb, S.K.; Tyagi, A.K. Order–disorder transition in Nd2−yGdyZr2O7 pyrochlore solid solution: An X-ray diffraction and Raman spectroscopic study. J. Solid State Chem. 2007, 180, 2643–2648. [Google Scholar] [CrossRef]
- Yamazaki, S.; Yamashita, T.; Matsui, T.; Nagasaki, T. Thermal expansion and solubility limits of plutonium-doped lanthanum zirconates. J. Nucl. Mater. 2001, 294, 183–187. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomie distances in halides and chaleogenides. Acta Crystallogr. Sect. A Cryst. Phys. Diffr. Theor. Gen. Cystallogr. 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Lian, J.; Zu, X.T.; Kutty, K.V.G.; Chen, J.; Wang, L.M.; Ewing, R.C. Ion-irradiation-induced amorphization of La2Zr2O7 pyrochlore. Phys. Rev. B 2002, 66, 054108. [Google Scholar] [CrossRef]
- Ravindran, P.; Fast, L.; Korzhavyi, P.A.; Johansson, B.; Wills, J.; Eriksson, O. Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2. J. Appl. Phys. 1998, 84, 4891–4904. [Google Scholar] [CrossRef]
- Lan, G.; Ouyang, B.; Song, J. The role of low-lying optical phonons in lattice thermal conductance of rare-earth pyrochlores: A first-principle study. Acta Mater. 2015, 91, 304–317. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Wang, B.T.; Zhao, X. Ground-state properties and high-pressure behavior of plutonium dioxide: Density functional theory calculations. Phys. Rev. B 2010, 82, 144110. [Google Scholar] [CrossRef] [Green Version]
- Van Dijk, M.P.; de Vries, K.J.; Burggraaf, A.J. Oxygen ion and mixed conductivity in compounds with the fluorite and pyrochlore structure. Solid State Ion. 1983, 9, 913–919. [Google Scholar] [CrossRef]
- Wu, J.; Wei, X.; Padtur, N.P.; Klemens, P.G.; Gell, M.; Garcia, E.; Miranzo, P.; Osendi, M.I. Low-thermal-conductivity rare-earth zirconates for potential thermal-barrier-coating applications. J. Am. Ceram. Soc. 2002, 85, 3031–3035. [Google Scholar] [CrossRef]
- Zhao, F.A.; Xiao, H.Y.; Bai, X.M.; Liu, Z.J.; Zu, X.T. Effects of Nd doping on the mechanical properties and electronic structures of Gd2Zr2O7: A first-principles-based study. J. Mater. Sci. 2018, 53, 16423–16438. [Google Scholar] [CrossRef]
- Chung, D.H. Elastic moduli of single crystal and polycrystalline MgO. Philos. Mag. 1963, 8, 833–841. [Google Scholar] [CrossRef]
- Voigt, W. Ueber die Beziehung zwischen den beiden Elasticitätsconstanten isotroper Körper. Annalen der Physik 1889, 274, 573–587. [Google Scholar] [CrossRef]
- Reuss, A. Account of the liquid limit of mixed crystals on the basis of the plasticity condition for single crystal. Z. Angew. Math. Mech. 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. Sect. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Liu, B.; Wang, J.Y.; Li, F.Z.; Zhou, Y.C. Theoretical elastic stiffness, structural stability and thermal conductivity of La2T2O7 (T = Ge, Ti, Sn, Zr, Hf) pyrochlore. Acta Mater. 2010, 58, 4369–4377. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, H.B.; Zhao, F.A.; Jiang, M.; Xiao, H.Y.; Liu, Z.J.; Zu, X.T. Impact of isovalent and aliovalent substitution on the mechanical and thermal properties of Gd2Zr2O7. Sci. Rep. 2017, 7, 6399. [Google Scholar] [CrossRef] [PubMed]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 2009, 45, 823–843. [Google Scholar] [CrossRef]
- Fine, M.E.; Brown, L.D.; Marcus, H.L. Elastic constants versus melting temperature in metals. Scr. Metall. 1984, 18, 951–956. [Google Scholar] [CrossRef]
- Ledbetter, H.; Migliori, A. A general elastic-anisotropy measure. J. Appl. Phys. 2006, 100, 063516. [Google Scholar] [CrossRef]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal elastic anisotropy index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Xiao, B.; Wan, C.L.; Qu, Z.X.; Huang, Z.C.; Chen, J.C.; Zhou, R.; Pan, W. Electronic structure, mechanical properties and thermal conductivity of Ln2Zr2O7 (Ln = La, Pr, Nd, Sm, Eu and Gd) pyrochlore. Acta Mater. 2011, 59, 1742–1760. [Google Scholar] [CrossRef]
- Zhao, F.A.; Xiao, H.Y.; Liu, Z.J.; Li, S.; Zu, X.T. A DFT study of mechanical properties, thermal conductivity and electronic structures of Th-doped Gd2Zr2O7. Acta Mater. 2016, 121, 299–309. [Google Scholar] [CrossRef]
Figure 1.
Schematic views of optimized geometrical structures for: (a) Gd2Zr2O7; (b) Gd1.5Pu0.5Zr2O7; (c) Gd1.0Pu1.0Zr2O7; (d) Gd0.5Pu1.5Zr2O7; (e) Pu2Zr2O7; (f) Gd2Zr1.5Pu0.5O7; (g) Gd2Zr1.0Pu1.0O7; (h) Gd2Zr0.5Pu1.5O7; (i) Gd2Pu2O7. The purple, dark grey, green and red spheres represent Gd, Pu, Zr and O atoms, respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Figure 1.
Schematic views of optimized geometrical structures for: (a) Gd2Zr2O7; (b) Gd1.5Pu0.5Zr2O7; (c) Gd1.0Pu1.0Zr2O7; (d) Gd0.5Pu1.5Zr2O7; (e) Pu2Zr2O7; (f) Gd2Zr1.5Pu0.5O7; (g) Gd2Zr1.0Pu1.0O7; (h) Gd2Zr0.5Pu1.5O7; (i) Gd2Pu2O7. The purple, dark grey, green and red spheres represent Gd, Pu, Zr and O atoms, respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
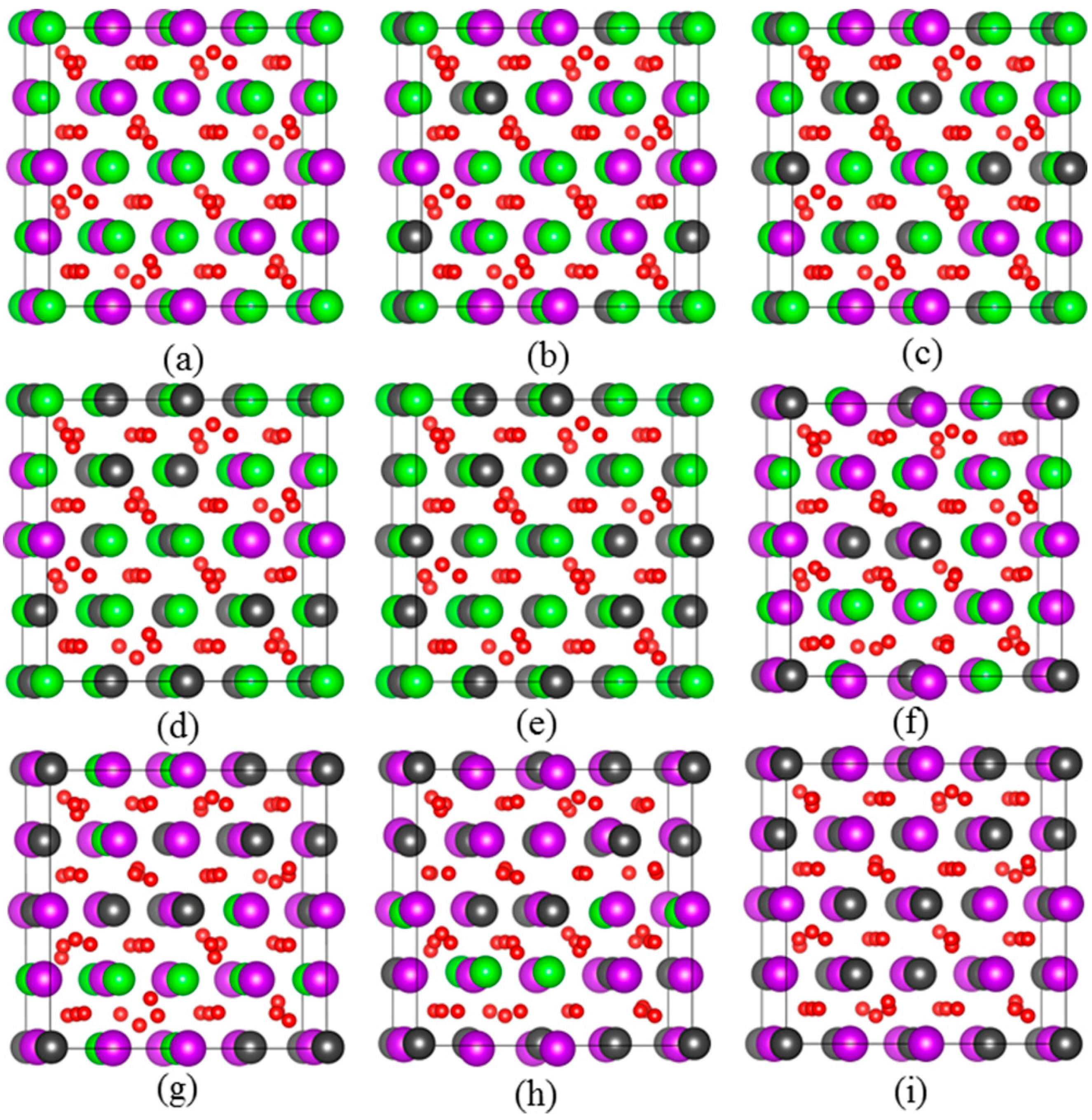
Figure 2.
Density of state distribution for PuO2 at (a) Ueff = 0 eV and (b) 4 eV and for Pu2O3 at (c) Ueff = 0 eV and (d) 4 eV obtained by GGA+U.
Figure 2.
Density of state distribution for PuO2 at (a) Ueff = 0 eV and (b) 4 eV and for Pu2O3 at (c) Ueff = 0 eV and (d) 4 eV obtained by GGA+U.

Figure 3.
Variation of (a) lattice constant and (b) for Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7 with Pu content. The calculated and fitted results are represented by symbols and dashed lines, respectively.
Figure 3.
Variation of (a) lattice constant and (b) for Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7 with Pu content. The calculated and fitted results are represented by symbols and dashed lines, respectively.

Figure 4.
Variation of elastic constants (C11, C12 and C44) for (a) Gd2−yPuyZr2O7 and (b) Gd2Zr2−yPuyO7 (0 ≤ y ≤2) with Pu content.
Figure 4.
Variation of elastic constants (C11, C12 and C44) for (a) Gd2−yPuyZr2O7 and (b) Gd2Zr2−yPuyO7 (0 ≤ y ≤2) with Pu content.
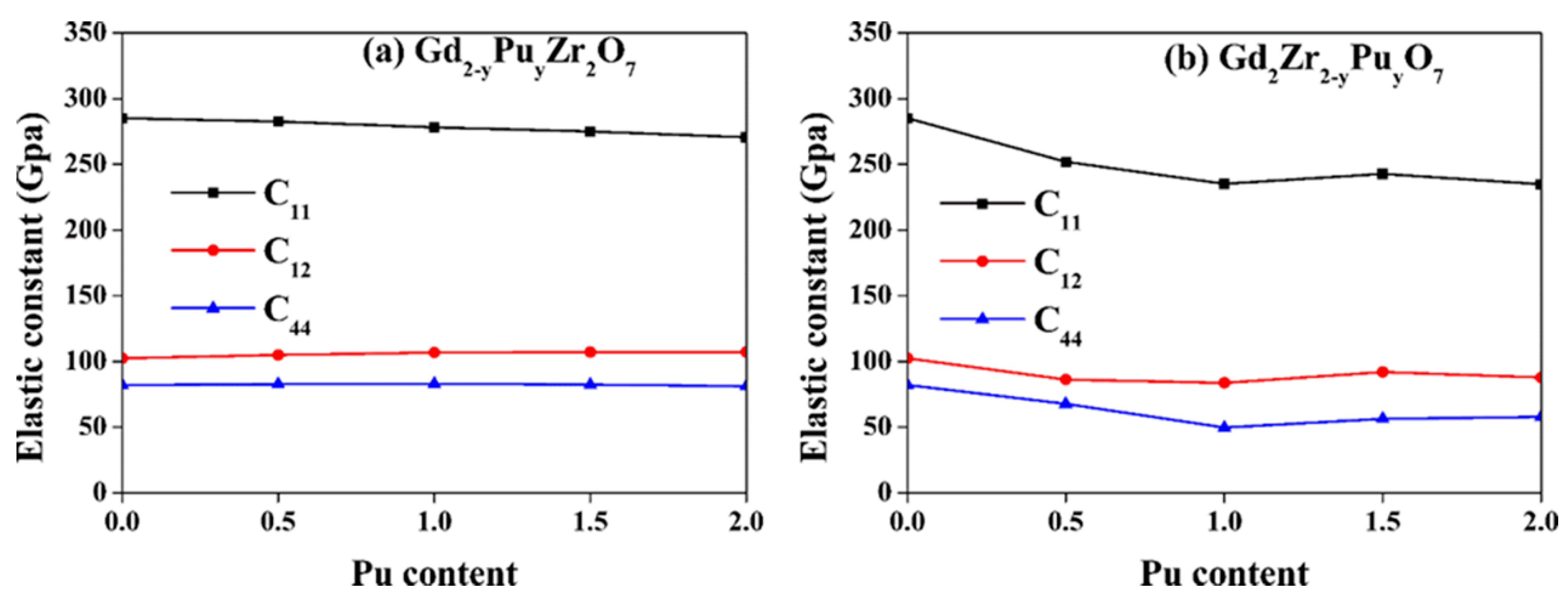
Figure 5.
Variation of elastic moduli for (a) Gd2−yPuyZr2O7 and (b) Gd2Zr2−yPuyO7 (0 ≤ y ≤2) as a function of Pu content. B: bulk modulus; G: shear modulus; E: Young’s modulus.
Figure 5.
Variation of elastic moduli for (a) Gd2−yPuyZr2O7 and (b) Gd2Zr2−yPuyO7 (0 ≤ y ≤2) as a function of Pu content. B: bulk modulus; G: shear modulus; E: Young’s modulus.
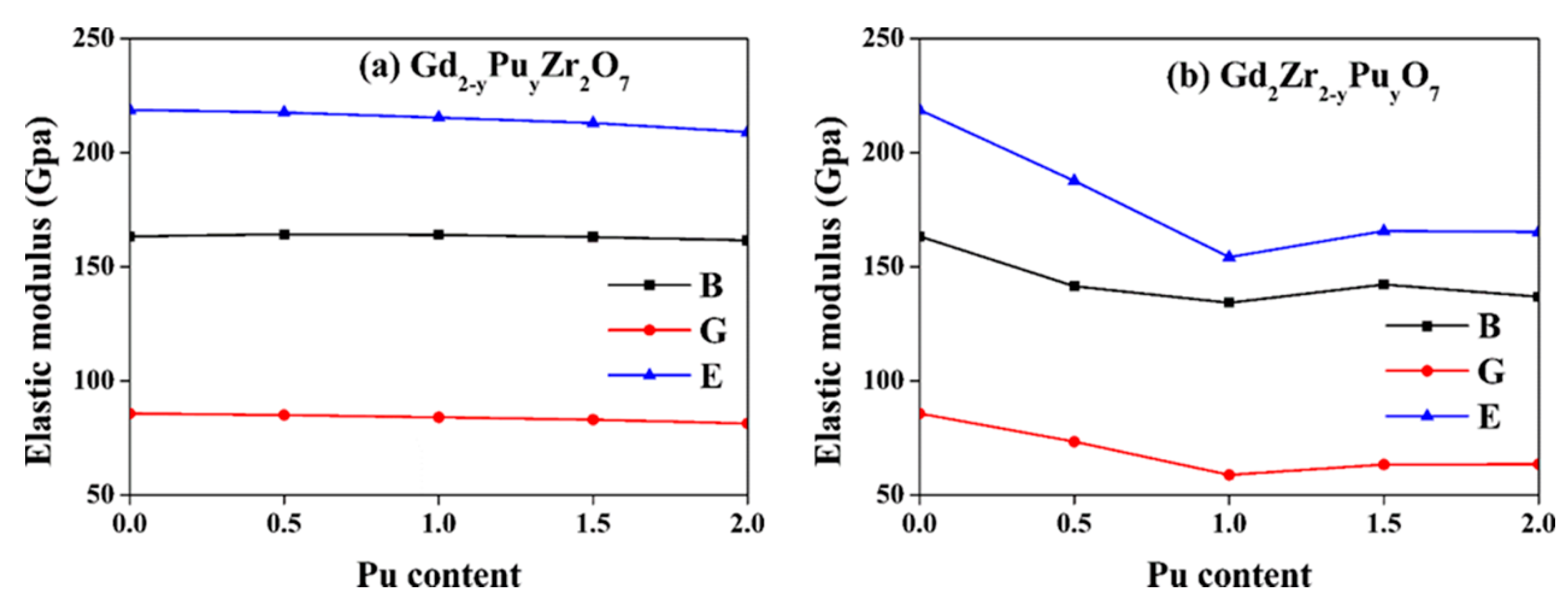
Figure 6.
Total and projected density of state distributions for: (a) Gd2Zr2O7; (b) Gd1.5Pu0.5Zr2O7; (c) Gd1.0Pu1.0Zr2O7; (d) Gd0.5Pu1.5Zr2O7; (e) Pu2Zr2O7.
Figure 6.
Total and projected density of state distributions for: (a) Gd2Zr2O7; (b) Gd1.5Pu0.5Zr2O7; (c) Gd1.0Pu1.0Zr2O7; (d) Gd0.5Pu1.5Zr2O7; (e) Pu2Zr2O7.

Figure 7.
Total and projected density of state distributions for: (a) Gd2Zr2O7; (b) Gd2Zr1.5Pu0.5O7; (c) Gd2Zr1.0Pu1.0O7; (d) Gd2Zr0.5Pu1.5O7; (e)Gd2Pu2O7.
Figure 7.
Total and projected density of state distributions for: (a) Gd2Zr2O7; (b) Gd2Zr1.5Pu0.5O7; (c) Gd2Zr1.0Pu1.0O7; (d) Gd2Zr0.5Pu1.5O7; (e)Gd2Pu2O7.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Lattice constant a0 (Å), O48f positional parameter x and bond distances (Å) for Gd2−yPuyZr2O7. Eg represents the band gap.
Table 1.
Lattice constant a0 (Å), O48f positional parameter x and bond distances (Å) for Gd2−yPuyZr2O7. Eg represents the band gap.
| a0 | xO48f | Eg (eV) | d<Gd-O48f> | d<Gd-O8b> | d<Pu-O48f> | d<Pu-O8b> | d<Zr-O48f> | |
|---|---|---|---|---|---|---|---|---|
| y = 0 | 10.666 | 0.339 | 2.86 | 2.553 | 2.309 | - | - | 2.109 |
| Exp. | 10.540 [38] | 0.345 [41] | 2.483 [38] | |||||
| Cal. | 10.66 [18] | 0.339 [18] | 2.548 [18] | 2.307 [18] | 2.110 [18] | |||
| y = 0.5 | 10.703 | 0.337 | 2.33 | 2.561 | 2.302 | 2.582 | 2.369 | 2.111 |
| y = 1.0 | 10.736 | 0.337 | 2.27 | 2.578 | 2.284 | 2.591 | 2.366 | 2.114 |
| y = 1.5 | 10.768 | 0.336 | 2.33 | 2.574 | 2.286 | 2.605 | 2.349 | 2.116 |
| y = 2.0 | 10.802 | 0.335 | 2.37 | - | - | 2.615 | 2.339 | 2.117 |
| Exp. | 10.70 [39] |
Table 2.
Lattice constant a0 (Å), O48f positional parameter x and bond distances (Å) for Gd2Zr2−yPuyO7. Eg represents the band gap.
Table 2.
Lattice constant a0 (Å), O48f positional parameter x and bond distances (Å) for Gd2Zr2−yPuyO7. Eg represents the band gap.
| a0 | xO48f | Eg (eV) | d<Gd-O48f> | d<Gd-O8b> | d<Pu-O48f> | d<Zr-O48f> | |
|---|---|---|---|---|---|---|---|
| y = 0 | 10.666 | 0.339 | 2.86 | 2.553 | 2.309 | - | 2.109 |
| Exp. | 10.540 [38] | 0.345 [41] | 2.483 [38] | ||||
| Cal. | 10.66 [18] | 0.339 [18] | 2.548 [18] | 2.307 [18] | 2.110 [18] | ||
| y = 0.5 | 10.750 | 0.342 | 2.33 | 2.513 | 2.338 | 2.296 | 2.110 |
| y = 1.0 | 10.836 | 0.344 | 1.99 | 2.555 | 2.347 | 2.228 | 2.114 |
| y = 1.5 | 10.909 | 0.350 | 1.68 | 2.508 | 2.364 | 2.273 | 2.121 |
| y = 2.0 | 11.003 | 0.350 | 1.75 | 2.552 | 2.382 | 2.234 | - |
Table 3.
Elastic constants (C11, C12, C44, in GPa), bulk modulus B (GPa), shear modulus G (GPa), Young’s modulus E (GPa) of Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7 (0≤ y ≤2).
Table 3.
Elastic constants (C11, C12, C44, in GPa), bulk modulus B (GPa), shear modulus G (GPa), Young’s modulus E (GPa) of Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7 (0≤ y ≤2).
| C11 | C12 | C44 | B | G | E | ||
|---|---|---|---|---|---|---|---|
| Gd2Zr2O7 | 285.1 | 102.5 | 82.1 | 163.4 | 85.7 | 218.8 | |
| Cal [43] | 289 | 103 | 85 | 165 | 88 | 224 | |
| Exp [45,46] | 153 | 80 | 205 | ||||
| Exp [20] | 174 | 92 | 236 | ||||
| Gd1.5Pu0.5Zr2O7 | 282.6 | 105.1 | 82.7 | 164.3 | 85.1 | 217.6 | |
| Gd1.0Pu1.0Zr2O7 | 278.1 | 106.9 | 83.1 | 164.0 | 84.1 | 215.4 | |
| Gd0.5Pu1.5Zr2O7 | 274.9 | 107.2 | 82.5 | 163.1 | 83.0 | 213.0 | |
| Pu2Zr2O7 | 270.6 | 107.3 | 81.2 | 161.7 | 81.4 | 209.1 | |
| Cal [2] | 306 | 131.8 | 90.2 | ||||
| Gd2Zr1.5Pu0.5O7 | 251.9 | 86.3 | 67.7 | 141.5 | 73.4 | 187.7 | |
| Gd2Zr1.0Pu1.0O7 | 235.2 | 83.8 | 49.8 | 134.3 | 58.9 | 154.2 | |
| Gd2Zr0.5Pu1.5O7 | 242.8 | 92.0 | 56.5 | 142.3 | 63.4 | 165.7 | |
| Gd2Pu2O7 | 234.8 | 87.9 | 57.8 | 136.9 | 63.6 | 165.3 |
Table 4.
Bader charge (|e|) for each ion in Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7 (y = 0, 0.5, 1.0, 1.5, 2.0).
Table 4.
Bader charge (|e|) for each ion in Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7 (y = 0, 0.5, 1.0, 1.5, 2.0).
| Gd | Pu | Zr | O48f | O8b | |
|---|---|---|---|---|---|
| Gd2Zr2O7 | 2.16 | - | 2.26 | −1.25 | −1.37 |
| Gd1.5Pu0.5Zr2O7 | 2.15 | 2.10 | 2.27 | −1.25 | −1.35 |
| Gd1.0Pu1.0Zr2O7 | 2.13 | 2.11 | 2.27 | −1.24 | −1.35 |
| Gd0.5Pu1.5Zr2O7 | 2.15 | 2.09 | 2.28 | −1.24 | −1.33 |
| Pu2Zr2O7 | - | 2.08 | 2.27 | −1.23 | −1.32 |
| Gd2Zr1.5Pu0.5O7 | 2.14 | 2.36 | 2.26 | −1.25 | −1.37 |
| Gd2Zr1.0Pu1.0O7 | 2.15 | 2.31 | 2.27 | −1.25 | −1.37 |
| Gd2Zr0.5Pu1.5O7 | 2.15 | 2.34 | 2.24 | −1.25 | −1.36 |
| Gd2Pu2O7 | 2.16 | 2.30 | - | −1.26 | −1.38 |
Table 5.
Pugh’s indicator (), elastic anisotropy index (), sound wave velocity (, in m/s), Debye temperature (, in K) and Poisson’s ratio () of Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7 (0 ≤ y ≤ 2).
Table 5.
Pugh’s indicator (), elastic anisotropy index (), sound wave velocity (, in m/s), Debye temperature (, in K) and Poisson’s ratio () of Gd2−yPuyZr2O7 and Gd2Zr2−yPuyO7 (0 ≤ y ≤ 2).
| Gd2Zr2O7 | 1.907 | 0.01355 | 4666.0 | 580.2 | 0.277 | |
| Exp. [45,46] | 1.913 | 0.276 | ||||
| Exp. [20] | 1.891 | 513.3 | 0.274 | |||
| Cal. [53] | 2.004 | 0.00420 | 4833.5 | 612.9 | 0.286 | |
| Cal. [43] | 0.273 | |||||
| Gd1.5Pu0.5Zr2O7 | 1.931 | 0.00598 | 4533.7 | 560.7 | 0.279 | |
| Gd1.0Pu1.0Zr2O7 | 1.950 | 0.00105 | 4367.8 | 540.2 | 0.281 | |
| Gd0.5Pu1.5Zr2O7 | 1.964 | 0.00032 | 4247.4 | 522.5 | 0.282 | |
| Pu2Zr2O7 | 1.987 | 0.00004 | 4106.4 | 503.8 | 0.285 | |
| Gd2Zr1.5Pu0.5O7 | 1.928 | 0.04881 | 4124.6 | 508.7 | 0.279 | |
| Gd2Zr1.0Pu1.0O7 | 2.278 | 0.21353 | 3591.7 | 439.5 | 0.309 | |
| Gd2Zr0.5Pu1.5O7 | 2.243 | 0.10062 | 3590.2 | 435.9 | 0.306 | |
| Gd2Pu2O7 | 2.151 | 0.06923 | 3471.3 | 418.3 | 0.299 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, P.; Zhao, F.; Xiao, H.; Zhang, H.; Gong, H.; Zhang, S.; Liu, Z.; Zu, X. First-Principles Study of Thermo-Physical Properties of Pu-Containing Gd2Zr2O7. Nanomaterials 2019, 9, 196. https://doi.org/10.3390/nano9020196
AMA Style
Li P, Zhao F, Xiao H, Zhang H, Gong H, Zhang S, Liu Z, Zu X. First-Principles Study of Thermo-Physical Properties of Pu-Containing Gd2Zr2O7. Nanomaterials. 2019; 9(2):196. https://doi.org/10.3390/nano9020196
Chicago/Turabian StyleLi, Pengcheng, Fengai Zhao, Haiyan Xiao, Haibin Zhang, Hengfeng Gong, Sa Zhang, Zijiang Liu, and Xiaotao Zu. 2019. "First-Principles Study of Thermo-Physical Properties of Pu-Containing Gd2Zr2O7" Nanomaterials 9, no. 2: 196. https://doi.org/10.3390/nano9020196
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.