Stealth Biocompatible Si-Based Nanoparticles for Biomedical Applications

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
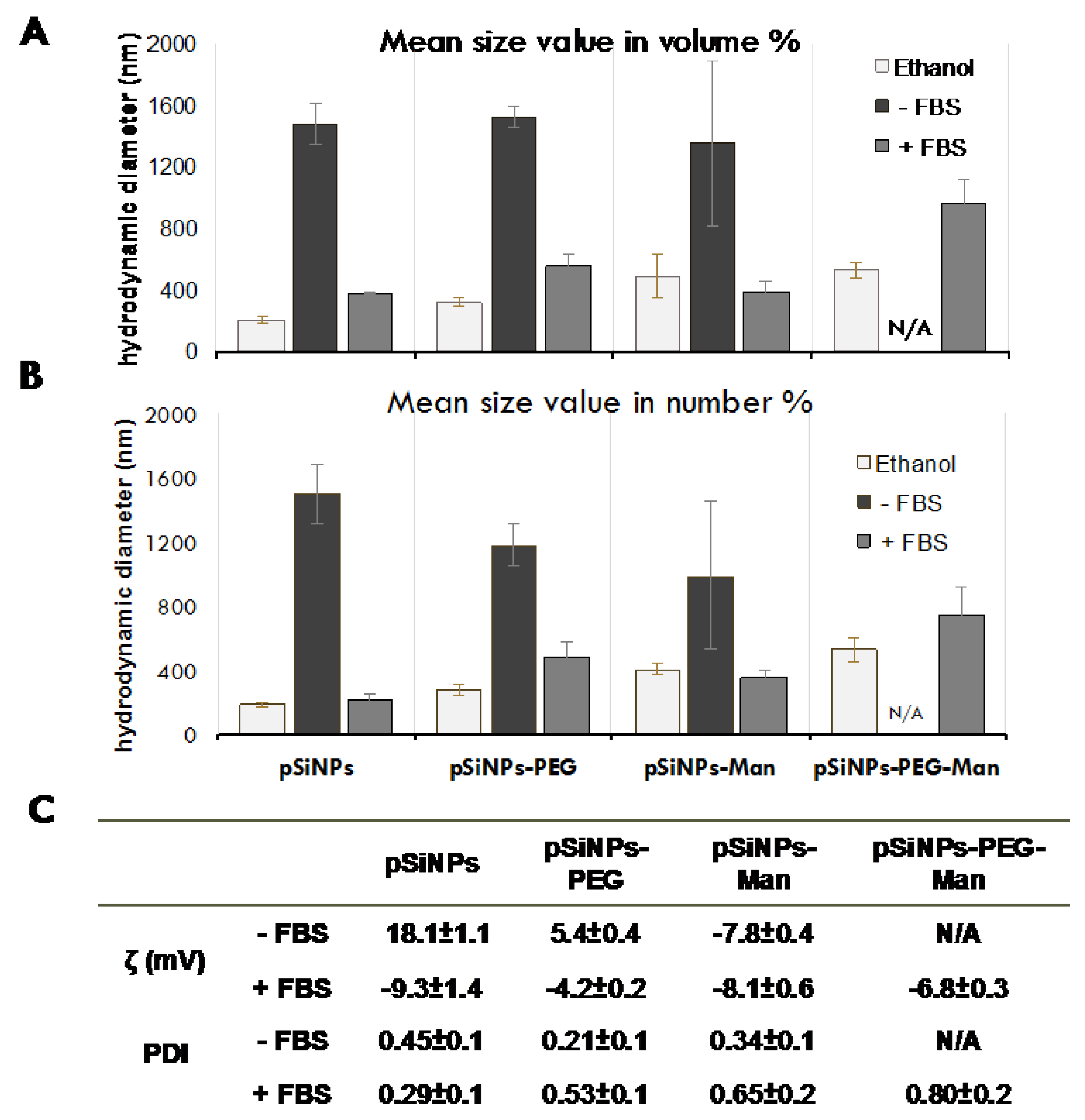
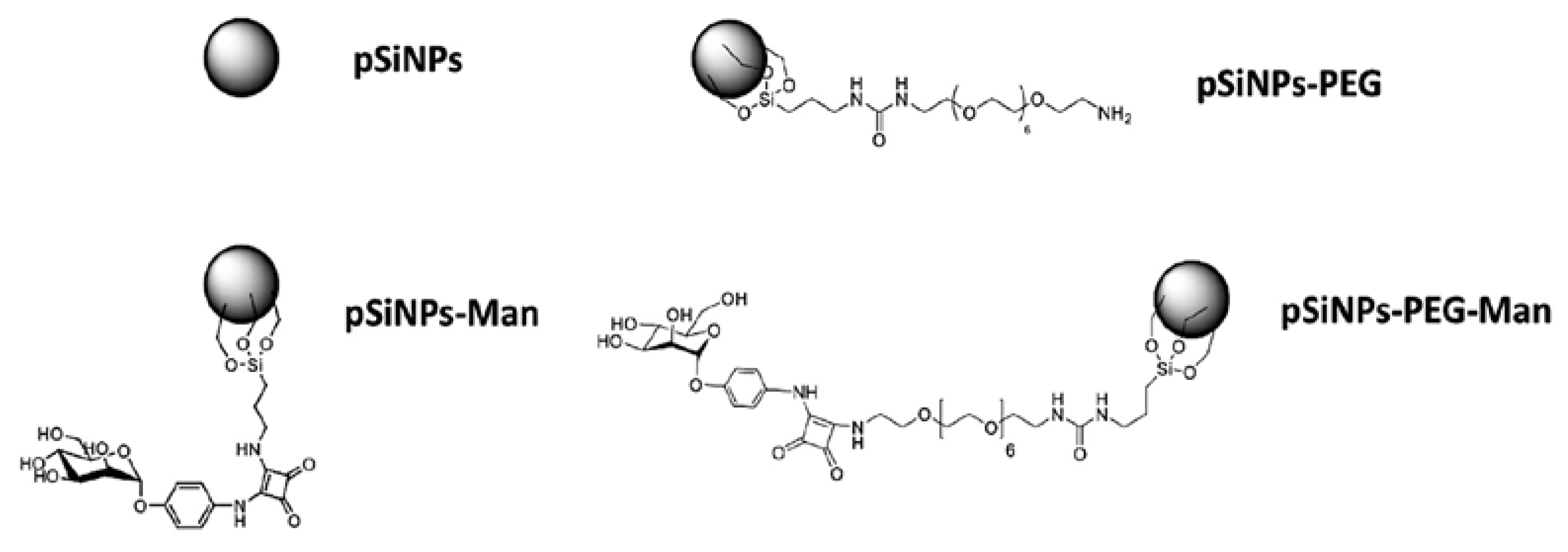
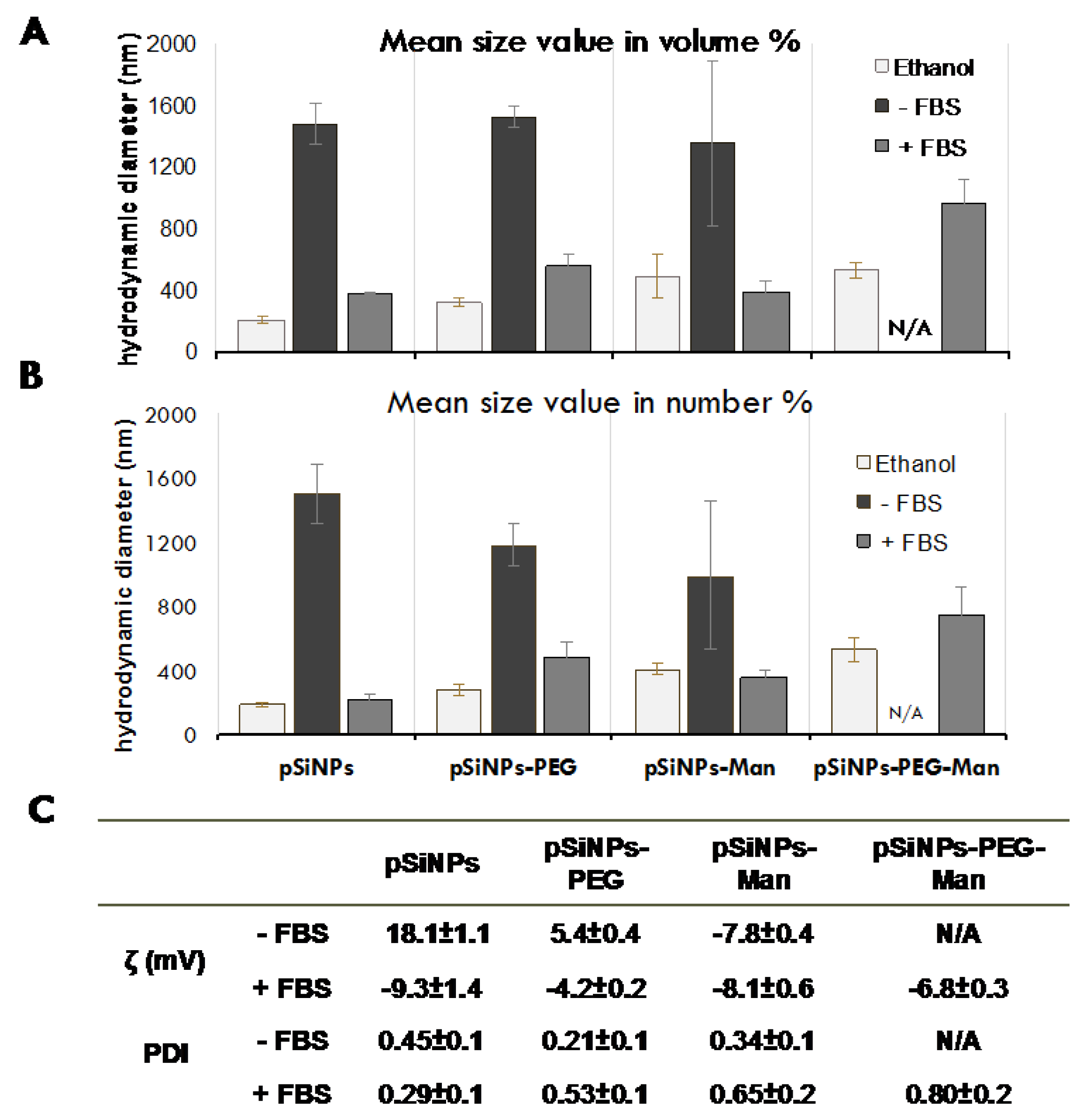
2.1. Physico-Chemical Properties of Pristine and Formulated pSiNPs in Stock Suspensions
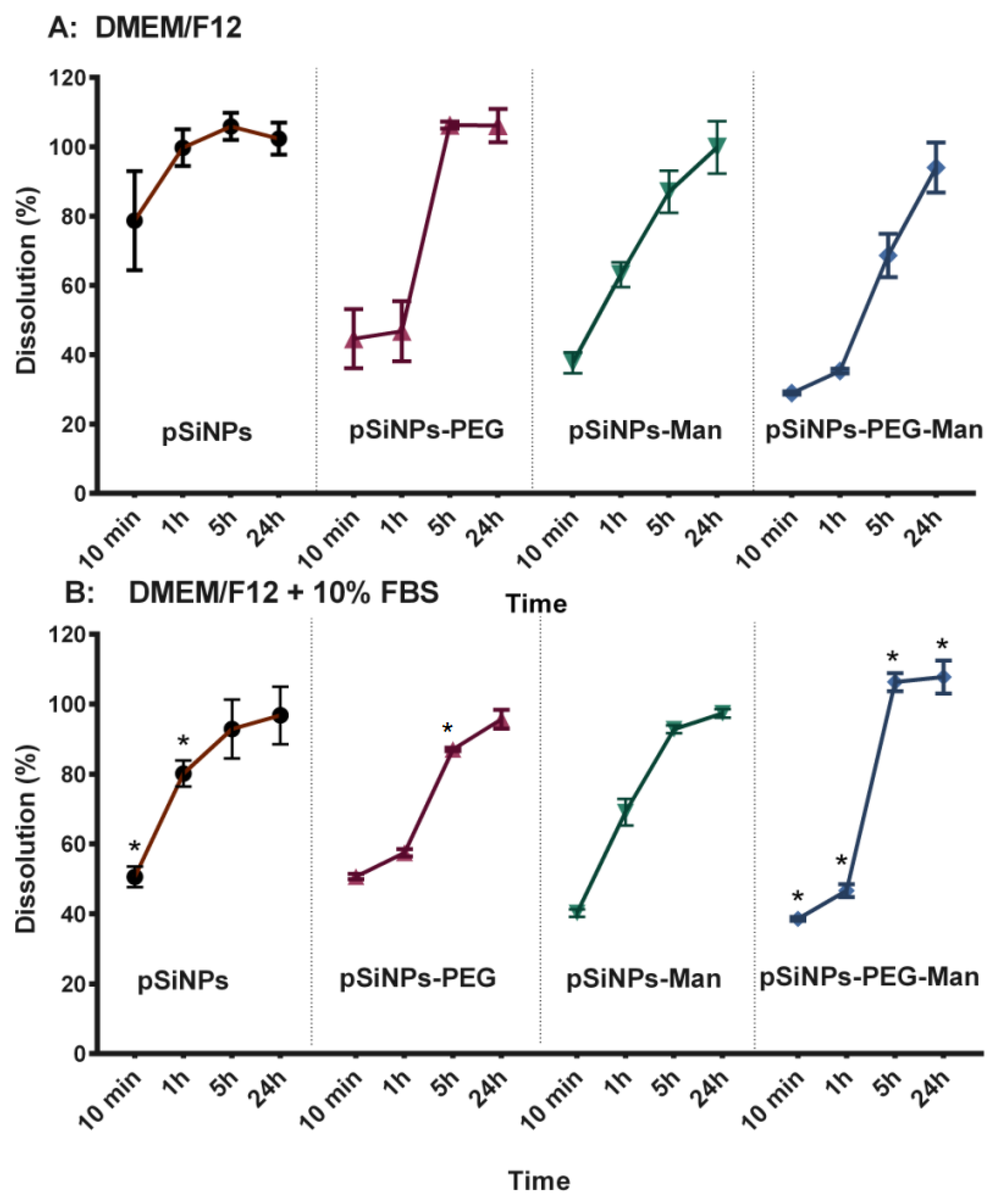
2.2. In Vitro Biodegradability Kinetics
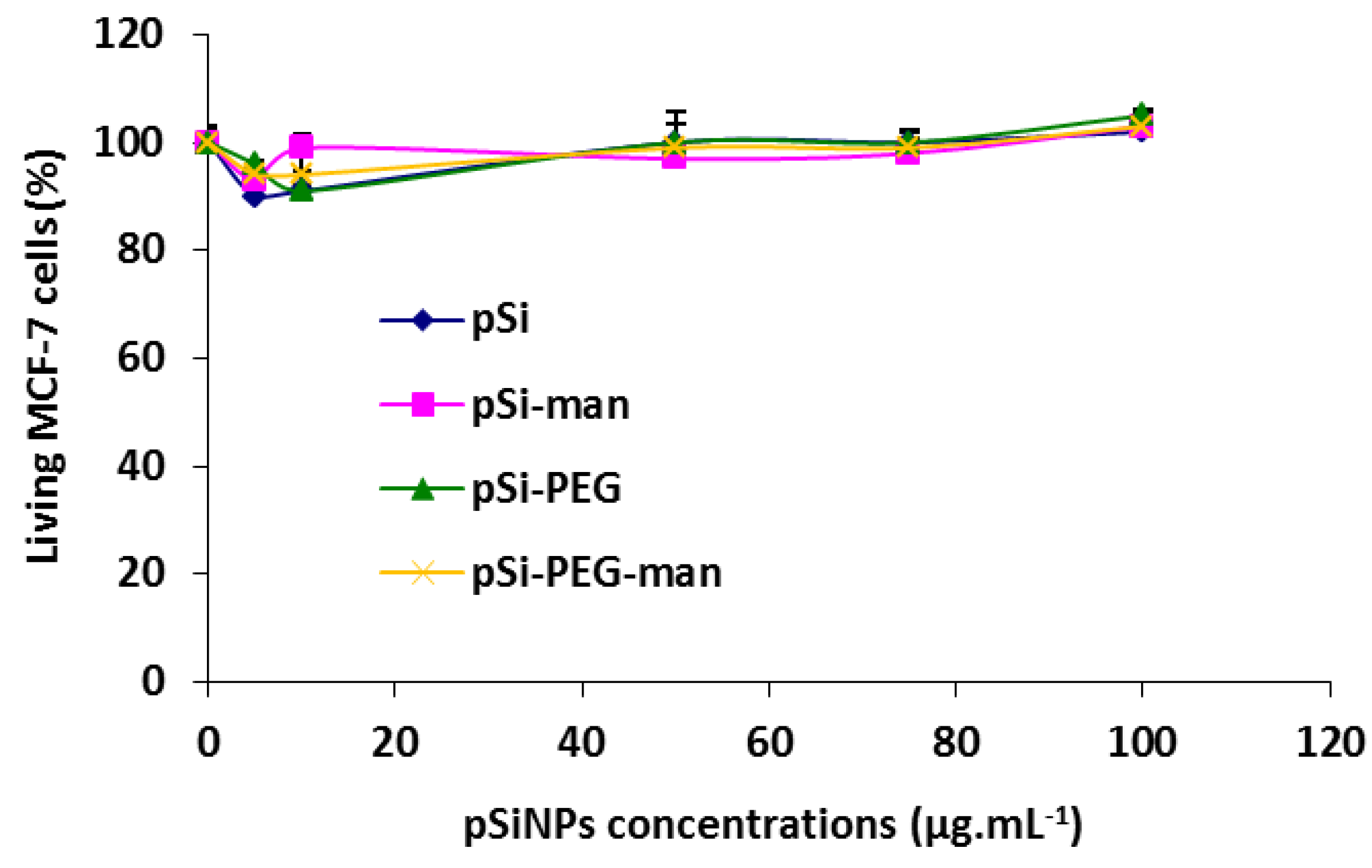
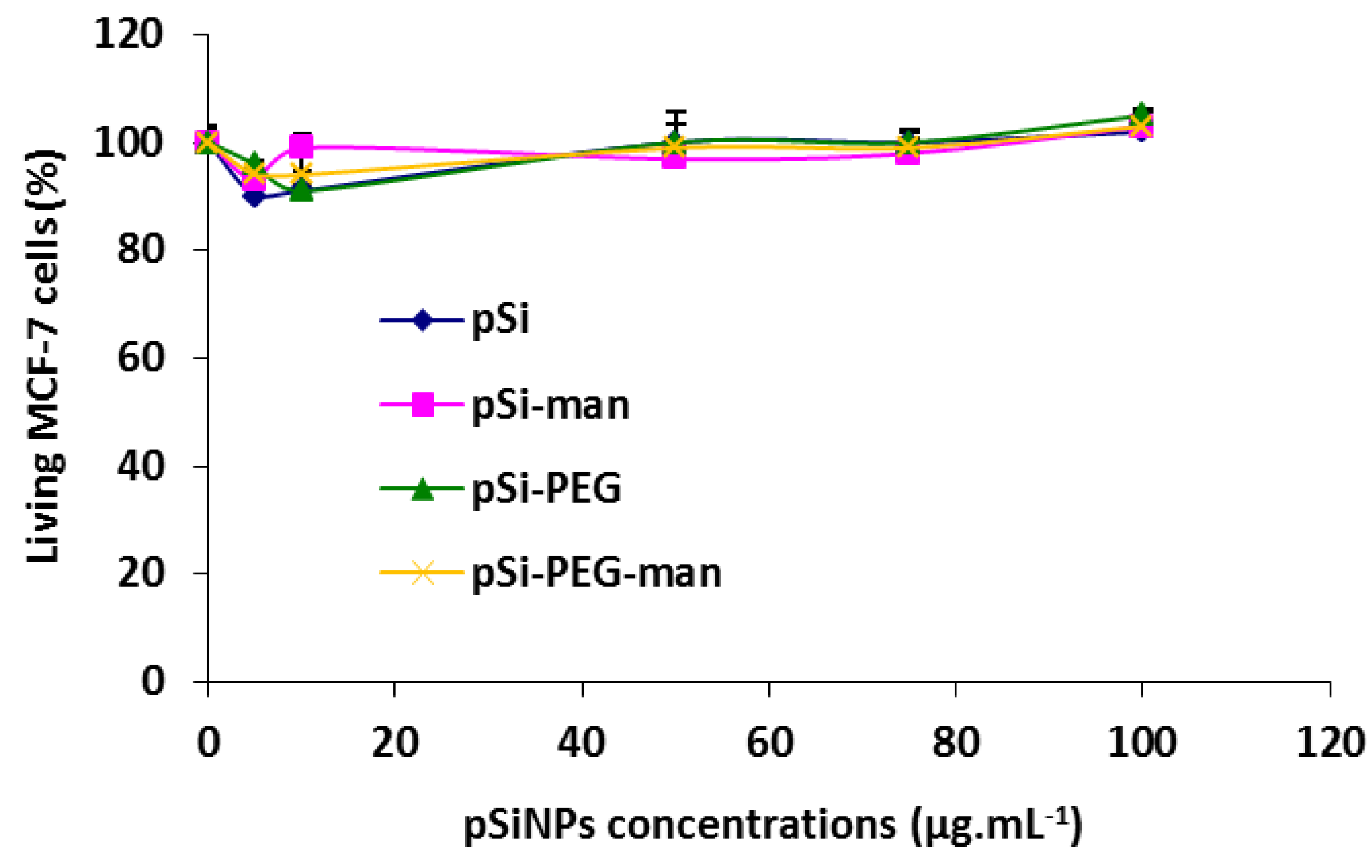
2.3. Biocompatibility Studies In Vitro
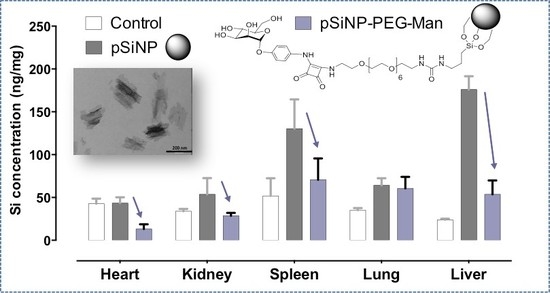
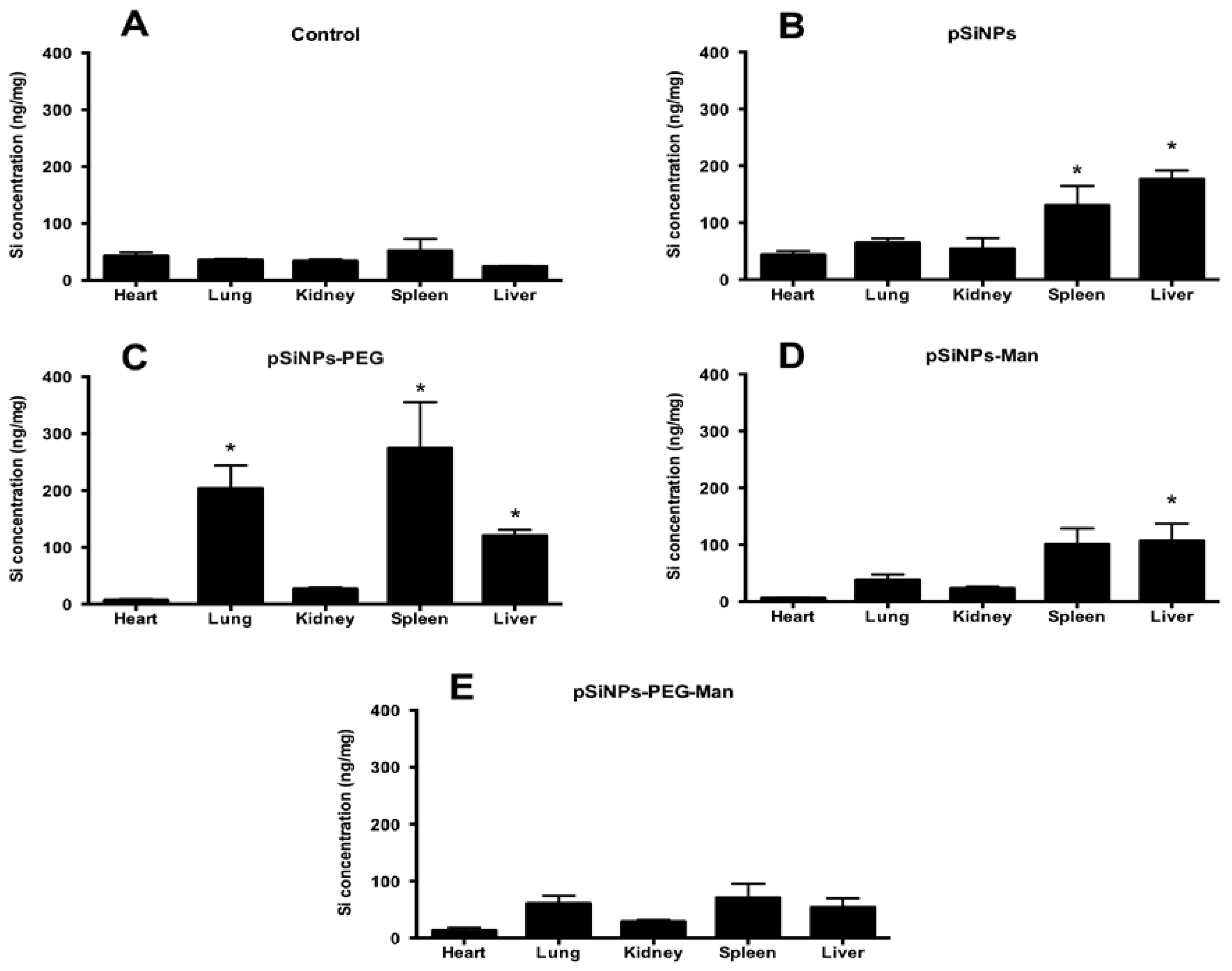
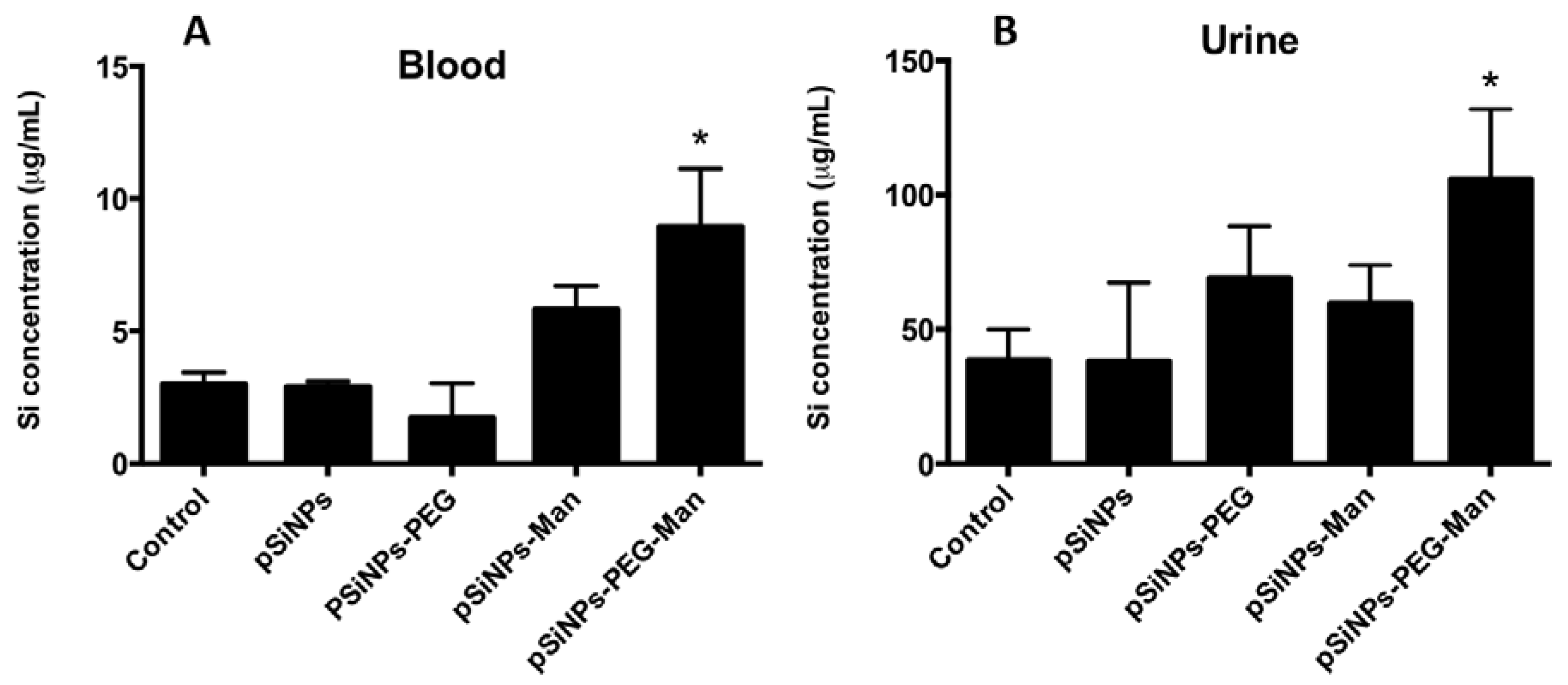
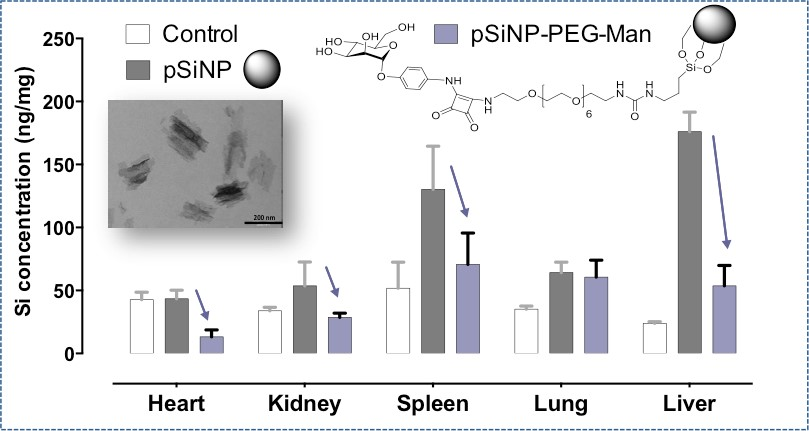
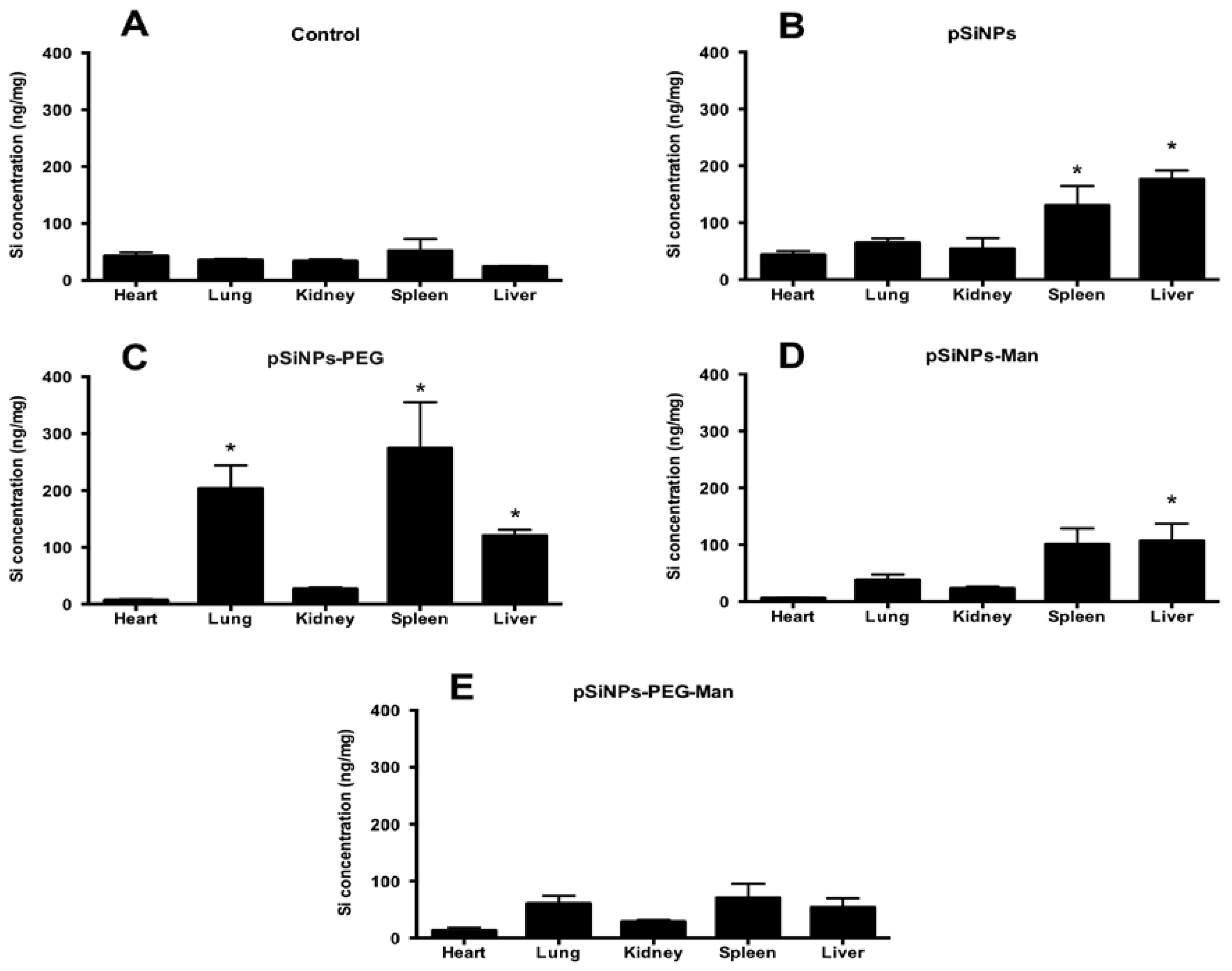
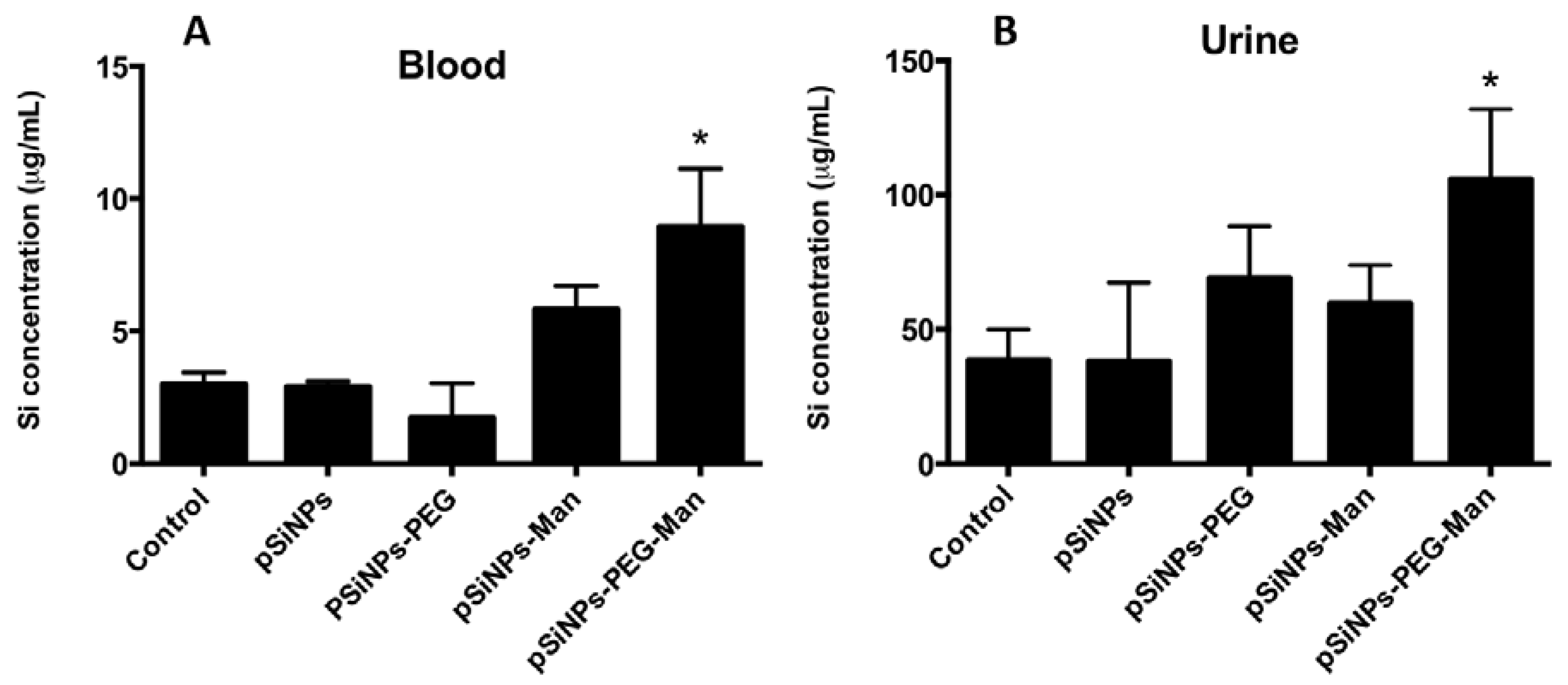
2.4. Biodistribution and Clearance of the pSiNps Formulation In Vivo
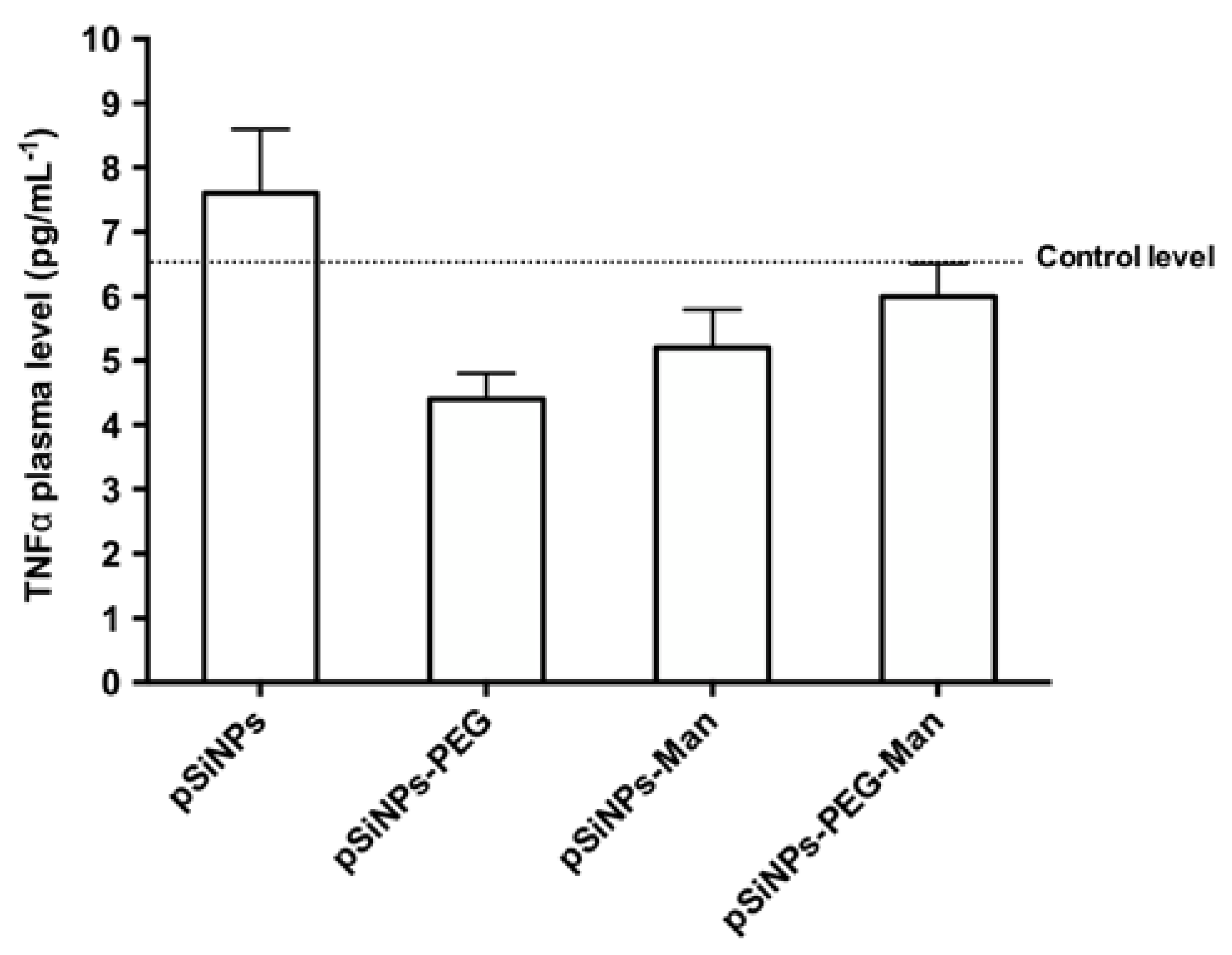
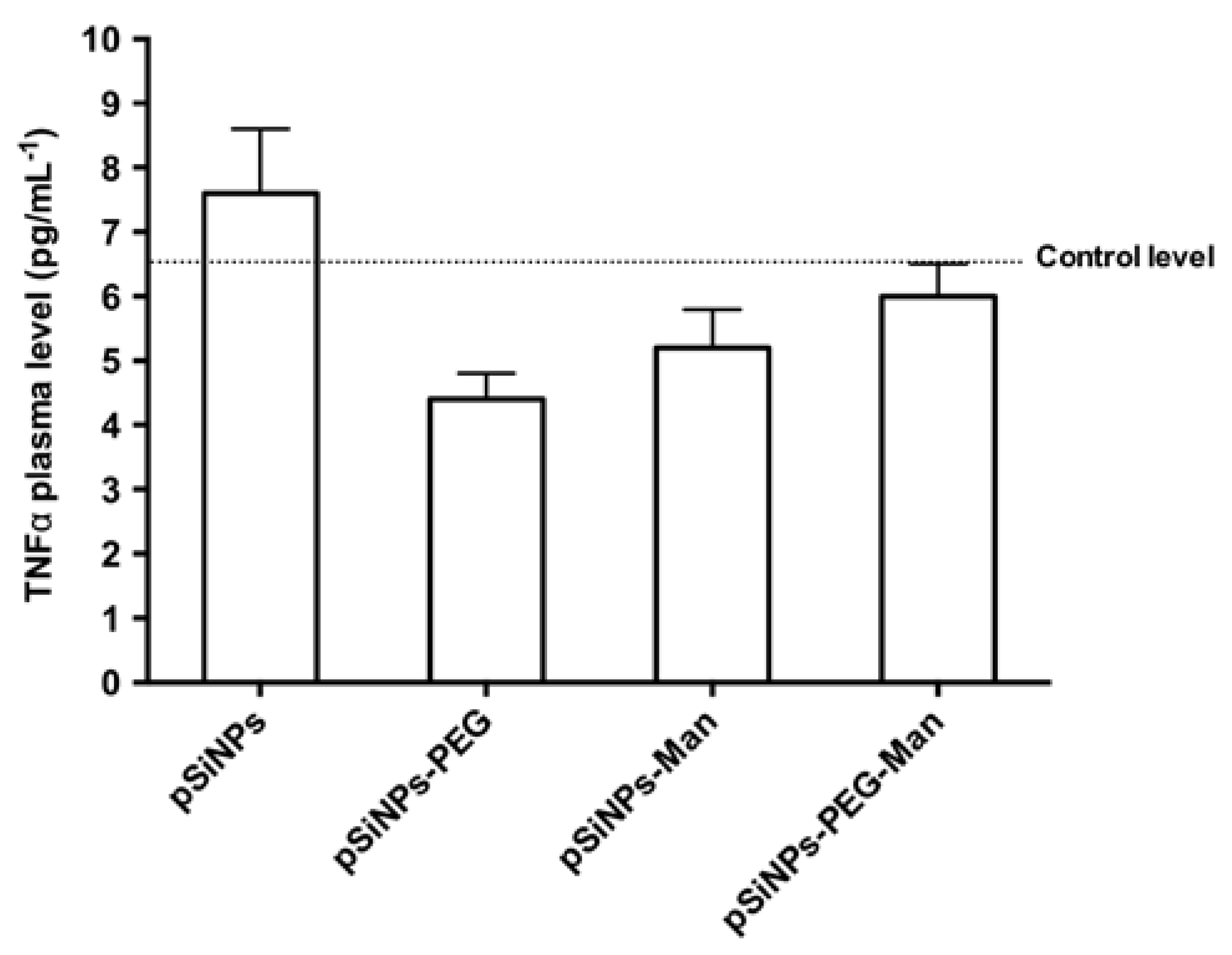
2.5. Biocompatibility Studies In Vivo
3. Materials and Methods
3.1. pSiNPs Synthesis
3.2. pSiNPs Functionalization With PEG
3.3. pSiNPs–PEG Functionalization With Man
3.4. pSiNPs Functionalization With Man
3.5. Characterizations of pSiNPs Initial Suspensions in Ethanol
3.6. Dosing of the Phenyl Squarate Mannose Grafted on the pSiNPs–Man and pSiNPs–PEG–Man Formulations
3.7. Physico-Chemical Stability In Vitro
3.8. In Vitro Cytotoxicity on Cell Line
3.9. Biodistribution in Mice
3.10. TNFα Plasma Levels Measurement
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lu, J.; Liong, M.; Li, Z.; Zink, J.I.; Tamanoi, F. Biocompatibility, biodistribution, and drug-delivery efficiency of mesoporous silica nanoparticles for cancer therapy in animals. Small 2010, 6, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Xue, M.; Xia, T.; Ji, Z.; Tarn, D.Y.; Zink, J.I.; Nel, A.E. Use of size and a copolymer design feature to improve the biodistribution and the enhanced permeability and retention effect of doxorubicin-loaded mesoporous silica nanoparticles in a murine xenograft tumor model. ACS Nano 2011, 5, 4131–4144. [Google Scholar] [CrossRef] [PubMed]
- Wilczewska, A.Z.; Niemirowicz, K.; Markiewicz, K.H.; Car, H. Nanoparticles as drug delivery systems. Pharmacol Rep. 2012, 64, 1020–1037. [Google Scholar] [CrossRef]
- Mamaeva, V.; Sahlgren, C.; Lindén, M. Mesoporous silica nanoparticles in medicine—Recent advances. Adv. Drug Deliv. Rev. 2013, 65, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Santos, H.A.; Makila, E.; Airaksinen, A.J.; Bimbo, L.M.; Hirvonen, J. Porous silicon nanoparticles for nanomedicine: Preparation and biomedical applications. Nanomedicine 2014, 9, 535–554. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Gu, L.; von Maltzahn, G.; Ruoslahti, E.; Bhatia, S.N.; Sailor, M.J. Biodegradable luminescent porous silicon nanoparticles for in vivo applications. Nat. Mater. 2009, 8, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, K.L.; Barnes, T.J.; Prestidge, C.A. Surface chemistry of porous silicon and implications for drug encapsulation and delivery applications. Adv. Colloid Interface Sci. 2012, 175, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Gobin, A.M.; Lee, M.H.; Halas, N.J.; James, W.D.; Drezek, R.A.; West, J.L. Near-infrared resonant nanoshells for combined optical imaging and photothermal cancer therapy. Nano Lett. 2007, 7, 1929–1934. [Google Scholar] [CrossRef] [PubMed]
- Sailor, M.J.; Wu, E.C. Photoluminescence-Based Sensing With Porous Silicon Films, Microparticles, and Nanoparticles. Adv. Funct. Mater. 2009, 19, 3195–3208. [Google Scholar] [CrossRef]
- Gu, L.; Hall, D.J.; Qin, Z.; Anglin, E.; Joo, J.; Mooney, D.J.; Howell, S.B.; Sailor, M.J. In vivo time-gated fluorescence imaging with biodegradable luminescent porous silicon nanoparticles. Nat. Commun. 2013, 4, 2326–2332. [Google Scholar] [CrossRef] [PubMed]
- Gary-Bobo, M.; Mir, Y.; Rouxel, C.; Brevet, D.; Basile, I.; Maynadier, M.; Vaillant, O.; Mongin, O.; Blanchard-Desce, M.; Morère, P.A. Mannose-functionalized mesoporous silica nanoparticles for efficient two-photon photodynamic therapy of solid tumors. Angew. Chem. 2011, 123, 11627–11631. [Google Scholar] [CrossRef]
- Monopoli, M.P.; Walczyk, D.; Campbell, A.G.; Elia, G.; Lynch, I.; Bombelli, F.B.; Dawson, K.A. Physical-chemical aspects of protein corona: Relevance to in vitro and in vivo biological impacts of nanoparticles. J. Am. Chem. Soc. 2011, 133, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Gary-Bobo, M.; Hocine, O.; Brevet, D.; Maynadier, M.; Raehm, L.; Richeter, S.; Charasson, V.; Loock, B.; Morère, A.; Maillard, P. Cancer therapy improvement with mesoporous silica nanoparticles combining targeting, drug delivery and PDT. Int. J. Pharm. 2012, 423, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Graf, C.; Gao, Q.; Schütz, I.; Noufele, C.N.; Ruan, W.; Posselt, U.; Korotianskiy, E.; Nordmeyer, D.; Rancan, F.; Hadam, S. Surface functionalization of silica nanoparticles supports colloidal stability in physiological media and facilitates internalization in cells. Langmuir 2012, 28, 7598–7613. [Google Scholar] [CrossRef] [PubMed]
- Perrier, M.; Gary-Bobo, M.; Lartigue, L.; Brevet, D.; Morère, A.; Garcia, M.; Maillard, P.; Raehm, L.; Guari, Y.; Larionova, J. Mannose-functionalized porous silica-coated magnetic nanoparticles for two-photon imaging or PDT of cancer cells. J. Nanopart. Res. 2013, 15, 1–17. [Google Scholar] [CrossRef]
- Secret, E.; Maynadier, M.; Gallud, A.; Gary-Bobo, M.; Chaix, A.; Belamie, E.; Maillard, P.; Sailor, M.J.; Garcia, M.; Durand, J.-O. Anionic porphyrin-grafted porous silicon nanoparticles for photodynamic therapy. Chem. Commun. 2013, 49, 4202–4204. [Google Scholar] [CrossRef] [PubMed]
- Secret, E.; Smith, K.; Dubljevic, V.; Moore, E.; Macardle, P.; Delalat, B.; Rogers, M.L.; Johns, T.G.; Durand, J.O.; Cunin, F. Antibody-Functionalized Porous Silicon Nanoparticles for Vectorization of Hydrophobic Drugs. Adv. Healthc. Mater. 2013, 2, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Vaillant, O.; Cheikh, K.E.; Warther, D.; Brevet, D.; Maynadier, M.; Bouffard, E.; Salgues, F.; Jeanjean, A.; Puche, P.; Mazerolles, C. Mannose-6-Phosphate Receptor: A Target for Theranostics of Prostate Cancer. Angew. Chem. 2015, 127, 6050–6054. [Google Scholar] [CrossRef]
- Mogoşanu, G.D.; Grumezescu, A.M.; Bejenaru, C.; Bejenaru, L.E. Polymeric protective agents for nanoparticles in drug delivery and targeting. Int. J. Pharm. 2016, 2, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Debele, T.A.; Peng, S.; Tsai, H.C. Drug Carrier for Photodynamic Cancer Therapy. Int. J. Mol. Sci. 2015, 16, 22094–22136. [Google Scholar] [CrossRef] [PubMed]
- Giret, S.; Wong Chi Man, M.; Carcel, C. Mesoporous-Silica-Functionalized Nanoparticles for Drug Delivery. Chem. Eur. J. 2015, 21, 13850–13865. [Google Scholar] [CrossRef] [PubMed]
- Nigam, P.; Sarkar, D. Multifunctional silica nanoparticles for pancreatic cancer specific drug delivery and bioimaging. J. Chem. Appl. Biochem. 2015, 2, 110–116. [Google Scholar]
- Xiao, L.; Gu, L.; Howell, S.B.; Sailor, M.J. Porous silicon nanoparticle photosensitizers for singlet oxygen and their phototoxicity against cancer cells. ACS Nano 2011, 5, 3651–3659. [Google Scholar] [CrossRef] [PubMed]
- Secret, E.; Maynadier, M.; Gallud, A.; Chaix, A.; Bouffard, E.; Gary-Bobo, M.; Marcotte, N.; Mongin, O.; El Cheikh, K.; Hugues, V.; et al. Two-photon excitation of porphyrin-functionalized porous silicon nanoparticles for photodynamic therapy. Adv Mater. 2014, 26, 7643–7648. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.E.; Madler, L.; Velegol, D.; Xia, T.; Hoek, E.M.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Niu, M.; O’Mary, H.; Cui, Z. Targeting of tumor-associated macrophages made possible by PEG-sheddable, mannose-modified nanoparticles. Mol. Pharm. 2013, 10, 3525–3530. [Google Scholar] [CrossRef] [PubMed]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297–315. [Google Scholar]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(ethylene glycol) in drug delivery: Pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. Engl. 2010, 49, 6288–6308. [Google Scholar] [CrossRef] [PubMed]
- Morelli, C.; Maris, P.; Sisci, D.; Perrotta, E.; Brunelli, E.; Perrotta, I.; Panno, M.L.; Tagarelli, A.; Versace, C.; Casula, M.F. PEG-templated mesoporous silica nanoparticles exclusively target cancer cells. Nanoscale 2011, 3, 3198–3207. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Lee, J.; Zheng, H.; Hong, S.-S.; Lee, C. Porous silicon nanoparticles for cancer photothermotherapy. Nanoscale Res. Lett. 2011, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Kuthati, Y.; Sung, P.-J.; Weng, C.-F.; Mou, C.-Y.; Lee, C.-H. Functionalization of mesoporous silica nanoparticles for targeting, biocompatibility, combined cancer therapies and theragnosis. J. Nanosci. Nanotechnol. 2013, 13, 2399–2430. [Google Scholar] [CrossRef] [PubMed]
- Brevet, D.; Gary-Bobo, M.; Raehm, L.; Richeter, S.; Hocine, O.; Amro, K.; Loock, B.; Couleaud, P.; Frochot, C.; Morère, A. Mannose-targeted mesoporous silica nanoparticles for photodynamic therapy. Chem. Commun. 2009, 1475–1477. [Google Scholar] [CrossRef] [PubMed]
- Warther, D.; Jimenez, C.M.; Raehm, L.; Gérardin, C.; Durand, J.-O.; Morère, A.; El Cheikh, K.; Gallud, A.; Gary-Bobo, M.; Maynadier, M. Small sized mesoporous silica nanoparticles functionalized with mannose for retinoblastoma cell imaging. RSC Adv. 2014, 4, 37171–37179. [Google Scholar] [CrossRef]
- Chaix, A.; El Cheikh, K.; Bouffard, E.; Maynadier, M.; Aggad, D.; Stojanovic, V.; Knezevic, N.; Garcia, M.; Maillard, P.; Morere, A.; et al. Mesoporous silicon nanoparticles for targeted two-photon theranostics of prostate cancer. J. Mater. Chem. B 2016, 4, 3639–3642. [Google Scholar] [CrossRef]
- Tang, F.; Li, L.; Chen, D. Mesoporous silica nanoparticles: Synthesis, biocompatibility and drug delivery. Adv. Mater. 2012, 24, 1504–1534. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Zhang, J.; Shi, J.; Zhu, Z.; Zhang, L.; Bu, W.; Guo, L.; Chen, Y. The effect of PEGylation of mesoporous silica nanoparticles on nonspecific binding of serum proteins and cellular responses. Biomaterials 2010, 31, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Zhang, Z.; Gao, F.; Li, Y.; Shi, J. In vivo biodistribution and urinary excretion of mesoporous silica nanoparticles: Effects of particle size and PEGylation. Small 2011, 7, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Elliott, H.; Wallis, D.; Canham, L.; Powell, J. Dissolution of different forms of partially porous silicon wafers under simulated physiological conditions. Physica Status Solidi 2003, 197, 331–335. [Google Scholar] [CrossRef]
- Borkovec, M.; Papastavrou, G. Interactions between solid surfaces with adsorbed polyelectrolytes of opposite charge. Curr. Opin. Colloid Interface Sci. 2008, 13, 429–437. [Google Scholar] [CrossRef]
- Lee, C.H.; Cheng, S.H.; Huang, I.; Souris, J.S.; Yang, C.S.; Mou, C.Y.; Lo, L.W. Intracellular pH-responsive mesoporous silica nanoparticles for the controlled release of anticancer chemotherapeutics. Angew. Chem. 2010, 122, 8390–8395. [Google Scholar] [CrossRef]
- Pelaz, B.; del Pino, P.; Maffre, P.; Hartmann, R.; Gallego, M.; Rivera-Fernandez, S.; de la Fuente, J.M.; Nienhaus, G.U.; Parak, W.J. Surface functionalization of nanoparticles with polyethylene glycol: Effects on protein adsorption and cellular uptake. ACS Nano 2015, 9, 6996–7008. [Google Scholar] [CrossRef] [PubMed]
- Milani, S.; Bombelli, F.B.; Pitek, A.S.; Dawson, K.A.; Radler, J. Reversible versus irreversible binding of transferrin to polystyrene nanoparticles: Soft and hard corona. ACS Nano 2012, 6, 2532–2541. [Google Scholar] [CrossRef] [PubMed]
- Lesniak, A.; Fenaroli, F.; Monopoli, M.P.; Aberg, C.; Dawson, K.A.; Salvati, A. Effects of the presence or absence of a protein corona on silica nanoparticle uptake and impact on cells. ACS Nano 2012, 6, 5845–5857. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Rose, J.; Plantevin, S.; Auffan, M.; Bottero, J.-Y.; Vidaud, C. Protein corona formation for nanomaterials and proteins of a similar size: Hard or soft corona? Nanoscale 2013, 5, 1658–1668. [Google Scholar] [CrossRef] [PubMed]
- Gref, R.; Lück, M.; Quellec, P.; Marchand, M.; Dellacherie, E.; Harnisch, S.; Blunk, T.; Müller, R. ‘Stealth’corona-core nanoparticles surface modified by polyethylene glycol (PEG): Influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf. B Biointerfaces 2000, 18, 301–313. [Google Scholar] [CrossRef]
- Aggarwal, P.; Hall, J.B.; McLeland, C.B.; Dobrovolskaia, M.A.; McNeil, S.E. Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Adv. Drug Deliv. Rev. 2009, 61, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Godin, B.; Gu, J.; Serda, R.; Bhavane, R.; Tasciotti, E.; Chiappini, C.; Liu, X.; Tanaka, T.; Decuzzi, P.; Ferrari, M. Tailoring the degradation kinetics of mesoporous silicon structures through PEGylation. J. Biomed. Mater. Res. A 2010, 94, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Liu, T.; Li, L.; Liu, H.; Chen, D.; Tang, F. The absorption, distribution, excretion and toxicity of mesoporous silica nanoparticles in mice following different exposure routes. Biomaterials 2013, 34, 2565–2575. [Google Scholar] [CrossRef] [PubMed]
- Faure, A.C.; Dufort, S.; Josserand, V.; Perriat, P.; Coll, J.L.; Roux, S.; Tillement, O. Control of the in vivo biodistribution of hybrid nanoparticles with different poly (ethylene glycol) coatings. Small 2009, 5, 2565–2575. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.; Jefferies, C.; Cryan, S.-A. Targeted liposomal drug delivery to monocytes and macrophages. J. Drug. Deliv. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.L.; Reuter, K.G.; Kai, M.P.; Herlihy, K.P.; Jones, S.W.; Luft, J.C.; Napier, M.; Bear, J.E.; De Simone, J.M. PEGylated PRINT nanoparticles: The impact of PEG density on protein binding, macrophage association, biodistribution, and pharmacokinetics. Nano Lett. 2012, 12, 5304–5310. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanoparticles | pSiNPs | pSiNPs–PEG | pSiNPs–Man | pSiNPs–PEG–Man |
|---|---|---|---|---|
| Zeta potential (mV) | −25 ± 1 | 28 ± 2 | −18 ± 1 | 10 ± 0.2 |
| Hydrodynamic diameter (nm) vol. % | 200 ± 20 | 315 ± 26 | 488 ± 103 | 530 ± 56 |
| Hydrodynamic diameter (nm) num. % | 185 ± 18 | 276 ± 35 | 404 ± 33 | 528 ± 77 |
| PDI | 0.16 ± 0.02 | 0.35 ± 0.2 | 0.22 ± 0.1 | 0.68 ± 0.3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, W.; Chaix, A.; Gary-Bobo, M.; Angeletti, B.; Masion, A.; Da Silva, A.; Daurat, M.; Lichon, L.; Garcia, M.; Morère, A.; et al. Stealth Biocompatible Si-Based Nanoparticles for Biomedical Applications. Nanomaterials 2017, 7, 288. https://doi.org/10.3390/nano7100288
Liu W, Chaix A, Gary-Bobo M, Angeletti B, Masion A, Da Silva A, Daurat M, Lichon L, Garcia M, Morère A, et al. Stealth Biocompatible Si-Based Nanoparticles for Biomedical Applications. Nanomaterials. 2017; 7(10):288. https://doi.org/10.3390/nano7100288
Chicago/Turabian StyleLiu, Wei, Arnaud Chaix, Magali Gary-Bobo, Bernard Angeletti, Armand Masion, Afitz Da Silva, Morgane Daurat, Laure Lichon, Marcel Garcia, Alain Morère, and et al. 2017. "Stealth Biocompatible Si-Based Nanoparticles for Biomedical Applications" Nanomaterials 7, no. 10: 288. https://doi.org/10.3390/nano7100288