
Non-Conventional Peptide Self-Assembly into a Conductive Supramolecular Rope
 , ,
, ,  , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Modelling
2.2. Materials
2.3. Methods
3. Results
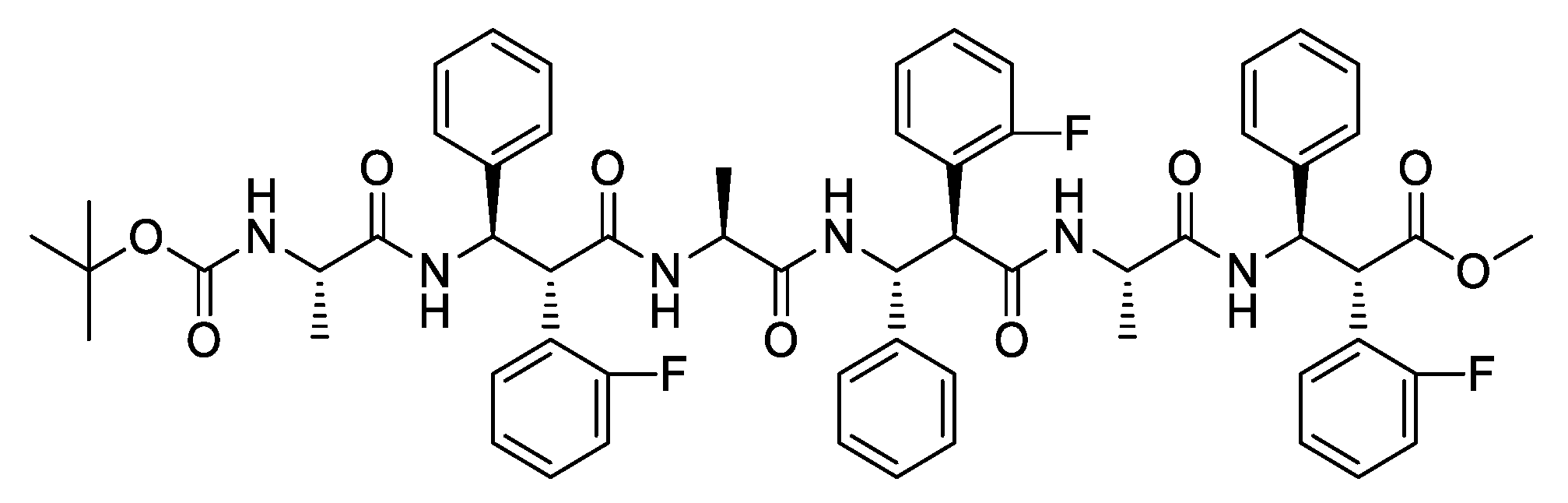
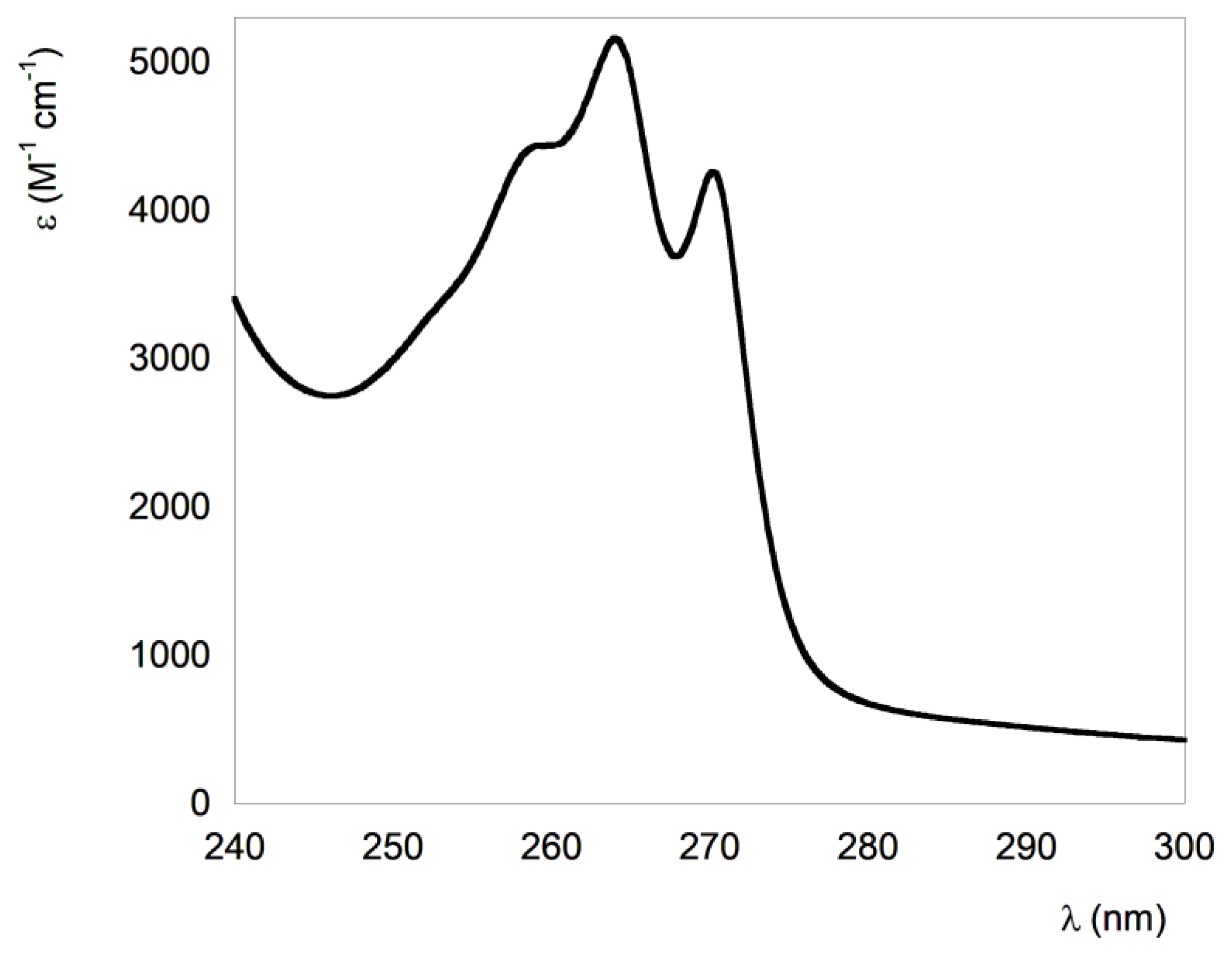
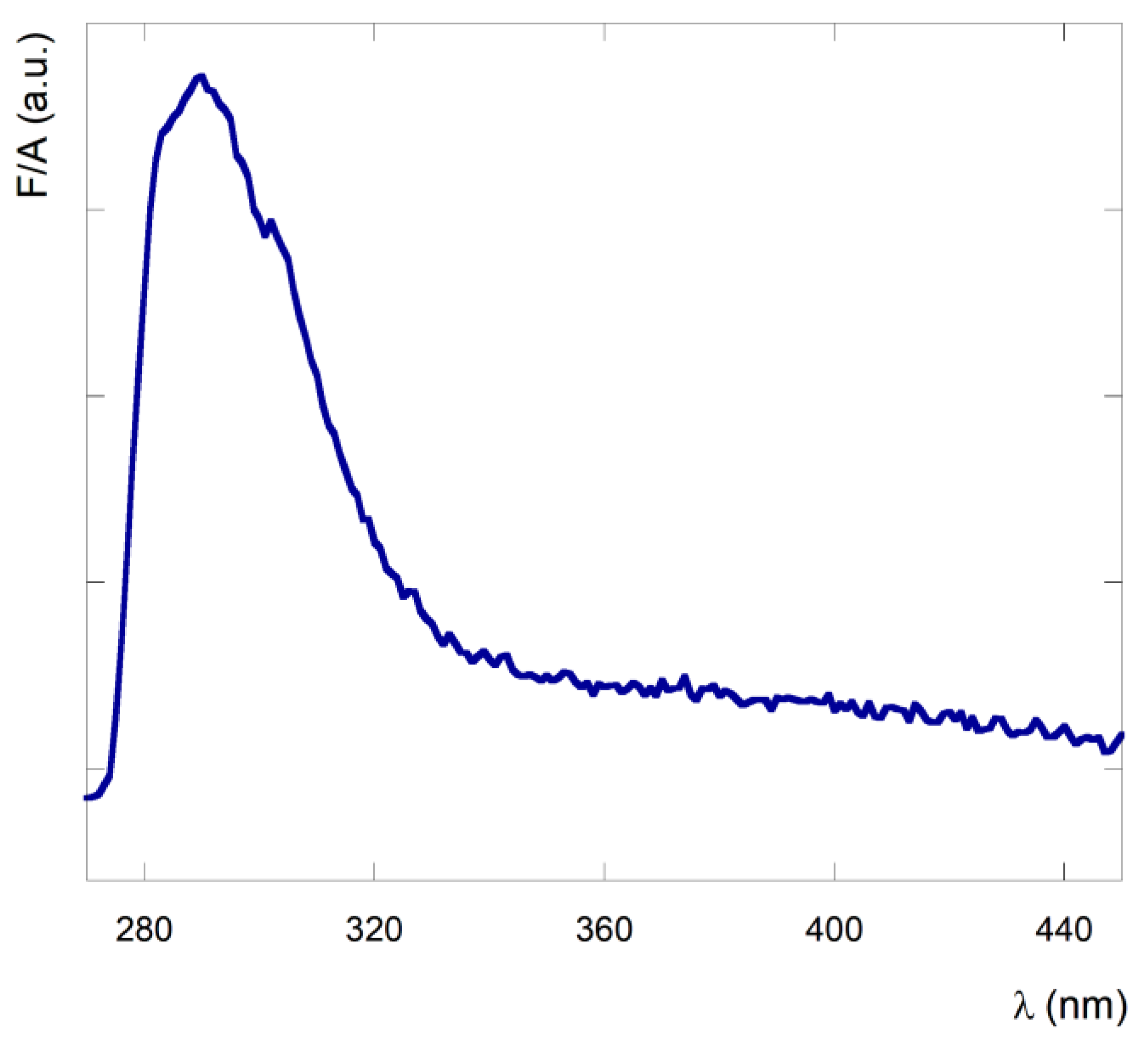
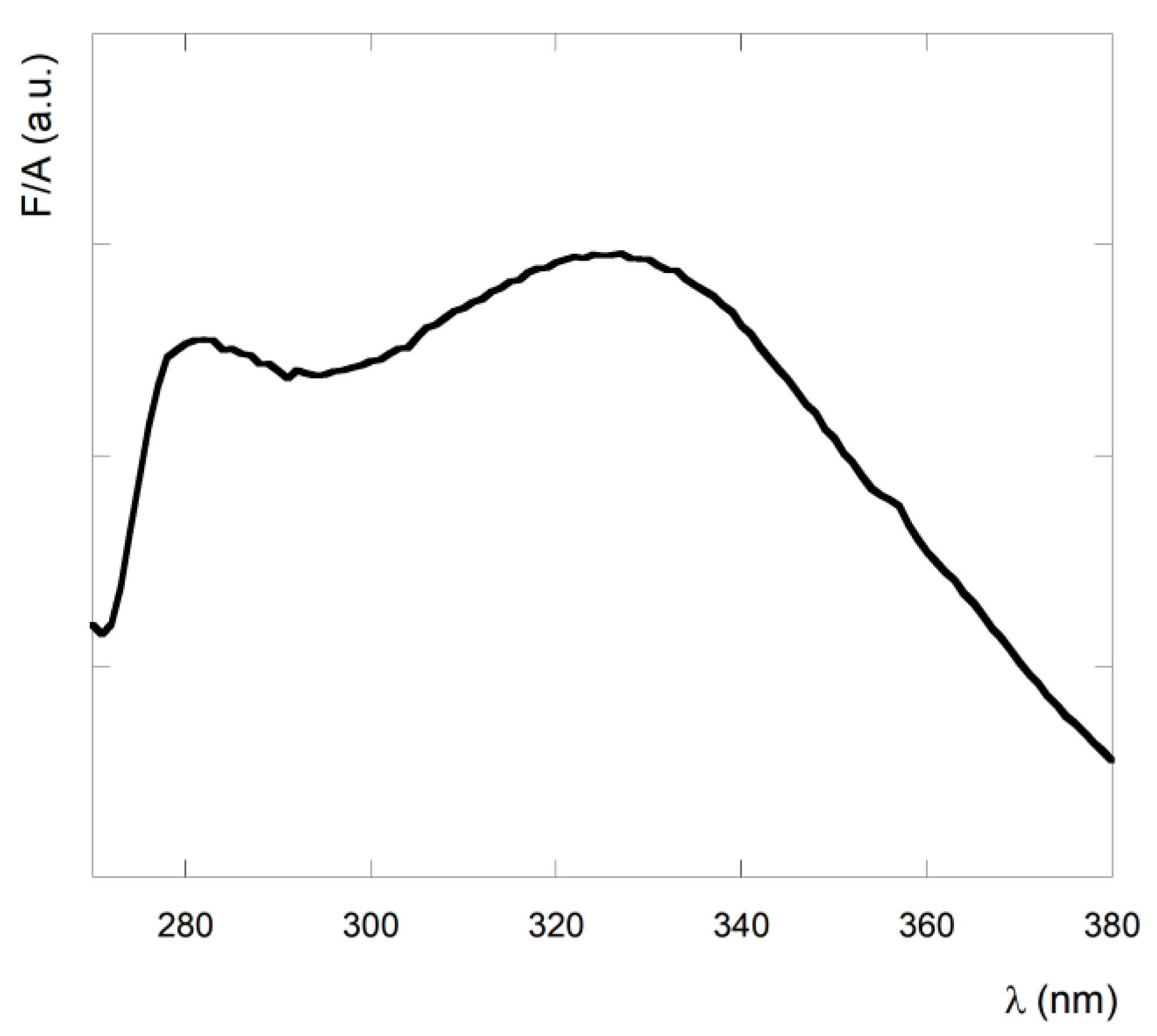
3.1. Spectrocopic Measurements
3.2. Morphology of the Supramolecular Assembly
3.3. Molecular Organization within the Assembly
3.4. Electronic Conduction Measurements
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martín, J.; Martín-González, M.; Francisco Fernández, J.; Caballero-Calero, O. Ordered three-dimensional interconnected nanoarchitectures in anodic porous alumina. Nat. Commun. 2014, 5, 5130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehn, J.-M. Supramolecular Chemistry: Receptors, Catalysts, and Carriers. Science 1985, 227, 849–856. [Google Scholar] [CrossRef]
- Lehn, J.-M. Supramolecular chemistry. Science 1993, 260, 1762–1763. [Google Scholar] [CrossRef]
- Philp, D.; Stoddart, J.F. Self-Assembly in Natural and Unnatural Systems. Angew. Chem. Int. Ed. Engl. 1996, 35, 1154–1196. [Google Scholar] [CrossRef]
- Bucci, R.; Bossi, A.; Erba, E.; Vaghi, F.; Saha, A.; Yuran, S.; Maggioni, D.; Gelmi, M.L.; Reches, M.; Pellegrino, S. Nucleobase morpholino β amino acids as molecular chimeras for the preparation of photoluminescent materials from ribonucleosides. Sci. Rep. 2020, 10, 19331. [Google Scholar] [CrossRef]
- Whitesides, G.M.; Grzybowski, B. Self-assembly at all scales. Science 2002, 295, 2418–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuraje, N.; Banerjee, I.A.; MacCuspie, R.I.; Yu, L.; Matsui, H. Biological Bottom-Up Assembly of Antibody Nanotubes on Patterned Antigen Arrays. J. Am. Chem. Soc. 2004, 126, 8088–8089. [Google Scholar] [CrossRef] [PubMed]
- Bucci, R.; Georgilis, E.; Bittner, A.M.; Gelmi, M.L.; Clerici, F. Peptide-Based Electrospun Fibers: Current Status and Emerging Developments. Nanomaterials 2021, 11, 1262. [Google Scholar] [CrossRef]
- Locarno, S.; Argentiere, S.; Ruffoni, A.; Maggioni, D.; Soave, R.; Bucci, R.; Erba, E.; Lenardi, C.; Gelmi, M.L.; Clerici, F. Self-assembled hydrophobic Ala-Aib peptide encapsulating curcumin: A convenient system for water insoluble drugs. RSC Adv. 2020, 10, 9964–9975. [Google Scholar] [CrossRef]
- Bucci, R.; Vaghi, F.; Erba, E.; Romanelli, A.; Gelmi, M.L.; Clerici, F. Peptide grafting strategies before and after electrospinning of nanofibers. Acta Biomater. 2021, 122, 82–100. [Google Scholar] [CrossRef]
- Levin, A.; Hakala, T.A.; Schnaider, L.; Bernardes, G.J.L.; Gazit, E.; Knowles, T.P.J. Biomimetic peptide self-assembly for functional materials. Nat. Rev. Chem. 2020, 4, 615–634. [Google Scholar] [CrossRef]
- Zhang, S.; Marini, D.M.; Hwang, W.; Santoso, S. Design of nanostructured biological materials through self-assembly of peptides and proteins. Curr. Opin. Chem. Biol. 2002, 6, 865–871. [Google Scholar] [CrossRef]
- Lee, S.; Trinh, T.H.T.; Yoo, M.; Shin, J.; Lee, H.; Kim, J.; Hwang, E.; Lim, Y.; Ryou, C. Self-Assembling Peptides and Their Application in the Treatment of Diseases. Int. J. Mol. Sci. 2019, 20, 5850. [Google Scholar] [CrossRef] [Green Version]
- Handelman, A.; Lapshina, N.; Apter, B.; Rosenman, G. Peptide Integrated Optics. Adv. Mater. 2018, 30, 1705776. [Google Scholar] [CrossRef]
- Gatto, E.; Porchetta, A.; Scarselli, M.; De Crescenzi, M.; Formaggio, F.; Toniolo, C.; Venanzi, M. Playing with peptides: How to build a supramolecular peptide nanostructure by exploiting helix···helix macrodipole interactions. Langmuir 2012, 28, 2817–2826. [Google Scholar] [CrossRef] [PubMed]
- Gatto, E.; Quatela, A.; Caruso, M.; Tagliaferro, R.; De Zotti, M.; Formaggio, F.; Toniolo, C.; Di Carlo, A.; Venanzi, M. Mimicking Nature: A Novel Peptide-based Bio-inspired Approach for Solar Energy Conversion. ChemPhysChem 2014, 15, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Gatto, E.; Kubitzky, S.; Schriever, M.; Cesaroni, S.; Mazzuca, C.; Marafon, G.; Venanzi, M.; De Zotti, M. Building Supramolecular DNA-Inspired Nanowires on Gold Surfaces: From 2D to 3D. Angew. Chem. Int. Ed. 2019, 58, 7308–7312. [Google Scholar] [CrossRef] [PubMed]
- Gatto, E.; Stella, L.; Baldini, C.; Venanzi, M.; Toniolo, C.; Formaggio, F. Photocurrent generation in peptide-based self-assembled monolayers on gold electrodes. Superlatt. Microstruct. 2009, 46, 34–39. [Google Scholar] [CrossRef]
- Venanzi, M.; Gatto, E.; Caruso, M.; Porchetta, A.; Formaggio, F.; Toniolo, C. Photoinduced Electron Transfer through Peptide-Based Self-Assembled Monolayers Chemisorbed on Gold Electrodes: Directing the Flow-in and Flow-out of Electrons through Peptide Helices. J. Phys. Chem. A 2014, 118, 6674–6684. [Google Scholar] [CrossRef]
- Gatto, E.; Venanzi, M.; Palleschi, A.; Stella, L.; Pispisa, B.; Lorenzelli, L.; Toniolo, C.; Formaggio, F.; Marletta, G. Self-assembled peptide monolayers on interdigitated gold microelectrodes. Mater. Sci. Eng. C 2007, 27, 1309–1312. [Google Scholar] [CrossRef]
- Gatto, E.; Stella, L.; Formaggio, F.; Toniolo, C.; Lorenzelli, L.; Venanzi, M. Electroconductive and photocurrent generation properties of self-assembled monolayers formed by functionalized, conformationally constrained peptides on gold electrodes. J. Pept. Sci. 2008, 14, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Gatto, E.; Caruso, M.; Porchetta, A.; Toniolo, C.; Formaggio, F.; Crisma, M.; Venanzi, M. Photocurrent generation through peptide-based self-assembled monolayers on a gold surface: Antenna and junction effects. J. Pept. Sci. 2011, 17, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Gatto, E.; Venanzi, M. Self-assembled monolayers formed by helical peptide building blocks: A new tool for bioinspired nanotechnology. Polym. J. 2013, 45, 468–480. [Google Scholar] [CrossRef] [Green Version]
- Kubitzky, S.; Venanzi, M.; Biondi, B.; Lettieri, R.; De Zotti, M.; Gatto, E. A pH-Induced Reversible Conformational Switch Able to Control the Photocurrent Efficiency in a Peptide Supramolecular System. Chem. Eur. J. 2021, 27, 2810–2817. [Google Scholar] [CrossRef]
- De Zotti, M.; Muzzi, B.; Gatto, E.; Di Napoli, B.; Mazzuca, C.; Palleschi, A.; Placidi, E.; Formaggio, F.; Toniolo, C.; Venanzi, M. Tuning the Morphology of Nanostructured Peptide Films by the Introduction of a Secondary Structure Conformational Constraint: A Case Study of Hierarchical Self-Assembly. J. Phys. Chem. B 2018, 122, 6305–6313. [Google Scholar] [CrossRef]
- Caruso, M.; Placidi, E.; Gatto, E.; Mazzuca, C.; Stella, L.; Bocchinfuso, G.; Palleschi, A.; Formaggio, F.; Toniolo, C.; Venanzi, M. Fibrils or Globules? Tuning the Morphology of Peptide Aggregates from Helical Building Blocks. J. Phys. Chem. B 2013, 117, 5448–5459. [Google Scholar] [CrossRef]
- Caruso, M.; Gatto, E.; Placidi, E.; Ballano, G.; Formaggio, F.; Toniolo, C.; Zanuy, D.; Alemán, C.; Venanzi, M. A single-residue substitution inhibits fibrillization of Ala-based pentapeptides. A spectroscopic and molecular dynamics investigation. Soft Matter 2014, 10, 2508–2519. [Google Scholar] [CrossRef] [Green Version]
- Gatto, E.; Porchetta, A.; Stella, L.; Guryanov, I.; Formaggio, F.; Toniolo, C.; Kaptein, B.; Broxterman, Q.B.; Venanzi, M. Conformational Effects on the Electron-Transfer Efficiency in Peptide Foldamers Based on α,α-Disubstituted Glycyl Residues. Chem. Biodivers. 2008, 5, 1263–1278. [Google Scholar] [CrossRef]
- Lim, Y.; Lee, M. Nanostructures of Beta-sheet peptides: Steps towards bioactive functional materials. J. Mater. Chem. 2008, 18, 723–727. [Google Scholar] [CrossRef]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sieyers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.W.; McFarlane, H.T.; et al. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef]
- Kim, S.; Kim, J.H.; Lee, J.S.; Park, C.B. Beta-Sheet-Forming, Self-Assembled Peptide Nanomaterials towards Optical, Energy, and Healthcare Applications. Small 2015, 30, 3623–3640. [Google Scholar] [CrossRef] [PubMed]
- Zapadka, K.L.; Becher, F.J.; Gomes dos Santos, A.L.; Jackson, S.E. Factors affecting the physical stability (aggregation) of peptide therapeutics. Interface Focus 2017, 7, 20170030. [Google Scholar] [CrossRef] [Green Version]
- Steer, D.L.; Lew, R.A.; Perlmutter, P.; Smith, A.I.; Aguilar, M.I. β-Amino Acids: Versatile Peptidomimetics. Current Medicinal Chemistry 2002, 9, 811–822. [Google Scholar] [CrossRef]
- Heck, T.; Limbach, M.; Geueke, B.; Zacharias, M.; Gardiner, J.; Kohler, H.-P.; Seebach, D. Enzymatic Degradation of β- and Mixed α,β-Oligopeptides. Chem. Biodivers. 2006, 3, 1325–1348. [Google Scholar] [CrossRef]
- Cheng, R.P.; Gellman, S.H.; DeGrado, W.F. β-peptides: From structure to function. Chem. Rev. 2001, 101, 3219–3232. [Google Scholar] [CrossRef]
- Misra, R.; Rudnick-Glick, S.; Adler-Abramovich, L. From Folding to Assembly: Functional Supramolecular Architectures of Peptides Comprised of Non-Canonical Amino Acids. Macromol. Biosci. 2021, 2100090. [Google Scholar] [CrossRef] [PubMed]
- Makam, P.; Gazit, E. Minimalistic peptide supramolecularco-assembly: Expanding the conformational space for nanotechnology. Chem. Soc. Rev. 2018, 47, 3406–3420. [Google Scholar] [CrossRef] [PubMed]
- Bucci, R.; Foschi, F.; Loro, C.; Erba, E.; Gelmi, M.L.; Pellegrino, S. Fishing in the Toolbox of Cyclic Turn Mimics: A Literature Overview of the Last Decade. Eur. J. Org. Chem. 2021, 20, 2887–2900. [Google Scholar] [CrossRef]
- Yu, J.; Horsley, J.R.; Abell, A.D. The Influence of Secondary Structure on Electron Transfer in Peptides. Austral. J. Chem. 2013, 66, 848–851. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Huang, D.M.; Shapter, J.G.; Abell, A.D. Electrochemical and Computational Studies on Intramolecular Dissociative Electron Transfer in β-Peptides. J. Phys. Chem. C 2012, 116, 26608–26617. [Google Scholar] [CrossRef]
- Bonetti, A.; Clerici, F.; Foschi, F.; Nava, D.; Pellegrino, S.; Penso, M.; Soave, R.; Gelmi, M.L. syn/anti Switching by Specific Heteroatom–Titanium Coordination in the Mannich-Like Synthesis of 2,3-Diaryl-β-amino Acid Derivatives. Eur. J. Org. Chem. 2014, 15, 3203–3209. [Google Scholar] [CrossRef]
- Bonetti, A.; Pellegrino, S.; Das, P.; Yuran, S.; Bucci, R.; Ferri, N.; Meneghetti, F.; Castellano, C.; Reches, M.; Gelmi, M.L. Dipeptide Nanotubes Containing Unnatural Fluorine-Substituted β2,3-Diarylamino Acid and l -Alanine as Candidates for Biomedical Applications. Org. Lett. 2015, 17, 4468–4471. [Google Scholar] [CrossRef]
- Bucci, R.; Das, P.; Iannuzzi, F.; Feligioni, M.; Gandolfi, R.; Gelmi, M.L.; Reches, M.; Pellegrino, S. Self-assembly of an amphipathic ααβ-tripeptide into cationic spherical particles for intracellular delivery. Org. Biomol. Chem. 2017, 15, 6773–6779. [Google Scholar] [CrossRef]
- Bucci, R.; Contini, A.; Clerici, F.; Beccalli, E.M.; Formaggio, F.; Maffucci, I.; Pellegrino, S.; Gelmi, M.L. Fluoro-Aryl Substituted α,β2,3-Peptides in the Development of Foldameric Antiparallel β-Sheets: A Conformational Study. Front. Chem. 2019, 7, 192. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.V.M.; Nagendar, P.; Jayaprakash, P.; Radha, P.; Krishna, P.; Ramakrishna, K.V.S.; Kunwar, A.C. 9/11 Mixed Helices in α/β Peptides Derived from C-Linked Carbo-β-Amino Acid and L-Ala Repeats. Angew. Chem. Int. Ed. 2005, 44, 5878–5882. [Google Scholar] [CrossRef]
- Srinivasulu, G.; Kiran Kumar, S.; Sharma, G.V.M.; Kunwar, A.C. 11/9-mixed helices in the alpha/beta-peptides derived from alternating alpha- and beta-amino acids with proteinogenic side chains. J. Org. Chem. 2006, 71, 8395–8400. [Google Scholar] [CrossRef] [PubMed]
- Angelici, G.; Luppi, G.; Kaptein, B.; Broxterman, Q.B.; Hofmann, H.J.; Tomasini, C. Synthesis and secondary structure of alternate α,β-hybrid peptides containing oxazolidin-2-one moieties. Eur. J. Org. Chem. 2007, 2713–2721. [Google Scholar] [CrossRef]
- Balamurugan, D.; Muraleedharan, K.M. Unprecedented Torsional Preferences in trans-β2,3-Amino Acid Residues and Formation of 11-Helices in α,β2,3-Hybrid Peptides. Chem. A Eur. J. 2012, 18, 9516–9520. [Google Scholar] [CrossRef] [PubMed]
- Basuroy, K.; Karuppiah, V.; Balaram, P. C11/C9 Helices in Crystals of αβ Hybrid Peptides and Switching Structures between Helix Types by Variation in the α-Residue. Org. Lett. 2014, 16, 4614–4617. [Google Scholar] [CrossRef] [PubMed]
- Bostick, C.D.; Mukhopadhyay, S.; Pecht, I.; Sheves, M.; Cahen, D.; Lederman, D. Protein bioelectronics: A review of what we do and do not know. Reports Prog. Phys. 2018, 81, 26601–26658. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; et al. Amber 2021; University of California: San Francisco, CA, USA, 2021. [Google Scholar]
- Goetz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Goetz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Babin, V.; Karpusenka, V.; Moradi, M.; Roland, C.; Sagui, C. Adaptively biased molecular dynamics: An umbrella sampling method with a time-dependent potential. Int. J. Quantum Chem. 2009, 109, 3666–3678. [Google Scholar] [CrossRef]
- Tabata, Y.; Uji, H.; Imai, T.; Kimura, S. Two one-dimensional arrays of naphthyl and anthryl groups along peptide nanotubes prepared from cyclic peptides comprising α- and β-amino acids. Soft Matter. 2018, 14, 7597–7604. [Google Scholar] [CrossRef]
- Del Mercato, L.L.; Pompa, P.P.; Maruccio, G.; Della Torre, A.; Sabella, S.; Tamburro, A.M.; Cingolani, R.; Rinaldi, R. Charge transport and intrinsic fluorescence in amyloid-like fibrils. PNAS 2007, 104, 18019–18024. [Google Scholar] [CrossRef] [Green Version]
- Chan, F.T.S.; Schierle, G.S.K.; Kumita, J.R.; Bertoncini, C.W.; Dobson, C.M.; Kaminski, C.F. Protein amyloids develop an intrinsic fluorescence signature during aggregation. Analyst 2013, 138, 2156–2162. [Google Scholar] [CrossRef] [Green Version]
- Sirangelo, I.; Borriello, M.; Irace, G.; Iannuzzi, C. Intrinsic blue-green fluorescence in amyloyd fibrils. AIMS Biophys. 2018, 5, 155–165. [Google Scholar] [CrossRef]
- Pinotsi, D.; Buell, A.K.; Dobson, C.M.; Schierle, G.S.K.; Kaminski, C.F. A label-free, quantitative assay of amyloid fibril growth based on intrinsic fluorescence. ChemBioChem 2013, 14, 846–850. [Google Scholar] [CrossRef]
- Pinotsi, D.; Grisanti, L.; Mahou, P.; Gebauer, R.; Kaminski, C.F.; Hassanali, A.; Schierle, G.S.G. Proton transfer and structure-specific fluorescence in hydrogen bond-rich protein structures. J. Am. Chem. Soc. 2016, 138, 3046–3057. [Google Scholar] [CrossRef] [Green Version]
- Villa, A.M.; Doglia, S.M.; De Gioia, L.; Bertini, L.; Natalello, A. Anomalous Intrinsic Fluorescence of HCl and NaOH Aqueous Solutions. J. Phys. Chem. Lett. 2019, 10, 7230–7236. [Google Scholar] [CrossRef]
- Bucci, R.; Dapiaggi, F.; Macut, H.; Pieraccini, S.; Sironi, M.; Gelmi, M.L.; Erba, E.; Pellegrino, S. On-resin multicomponent 1,3-dipolar cycloaddition of cyclopentanone–proline enamines and sulfonylazides as an efficient tool for the synthesis of amidino depsipeptide mimics. Amino Acids 2020, 52, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Bucci, R.; Contini, A.; Clerici, F.; Pellegrino, S.; Gelmi, M.L. From glucose to enantiopure morpholino β-amino acid: A new tool for stabilizing γ-turns in peptides. Org. Chem. Front. 2019, 6, 972–982. [Google Scholar] [CrossRef] [Green Version]
- Oliva, F.; Bucci, R.; Tamborini, L.; Pieraccini, S.; Pinto, A.; Pellegrino, S. Bicyclic Pyrrolidine-Isoxazoline γ Amino Acid: A Constrained Scaffold for Stabilizing α-Turn Conformation in Isolated Peptides. Front. Chem. 2019, 7, 133. [Google Scholar] [CrossRef] [Green Version]
- Vaghi, F.; Bucci, R.; Clerici, F.; Contini, A.; Gelmi, M.L. Non-natural 3-Arylmorpholino-β-amino Acid as a PPII Helix Inducer. Org. Lett. 2020, 22, 6197–6202. [Google Scholar] [CrossRef] [PubMed]
- Pierce, L.C.T.; Salomon-Ferrer, R.; Augusto, F. de Oliveira, C.; McCammon, J.A.; Walker, R.C.Routine access to millisecond time scale events with accelerated molecular dynamics. J. Chem. Theory Comput. 2012, 8, 2997–3002. [Google Scholar] [CrossRef]
- Piovesan, D.; Minervini, G.; Tosatto, S.C.E. The RING 2.0 web server for high quality residue interaction networks. Nucleic Acids Research 2016, 44, W367–W374. [Google Scholar] [CrossRef]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross- β spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anthony, W.P.; Fitzpatricka, A.W.P.; Galia, T.; Debelouchinac, G.T.; Marvin, J.; Bayroc, M.J.; Daniel, K.; Clared, D.K.; Marc, A.; Caporinic, M.A.; et al. Atomic structure and hierarchical assembly of a cross-β amyloid fibril. PNAS 2013, 110, 5468–5473. [Google Scholar]
- Tao, K.; Makam, P.; Aizen, R.; Gazit, E. Self-assembling peptide semiconductors. Science 2017, 358, eaam9756. [Google Scholar] [CrossRef]
- Evans, M.G.; Gergely, J. A discussion of the possibility of bands of energy levels in proteins electronic interaction in non bonded systems. Biochim. Biophys. Acta 1949, 3, 188–197. [Google Scholar] [CrossRef]
- Ladik, J. Energy band structure of proteins. Nature 1964, 202, 1208–1209. [Google Scholar] [CrossRef]
- Endres, R.G.; Cox, D.L.; Singh, R.R.P. Colloquium: The quest for high-conductance DNA. Rev. Mod. Phys. 2004, 76, 195–214. [Google Scholar] [CrossRef]
- Ron, I.; Pecht, I.; Sheves, M.; Cahen, D. Proteins as Solid-State Electronic Conductors. Acc. Chem. Res. 2010, 43, 945–953. [Google Scholar] [CrossRef]
- Blumberger, J. Recent Advances in the Theory and Molecular Simulation of Biological Electron Transfer Reactions. Chem. Rev. 2015, 115, 11191–11238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.; Adhikari, B.; Martic, S.; Munir, A.; Shahzad, S.; Ahmad, K.; Kraatz, H.-B. Electron transfer in peptides. Chem. Soc. Rev. 2015, 44, 1015–1027. [Google Scholar] [CrossRef]
- Williamson, H.R.; Dow, B.A.; Davidson, V.L. Mechanisms for control of biological electron transfer reactions. Bioorg. Chem. 2014, 57, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amdursky, N. Electron Transfer across Helical Peptides. Chempluschem 2015, 80, 1075–1095. [Google Scholar] [CrossRef] [PubMed]
- Wall, B.D.; Diegelmann, S.R.; Zhang, S.; Dawidczyk, T.J.; Wilson, W.L.; Katz, H.E.; Mao, H.-Q.; Tovar, J.D. Aligned Macroscopic Domains of Optoelectronic Nanostructures Prepared via Shear-Flow Assembly of Peptide Hydrogels. Adv. Mater. 2011, 23, 5009–5014. [Google Scholar] [CrossRef]
- Hartgerink, J.D.; Granja, J.R.; Milligan, R.A.; Ghadiri, M.R. Self-Assembling Peptide Nanotubes. J. Am. Chem. Soc. 1996, 118, 43–50. [Google Scholar] [CrossRef]
- Channon, K.J.; Devlin, G.L.; Magennis, S.W.; Finlayson, C.E.; Tickler, A.K.; Silva, C.; MacPhee, C.E. Modification of Fluorophore Photophysics through Peptide-Driven Self-Assembly. J. Am. Chem. Soc. 2008, 130, 5487–5491. [Google Scholar] [CrossRef] [PubMed]
- Meidanshahi, R.V.; Mazinani, S.K.S.; Mujica, V.; Tarakeshwas, P. Electronic transport across hydrogen bonds in organic electronics. Int. J. Nanotechnol. 2015, 12, 297–312. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forlano, N.; Bucci, R.; Contini, A.; Venanzi, M.; Placidi, E.; Gelmi, M.L.; Lettieri, R.; Gatto, E. Non-Conventional Peptide Self-Assembly into a Conductive Supramolecular Rope. Nanomaterials 2023, 13, 333. https://doi.org/10.3390/nano13020333
Forlano N, Bucci R, Contini A, Venanzi M, Placidi E, Gelmi ML, Lettieri R, Gatto E. Non-Conventional Peptide Self-Assembly into a Conductive Supramolecular Rope. Nanomaterials. 2023; 13(2):333. https://doi.org/10.3390/nano13020333
Chicago/Turabian StyleForlano, Nicola, Raffaella Bucci, Alessandro Contini, Mariano Venanzi, Ernesto Placidi, Maria Luisa Gelmi, Raffaella Lettieri, and Emanuela Gatto. 2023. "Non-Conventional Peptide Self-Assembly into a Conductive Supramolecular Rope" Nanomaterials 13, no. 2: 333. https://doi.org/10.3390/nano13020333