
Polyoxometalate-Modified Amphiphilic Polystyrene-block-poly(2-(dimethylamino)ethyl methacrylate) Membranes for Heterogeneous Glucose to Formic Acid Methyl Ester Oxidation
 , , and
, , and
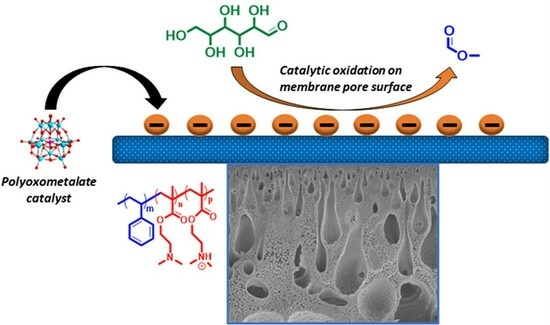
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Synthesis of the HPA-5 Heteropolyoxometalate
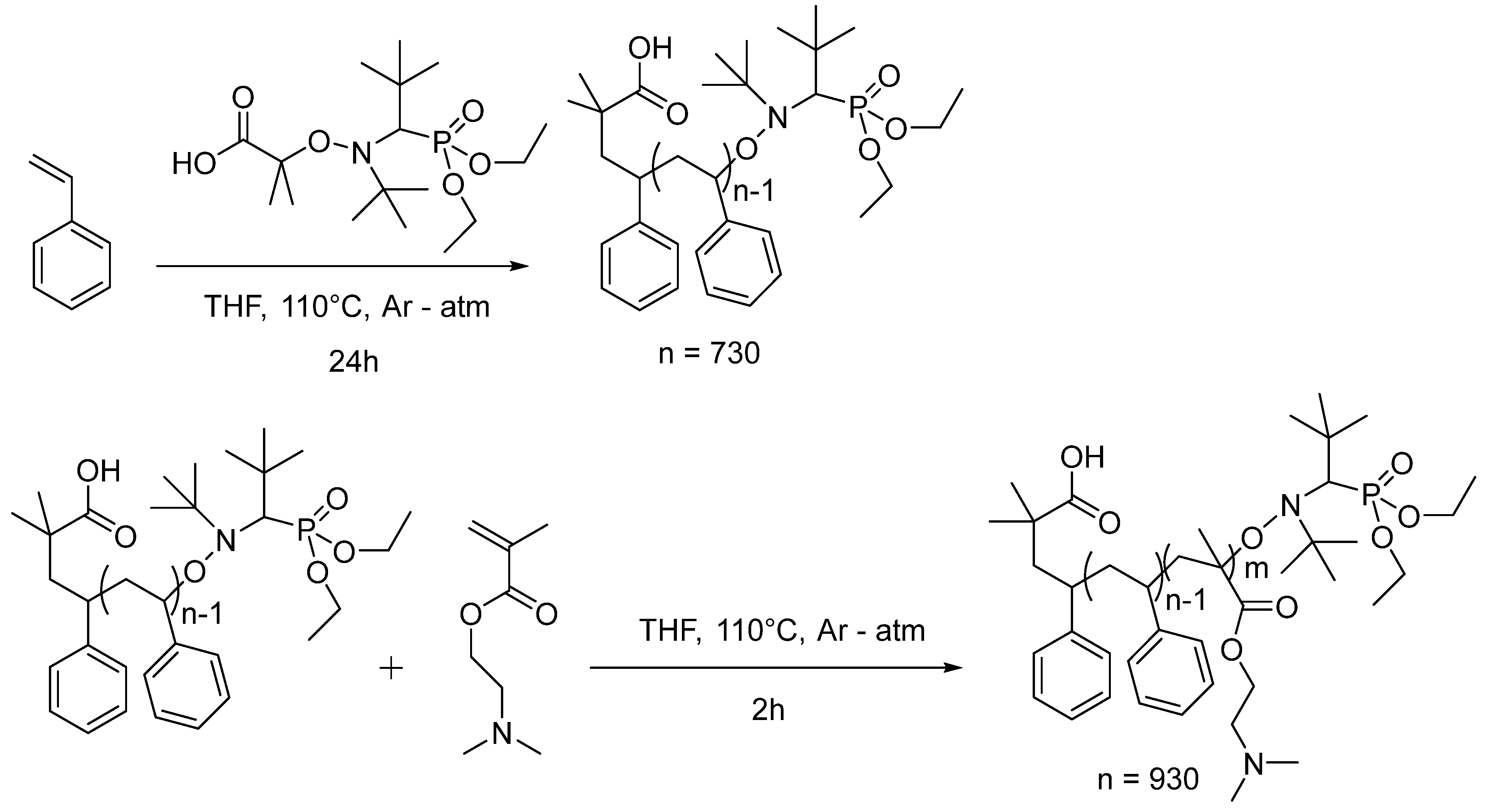
2.2. Synthesis of PS
2.3. Synthesis of PS-b-PDMAEMA Block Copolymer
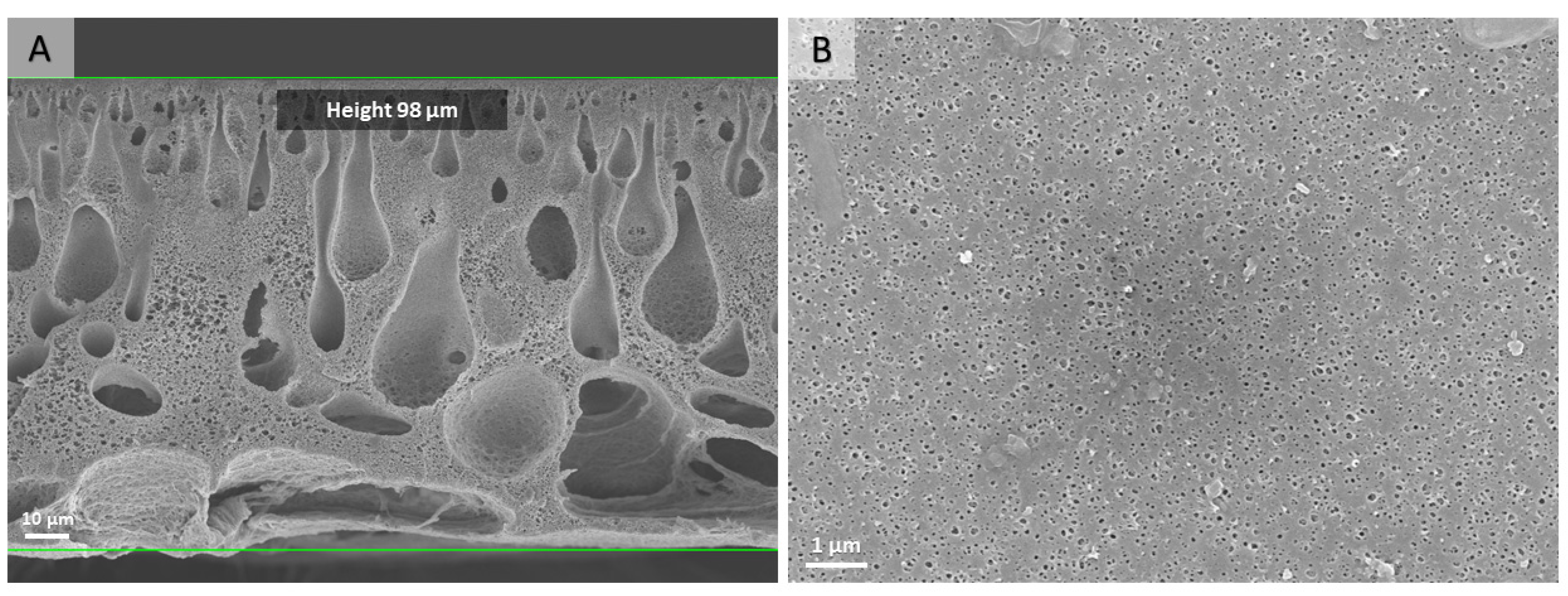
2.4. Preparation of the PS-b-PDMAEMA Porous Membranes
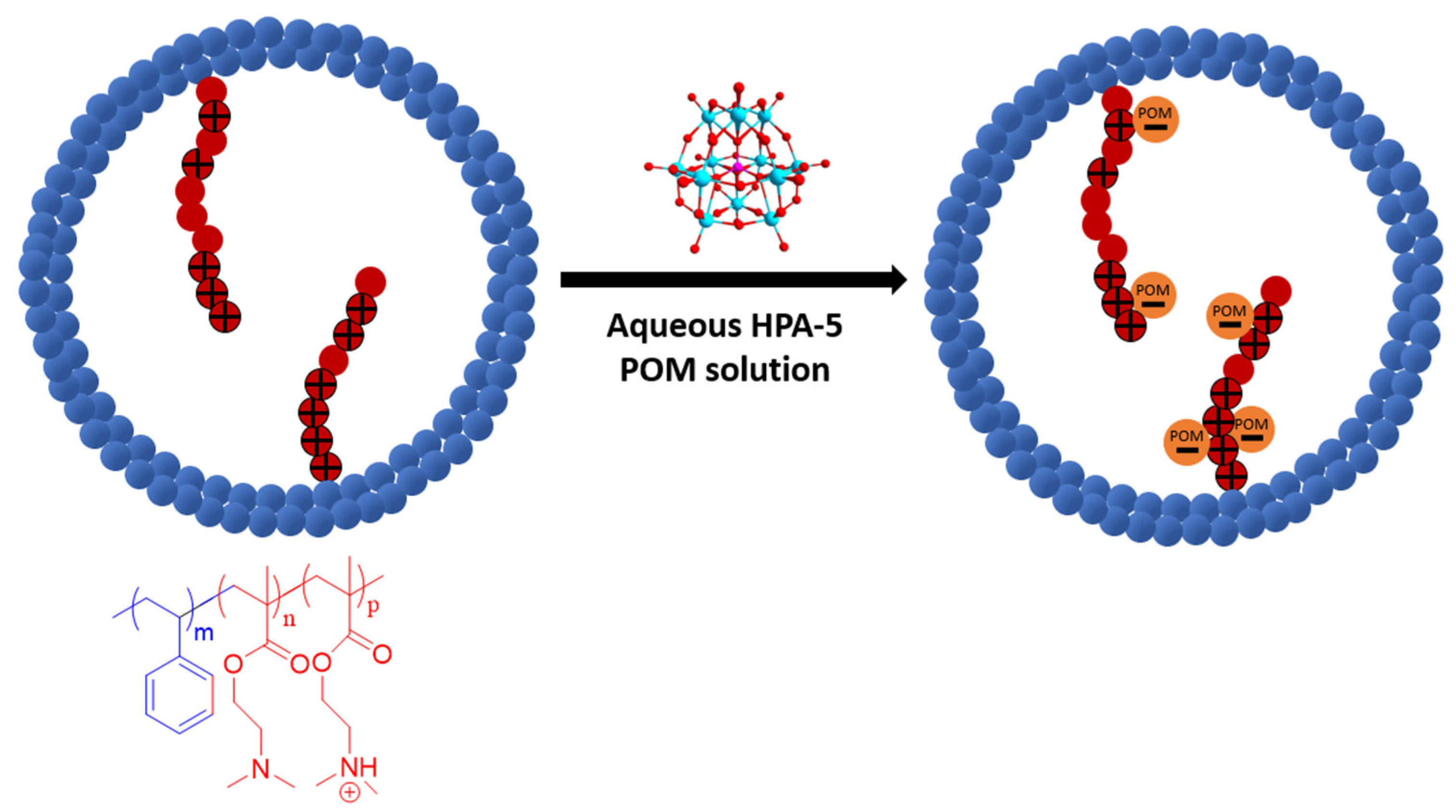
2.5. Preparation of the Catalytically Active Membranes
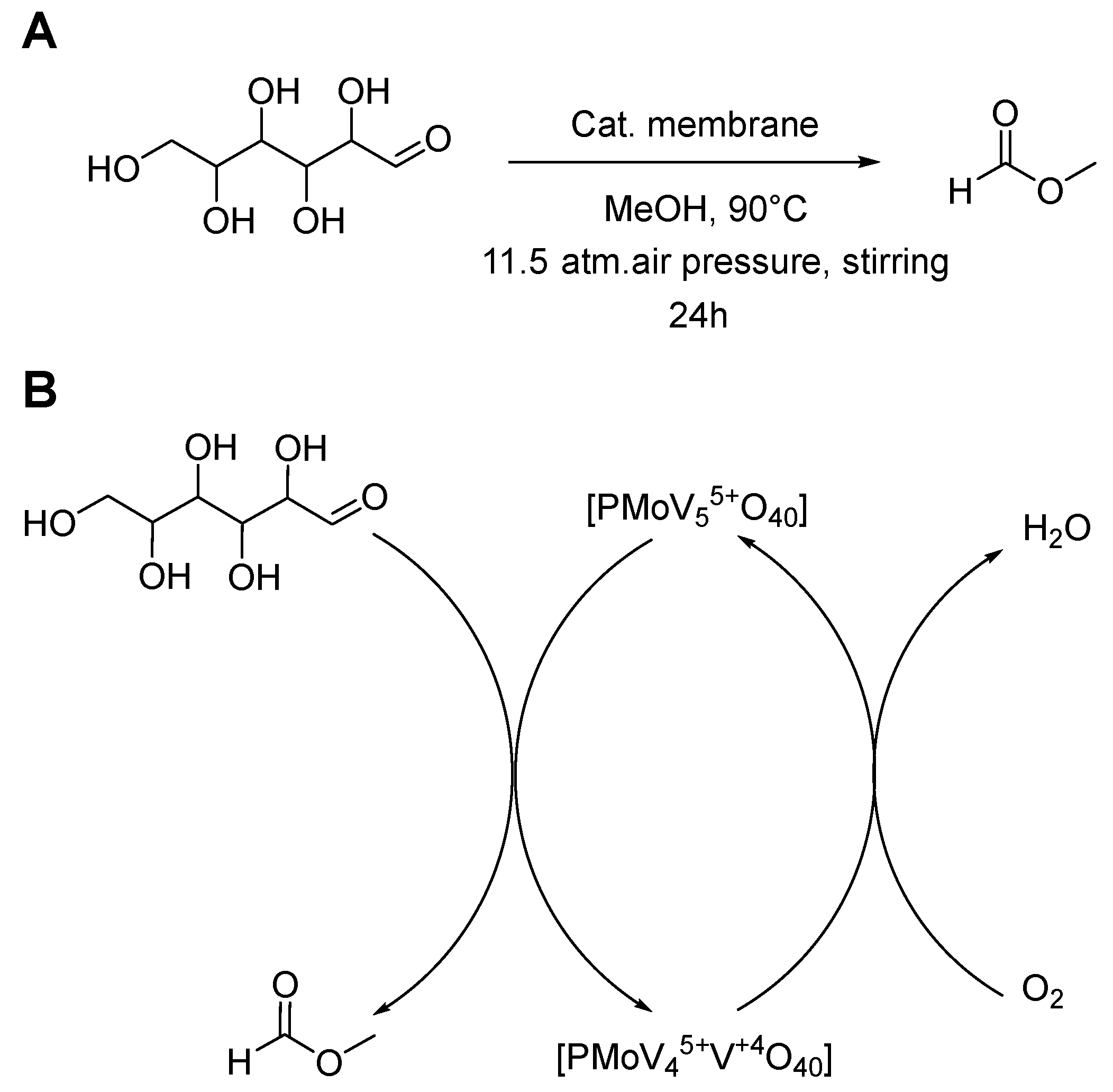
2.6. Catalytic Heterogeneous Glucose to Formic Acid Methyl Ester Oxidation
2.7. Instrumentation
2.7.1. NMR
2.7.2. SEC
2.7.3. TGA
2.7.4. ICP
2.7.5. SEM
2.7.6. XPS
2.7.7. Nitrogen Physisorption
2.7.8. Head-Space GC–MS
2.7.9. Chemicals
3. Results and Discussion
3.1. Preparation of the PS-b-PDMAEMA Block Copolymer
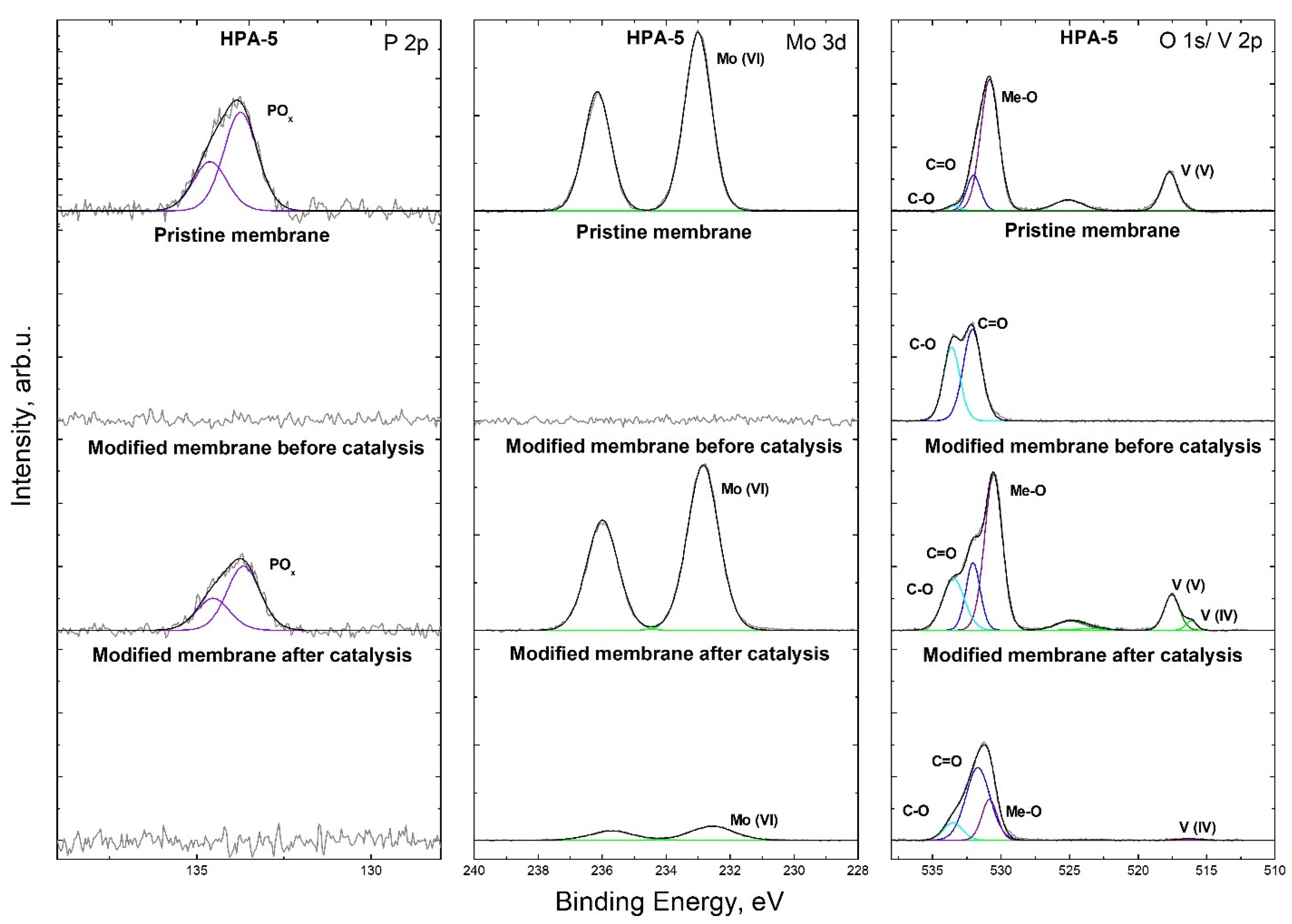
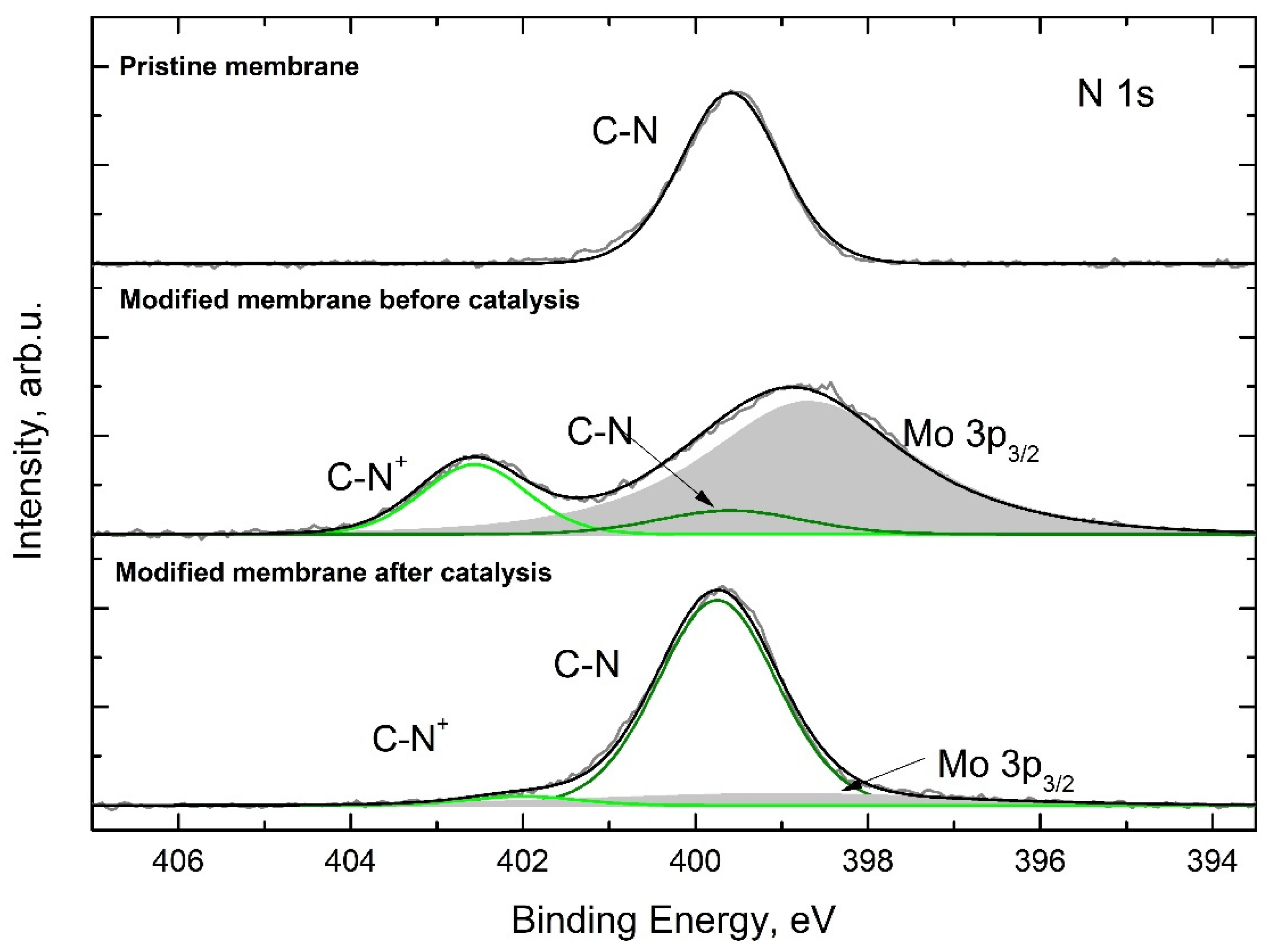
3.2. Preparation of Catalytically Active Membranes
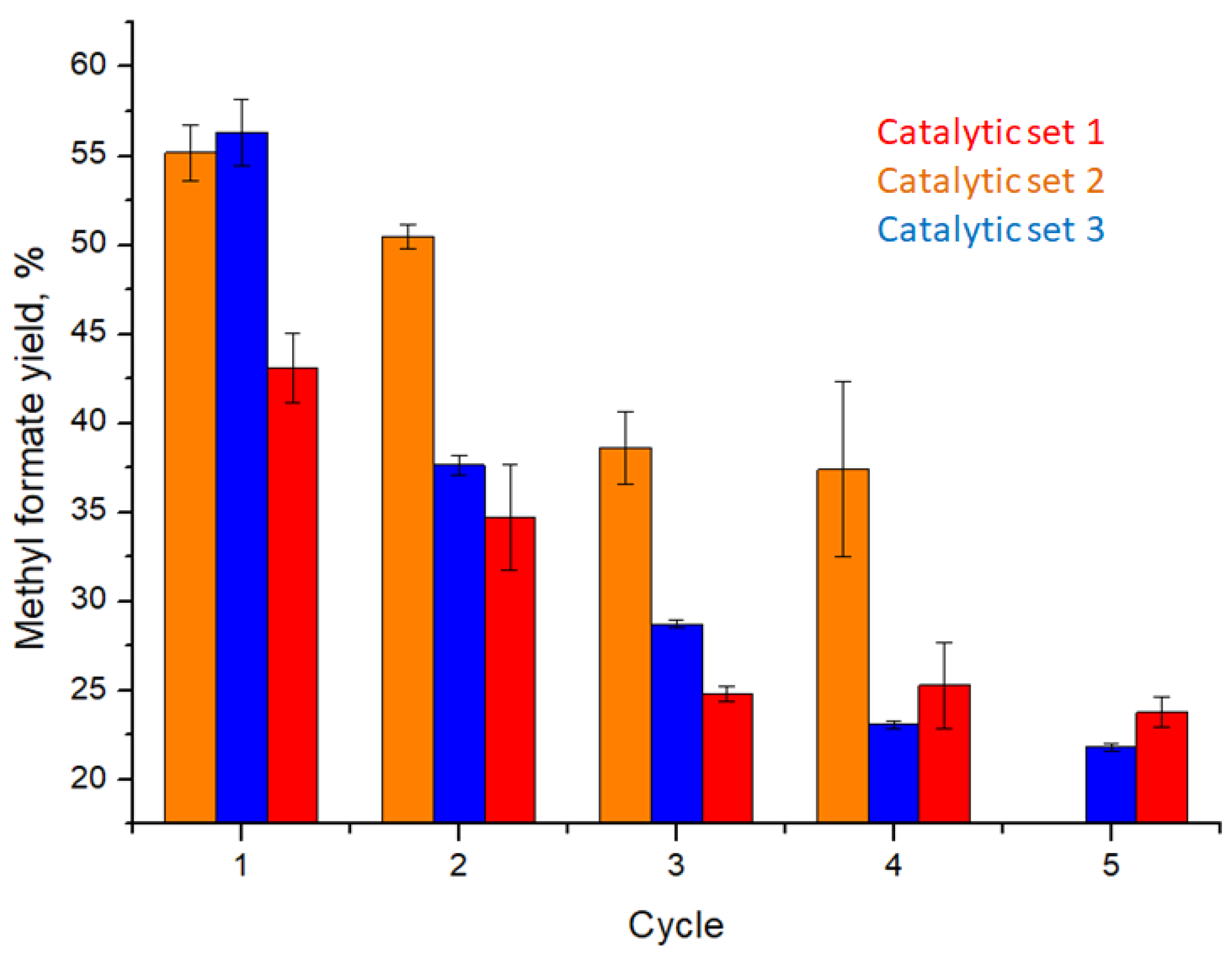
3.3. Catalytic Performance
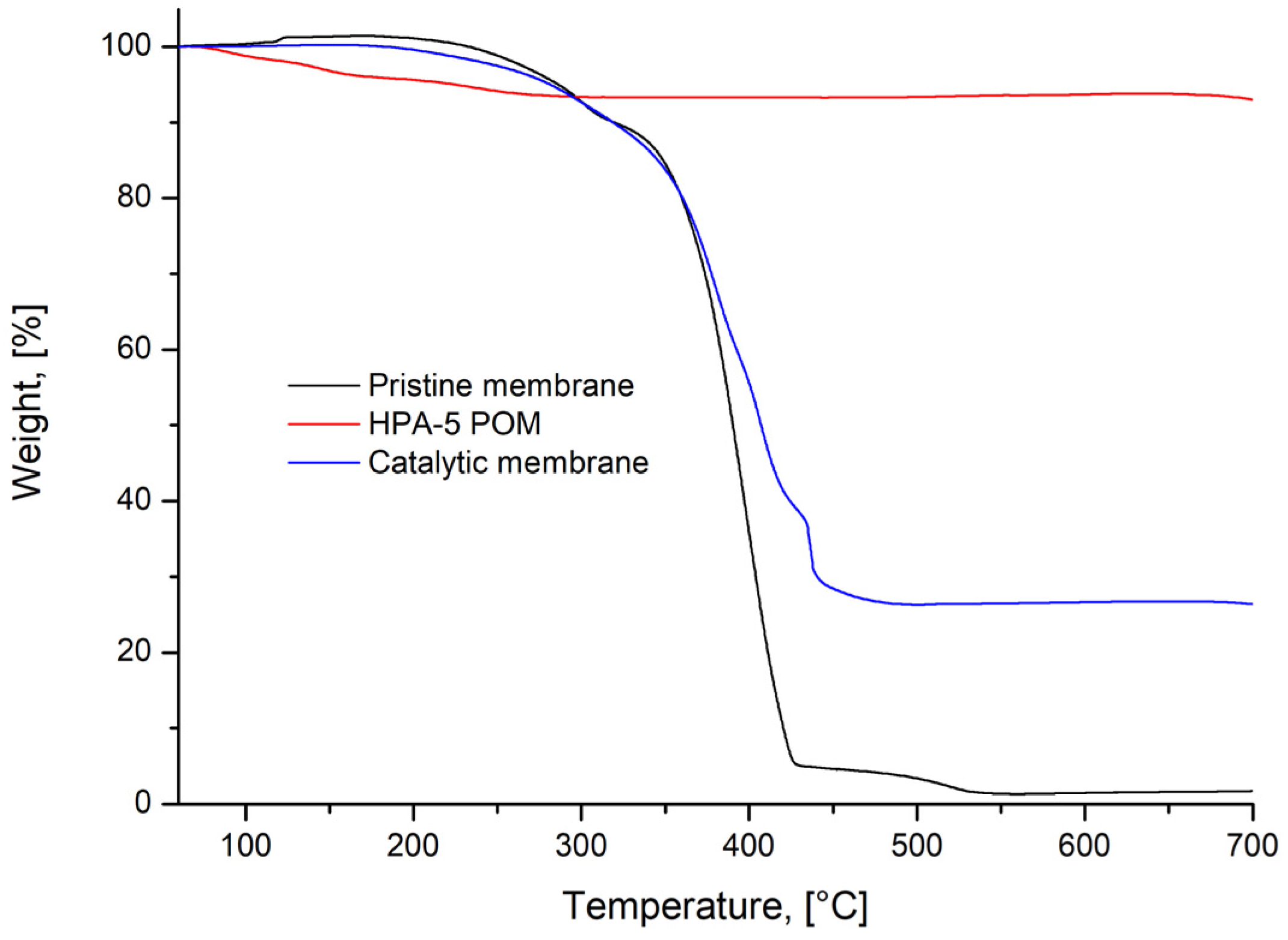
3.4. Catalytic Membrane Stability and Possibility of Reactivation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schacher, F.; Rudolph, T.; Wieberger, F.; Ulbricht, M.; Muller, A.H.E. Double Stimuli-Responsive Ultrafiltration Membranes from Polystyrene-block-poly(N,N-dimethylaminoethyl methacrylate) Diblock Copolymers. ACS Appl. Mater. Interfaces 2009, 1, 1492–1503. [Google Scholar] [CrossRef] [PubMed]
- Peinemann, K.V.; Abetz, V.; Simon, P.F.W. Asymmetric superstructure formed in a block copolymer via phase separation. Nat. Mater. 2007, 6, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.P. Recent advances in the controlled formation of pores in membranes. Trends Polym. Sci. 1997, 5, 187–192. [Google Scholar]
- Vriezekolk, E.J.; Nijmeijer, K.; de Vos, W.M. Dry-wet phase inversion block copolymer membranes with a minimum evaporation step from NMP/THF mixtures. J. Membr. Sci. 2016, 504, 230–239. [Google Scholar] [CrossRef]
- Nunes, S.P.; Sougrat, R.; Hooghan, B.; Anjum, D.H.; Behzad, A.R.; Zhao, L.; Pradeep, N.; Pinnau, I.; Vainio, U.; Peinemann, K.V. Ultraporous Films with Uniform Nanochannels by Block Copolymer Micelles Assembly. Macromolecules 2010, 43, 8079–8085. [Google Scholar] [CrossRef]
- Phillip, W.A.; Dorin, R.M.; Werner, J.; Hoek, E.M.V.; Wiesner, U.; Elimelech, M. Tuning Structure and Properties of Graded Triblock Terpolymer-Based Mesoporous and Hybrid Films. Nano Lett. 2011, 11, 2892–2900. [Google Scholar] [CrossRef]
- Gu, Y.B.; Wiesner, U. Tailoring Pore Size of Graded Mesoporous Block Copolymer Membranes: Moving from Ultrafiltration toward Nanofiltration. Macromolecules 2015, 48, 6153–6159. [Google Scholar] [CrossRef]
- Horenz, C.; Pietsch, C.; Goldmann, A.S.; Barner-Kowollik, C.; Schacher, F.H. Phase Inversion Membranes from Amphiphilic Diblock Terpolymers. Adv. Mater. Interfaces 2015, 2, 1500042. [Google Scholar] [CrossRef]
- Schacher, F.; Ulbricht, M.; Muller, A.H.E. Self-Supporting, Double Stimuli-Responsive Porous Membranes From Polystyrene-block-poly (N,N-dimethylaminoethyl methacrylate) Diblock Copolymers. Adv. Funct. Mater. 2009, 19, 1040–1045. [Google Scholar] [CrossRef]
- Cetintas, M.; de Grooth, J.; Hofman, A.H.; van der Kooij, H.M.; Loos, K.; de Vos, W.M.; Kamperman, M. Free-standing thermo-responsive nanoporous membranes from high molecular weight PS-PNIPAM block copolymers synthesized via RAFT polymerization. Polym. Chem. 2017, 8, 2235–2243. [Google Scholar] [CrossRef]
- Zhang, Z.Z.; Rahman, M.M.; Bajer, B.; Scharnagl, N.; Abetz, V. Highly selective isoporous block copolymer membranes with tunable polyelectrolyte brushes in soft nanochannels. J. Membr. Sci. 2022, 646, 120266. [Google Scholar] [CrossRef]
- Zhang, Z.Z.; Rahman, M.M.; Abetz, C.; Bajer, B.; Wang, J.L.; Abetz, V. Quaternization of a Polystyrene-block-poly(4-vinylpyridine) Isoporous Membrane: An Approach to Tune the Pore Size and the Charge Density. Macromol. Rapid Comm. 2019, 40, e1800729. [Google Scholar] [CrossRef] [PubMed]
- Schottner, S.; Brodrecht, M.; Uhlein, E.; Dietz, C.; Breitzke, H.; Tietze, A.A.; Buntkowsky, G.; Gallei, M. Amine-Containing Block Copolymers for the Bottom-Up Preparation of Functional Porous Membranes. Macromolecules 2019, 52, 2631–2641. [Google Scholar] [CrossRef]
- Romanenko, I.; Lechner, M.; Wendler, F.; Horenz, C.; Streb, C.; Schacher, F.H. POMbranes: Polyoxometalate-functionalized block copolymer membranes for oxidation catalysis. J. Mater. Chem. A 2017, 5, 15789–15796. [Google Scholar] [CrossRef]
- Romanenko, I.; Rajagopal, A.; Neumann, C.; Turchanin, A.; Streb, C.; Schacher, F.H. Embedding molecular photosensitizers and catalysts in nanoporous block copolymer membranes for visible-light driven hydrogen evolution. J. Mater. Chem. A 2020, 8, 6238–6244. [Google Scholar] [CrossRef]
- Chettri, A.; Kruse, J.H.; Jha, K.K.; Droge, L.; Romanenko, I.; Neumann, C.; Kupfer, S.; Turchanin, A.; Rau, S.; Schacher, F.H.; et al. A Molecular Photosensitizer in a Porous Block Copolymer Matrix-Implications for the Design of Photocatalytically Active Membranes. Chem.-Eur. J. 2021, 27, 17049–17058. [Google Scholar] [CrossRef]
- Kund, J.; Kruse, J.H.; Gruber, A.; Trentin, I.; Langer, M.; Read, C.; Neusser, G.; Blaimer, D.; Rupp, U.; Streb, C.; et al. Multimodal Analysis of Light-Driven Water Oxidation in Nanoporous Block Copolymer Membranes. Angew. Chem. Int. Ed. 2023, 62, e202217196. [Google Scholar] [CrossRef]
- Maerten, S.; Kumpidet, C.; Voss, D.; Bukowski, A.; Wasserscheid, P.; Albert, J. Glucose oxidation to formic acid and methyl formate in perfect selectivity. Green Chem. 2020, 22, 4311–4320. [Google Scholar] [CrossRef]
- Weinstock, I.A.; Schreiber, R.E.; Neumann, R. Dioxygen in Polyoxometalate Mediated Reactions. Chem. Rev. 2018, 118, 2680–2717. [Google Scholar] [CrossRef]
- Mizuno, N.; Kamata, K.; Yamaguchi, K. Green Oxidation Reactions by Polyoxometalate-Based Catalysts: From Molecular to Solid Catalysts. Top. Catal. 2010, 53, 876–893. [Google Scholar] [CrossRef]
- Mizuno, N.; Yamaguchi, K. Polyoxometalate catalysts: Toward the development of green H2O2-based epoxidation systems. Chem. Rec. 2006, 6, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Stracke, J.J.; Finke, R.G. Distinguishing Homogeneous from Heterogeneous Water Oxidation Catalysis when Beginning with Polyoxometalates. ACS Catal. 2014, 4, 909–933. [Google Scholar] [CrossRef]
- Sartorel, A.; Bonchio, M.; Campagna, S.; Scandola, F. Tetrametallic molecular catalysts for photochemical water oxidation. Chem. Soc. Rev. 2013, 42, 2262–2280. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.J.; Geletii, Y.V.; Zhao, C.C.; Vickers, J.W.; Zhu, G.B.; Luo, Z.; Song, J.; Lian, T.Q.; Musaev, D.G.; Hill, C.L. Polyoxometalate water oxidation catalysts and the production of green fuel. Chem. Soc. Rev. 2012, 41, 7572–7589. [Google Scholar] [CrossRef]
- Berger, M.E.M.; Assenbaum, D.; Taccardi, N.; Spiecker, E.; Wasserscheid, P. Simple and recyclable ionic liquid based system for the selective decomposition of formic acid to hydrogen and carbon dioxide. Green Chem. 2011, 13, 1411–1415. [Google Scholar] [CrossRef]
- Himeda, Y. Conversion of CO2 into formate by homogeneously catalyzed hydrogenation in water: Tuning catalytic activity and water solubility through the acid-base equilibrium of the ligand. Eur. J. Inorg. Chem. 2007, 2007, 3927–3941. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Y.; Wu, J.W. Sustainable production of formic acid from biomass and carbon dioxide. Mol. Catal. 2020, 483, 110716. [Google Scholar] [CrossRef]
- Odyakov, V.F.; Zhizhina, E.G. A Novel Method of the Synthesis of Molybdovanadophosphoric Heteropoly Acid Solutions. React. Kinet. Catal. L 2008, 95, 21–28. [Google Scholar] [CrossRef]
- Albert, J.; Luders, D.; Bosmann, A.; Guldi, D.M.; Wasserscheid, P. Spectroscopic and electrochemical characterization of heteropoly acids for their optimized application in selective biomass oxidation to formic acid. Green Chem. 2014, 16, 226–237. [Google Scholar] [CrossRef]
- Abetz, V. Isoporous Block Copolymer Membranes. Macromol. Rapid Comm. 2015, 36, 10–22. [Google Scholar] [CrossRef]
- Zhizhina, E.G.; Odyakov, V.F.; Simonova, M.V.; Matveev, K.I. Inetics of oxidation of reduced phosphorus-molybdenum-vanadium heteropoly acid species with dioxygen in aqueous solutions. Kinet. Catal. 2005, 46, 354–363. [Google Scholar] [CrossRef]
- Kozhevnikov, I.V. Catalysis by heteropoly acids and multicomponent polyoxometalates in liquid-phase reactions. Chem. Rev. 1998, 98, 171–198. [Google Scholar] [CrossRef] [PubMed]
- Wesinger, S.; Mendt, M.; Albert, J. Alcohol-Activated Vanadium-Containing Polyoxometalate Complexes in Homogeneous Glucose Oxidation Identified with V-51-NMR and EPR Spectroscopy. Chemcatchem 2021, 13, 3662–3670. [Google Scholar] [CrossRef]
- Lu, T.; Hou, Y.C.; Wu, W.Z.; Niu, M.G.; Ren, S.H.; Lin, Z.Q.; Ramani, V.K. Catalytic oxidation of biomass to oxygenated chemicals with exceptionally high yields using H5PV2Mo10O40. Fuel 2018, 216, 572–578. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalytic set 1 | |||||
| Cycle | 1 | 2 | 3 | 4 | 5 |
| Yield, % | 43.1 ± 1.9 | 34.7 ± 2.9 | 24.8 ± 0.4 | 25.3 ± 2.4 | 23.8 ± 0.8 |
| Catalytic set 2 | |||||
| Cycle | 1 | 2 | 3 | 4 | 5 |
| Yield, % | 55.2 ± 1.6 | 50.5 ± 0.7 | 38.6 ± 2.0 | 37.4 ± 4.9 | - |
| Catalytic set 3 | |||||
| Cycle | 1 | 2 | 3 | 4 | 5 |
| Yield, % | 56.3 ± 1.9 | 37.7 ± 0.6 | 28.7 ± 0.2 | 23.1 ± 0.2 | 21.8 ± 0.2 |
| Catalytic Set 2 | Catalytic Set 3 | |||||
|---|---|---|---|---|---|---|
| Element | Mo Loss, % | V Loss, % | P Loss, % | Mo Loss, % | V Loss, % | P Loss, % |
| Cycle 1 | 13.0 | 44.5 | 13.7 | 11.2 | 39.6 | 0 |
| Cycle 2 | 12.3 | 17.7 | 10.5 | 9.9 | 17.2 | 0 |
| Cycle 3 | 11.7 | 10.6 | 0 | 9.9 | 9.7 | 0 |
| Cycle 4 | 11.9 | 7.6 | 0 | 10.4 | 7.1 | 0 |
| Cycle 5 | 12.4 | 6.9 | 0 | 11.6 | 5.9 | 0 |
| Overall Loss | 61.3 | 87.2 | 24.2 | 53.1 | 79.4 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Utievskyi, Y.; Neumann, C.; Sindlinger, J.; Schutjajew, K.; Oschatz, M.; Turchanin, A.; Ueberschaar, N.; Schacher, F.H. Polyoxometalate-Modified Amphiphilic Polystyrene-block-poly(2-(dimethylamino)ethyl methacrylate) Membranes for Heterogeneous Glucose to Formic Acid Methyl Ester Oxidation. Nanomaterials 2023, 13, 2498. https://doi.org/10.3390/nano13182498
Utievskyi Y, Neumann C, Sindlinger J, Schutjajew K, Oschatz M, Turchanin A, Ueberschaar N, Schacher FH. Polyoxometalate-Modified Amphiphilic Polystyrene-block-poly(2-(dimethylamino)ethyl methacrylate) Membranes for Heterogeneous Glucose to Formic Acid Methyl Ester Oxidation. Nanomaterials. 2023; 13(18):2498. https://doi.org/10.3390/nano13182498
Chicago/Turabian StyleUtievskyi, Yurii, Christof Neumann, Julia Sindlinger, Konstantin Schutjajew, Martin Oschatz, Andrey Turchanin, Nico Ueberschaar, and Felix H. Schacher. 2023. "Polyoxometalate-Modified Amphiphilic Polystyrene-block-poly(2-(dimethylamino)ethyl methacrylate) Membranes for Heterogeneous Glucose to Formic Acid Methyl Ester Oxidation" Nanomaterials 13, no. 18: 2498. https://doi.org/10.3390/nano13182498