
Liquid-Phase Hydrogenation of 1-Phenyl-1-propyne on the Pd1Ag3/Al2O3 Single-Atom Alloy Catalyst: Kinetic Modeling and the Reaction Mechanism

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Catalyst Preparation
2.3. Catalyst Characterization
2.4. Catalytic Tests
3. Results and Discussion
3.1. Catalyst Characterization
3.1.1. Temperature-Programmed Pd Hydride Decomposition
3.1.2. Transmission Electron Microscopy
3.1.3. BET Surface Area Analysis
3.1.4. DRIFTS-CO
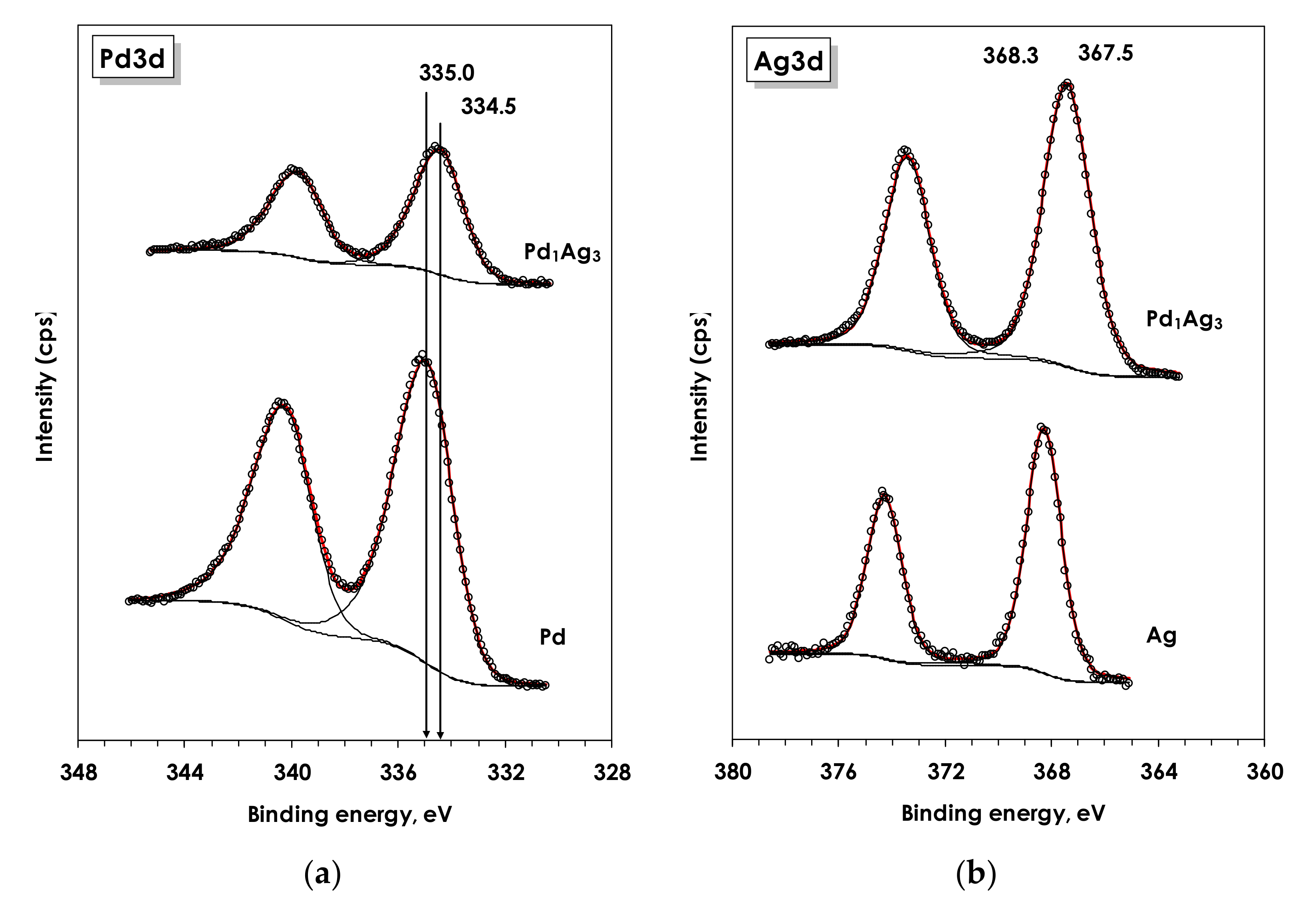
3.1.5. X-ray Photoelectron Spectroscopy
3.2. Catalytic Performance of Pd/Al2O3 and Pd1Ag3/Al2O3 Catalysts in Alkyne Hydrogenation
3.2.1. Comparison of Kinetic Profiles
3.2.2. Effect of the Substrate Concentration
3.2.3. Hydrogen Pressure Effect
3.3. Kinetic Modelling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Delgado, J.A.; Godard, C. Progress in the Selective Semi-hydrogenation of Alkynes by Nanocatalysis. In Recent Advances in Nanoparticle Catalysis; Van Leeuwen, P., Claver, C., Eds.; Springer: Cham, Switzerland, 2020; pp. 303–344. [Google Scholar] [CrossRef]
- Török, B.; Schäfer, C.; Kokel, A. Hydrogenation. In Heterogeneous Catalysis in Sustainable Synthesis; Elsevier: Amsterdam, The Netherlands, 2022; pp. 85–156. [Google Scholar]
- Hou, R. Introduction, Catalytic and Process Study of the Selective Hydrogenation of Acetylene and 1,3-Butadiene.; Springer: Singapore, 2017. [Google Scholar]
- Takht Ravanchi, M.; Sahebdelfar, S.; Komeili, S. Acetylene selective hydrogenation: A technical review on catalytic aspects. Rev. Chem. Eng. 2018, 34, 215–237. [Google Scholar] [CrossRef]
- Osswald, J. Active-Site Isolation for the Selective Hydrogenation of Acetylene: The Pd-Ga and Pd-Sn Intermetallic Compounds. Ph.D. Thesis, Fakultät II—Mathematik und Naturwissenschaften, Technische Universitat Berlin, Berlin, Germany, 2005; p. 10. [Google Scholar]
- Bonrath, W.; Medlock, J.; Schütz, J.; Wüstenberg, B.; Netscher, T. Hydrogenation in the Vitamins and Fine Chemicals Industry—An Overview, Hydrogenation, Iyad Karamé, IntechOpen. 2012. Available online: https://www.intechopen.com/books/hydrogenation/hydrogenation-in-the-vitamins-and-fine-chemicals-industry-an-overview (accessed on 10 October 2012).
- Eggersdorfer, M.; Laudert, D.; Létinois, U.; McClymont, T.; Medlock, J.; Netscher, T.; Bonrath, W. One Hundred Years of Vitamins—A Success Story of the Natural Sciences. Angew. Chem. Int. Ed. 2012, 51, 12960–12990. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Mao, S.; Li, J.; Li, M.; Chen, P.; Li, H.; Chen, Z.; Wang, Y. PdZn Intermetallic on a CN@ZnO Hybrid as an Efficient Catalyst for the Semi-Hydrogenation of Alkynols. J. Catal. 2017, 350, 13–20. [Google Scholar] [CrossRef]
- Teschner, D.; Borsodi, J.; Wootsch, A.; Révay, Z.; Hävecker, M.; Knop-Gericke, A.; Jackson, S.D.; Schlögl, R. The Roles of Subsurface Carbon and Hydrogen in Palladium-Catalyzed Alkyne Hydrogenation. Science 2008, 320, 86–89. [Google Scholar] [CrossRef]
- Rassolov, A.V.; Bragina, G.O.; Baeva, G.N.; Smirnova, N.S.; Kazakov, A.V.; Mashkovsky, I.S.; Bukhtiyarov, A.V.; Zubavichus, Y.V.; Stakheev, A.Y. Formation of Isolated Single-Atom Pd1 Sites on the Surface of Pd–Ag/Al2O3 Bimetallic Catalysts. Kinet. Catal. 2020, 61, 758–767. [Google Scholar] [CrossRef]
- Markov, P.V.; Mashkovsky, I.S.; Bragina, G.O.; Wärnå, J.; Gerasimov, E.Y.; Bukhtiyarov, V.I.; Stakheev, A.Y.; Murzin, D.Y. Particle size effect in liquid-phase hydrogenation of phenylacetylene over Pd catalysts: Experimental data and theoretical analysis. Chem. Eng. J. 2019, 358, 520–530. [Google Scholar] [CrossRef]
- Rassolov, A.V.; Mashkovsky, I.S.; Bragina, G.O.; Baeva, G.N.; Markov, P.V.; Smirnova, N.S.; Wärnå, J.; Stakheev, A.Y.; Murzin, D.Y. Kinetics of liquid-phase diphenylacetylene hydrogenation on “single-atom alloy” Pd-Ag catalyst: Experimental study and kinetic analysis. Mol. Catal. 2021, 506, 111550. [Google Scholar] [CrossRef]
- Markov, P.V.; Mashkovsky, I.S.; Bragina, G.O.; Wärnä, J.; Bukhtiyarov, V.I.; Stakheev, A.Y.; Murzin, D.Y. Experimental and theoretical analysis of particle size effect in liquid-phase hydrogenation of diphenylacetylene. Chem. Eng. J. 2021, 404, 126409. [Google Scholar] [CrossRef]
- Hannagan, R.T.; Giannakakis, G.; Flytzani-Stephanopoulos, M.; Sykes, E.C.H. Single-atom alloy catalysis. Chem. Rev. 2020, 120, 12044–12088. [Google Scholar] [CrossRef] [PubMed]
- Darby, M.T.; Stamatakis, M.; Michaelides, A.; Sykes, E.C.H. Lonely atoms with special gifts: Breaking linear scaling relationships in heterogeneous catalysis with single-atom alloys. J. Phys. Chem. Lett. 2018, 9, 5636–5646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannakakis, G.; Flytzani-Stephanopoulos, M.; Sykes, E.C.H. Single-atom alloys as a reductionist approach to the rational design of heterogeneous catalysts. Acc. Chem. Res. 2019, 52, 237–247. [Google Scholar] [CrossRef]
- Han, J.; Lu, J.; Wang, M.; Wang, Y.; Wang, F. Single atom alloy preparation and applications in heterogeneous catalysis. Chin. J. Chem. 2019, 37, 977–988. [Google Scholar] [CrossRef]
- Kyriakou, G.; Boucher, M.B.; Jewell, A.D.; Lewis, E.A.; Lawton, T.J.; Baber, A.E.; Tierney, H.L.; Flytzani-Stephanopoulos, M.; Sykes, E.C. Isolated Metal Atom Geometries as a Strategy for Selective Heterogeneous Hydrogenations. Science 2012, 335, 1209–1212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, A.; Miller, J.T.; Liu, X.; Yang, X.; Wang, W.; Li, L.; Huang, Y.; Mou, C.-Y.; Zhang, T. Efficient and Durable Au Alloyed Pd Single-Atom Catalysts for the Ulmann Reaction of Aryl Chlorides in Water. ACS Catal. 2014, 4, 1546–1553. [Google Scholar] [CrossRef]
- Pei, G.X.; Liu, X.Y.; Wang, A.; Lee, A.F.; Isaacs, M.A.; Li, L.; Pan, X.; Yang, X.; Wang, X.; Tai, Z.; et al. Ag Alloyed Pd Single-Atom Catalysts for Efficient Selective Hydrogenation of Acetylene to Ethylene in Excess Ethylene. ACS Catal. 2015, 5, 3717–3725. [Google Scholar] [CrossRef]
- Pei, G.; Liu, X.; Chai, M.; Wang, A.; Zhang, T. Isolation of Pd atoms by Cu for semi-hydrogenation of acetylene: Effects of Cu loading. Chin. J. Catal. 2017, 38, 1540–1548. [Google Scholar] [CrossRef]
- Pei, G.; Liu, X.Y.; Yang, X.; Zhang, L.; Wang, A.; Li, L.; Wang, H.; Wang, X.; Zhang, T. Performance of Cu-Alloyed Pd single-atom catalyst for semihydrogenation of acetylene under simulated front-end conditions. ACS Catal. 2017, 7, 1491–1500. [Google Scholar] [CrossRef]
- Pei, G.X.; Liu, X.Y.; Wang, A.Q.; Li, L.; Huang, Y.Q.; Zhang, T.; Lee, J.W.; Jang, B.W.L.; Mou, C.Y. Promotional effect of Pd single atoms on Au nanoparticles supported on silica for the selective hydrogenation of acetylene in excess ethylene. New J. Chem. 2014, 38, 2043–2051. [Google Scholar] [CrossRef]
- Lucci, F.R.; Liu, J.; Marcinkowski, M.P.; Yang, M.; Allard, L.F.; Flytzani-Stephanopoulos, M.; Sykes, C.E.H. Selective hydrogenation of 1,3-butadiene on platinum–copper alloys at the single-atom limit. Nat. Commun. 2015, 6, 8550. [Google Scholar] [CrossRef] [Green Version]
- Lv, C.-Q.; Liu, J.-H.; Guo, Y.; Wang, G.-C. Selective hydrogenation of 1,3-butadiene over single Pt1/Cu(1 1 1) model catalysts: A DFT study. Appl. Surf. Sci. 2019, 466, 946–955. [Google Scholar] [CrossRef]
- Yang, K.; Yang, B. Identification of the Active and Selective Sites over a Single Pt Atom-Alloyed Cu Catalyst for the Hydrogenation of 1,3-butadiene: A Combined DFT and Microkinetic Modelling Study. J. Phys. Chem. C 2018, 122, 10883–10891. [Google Scholar] [CrossRef]
- Liu, D.; Chen, H.Y.; Zhang, J.Y.; Huang, J.Y.; Li, Y.M.; Peng, Q.M. Theoretical investigation of selective hydrogenation of 1,3-butadiene on Pt doping Cu nanoparticles. Appl. Surf. Sci. 2018, 456, 59–68. [Google Scholar] [CrossRef]
- Wencka, M.; Hahne, M.; Kocjan, A.; Vrtnik, S.; Koželj, P.; Korže, D.; Jagličić, Z.; Sorić, M.; Popčević, P.; Ivkov, J.; et al. Physical properties of the InPd intermetallic catalyst. Intermetallics 2014, 55, 56–65. [Google Scholar] [CrossRef]
- Langmuir, I. The Dissociation of Hydrogen into Atoms. III. The Mechanism of the Reaction. J. Am. Chem. Soc. 1916, 38, 1145–1156. [Google Scholar] [CrossRef]
- Groß, A.; Dianat, A. Hydrogen dissociation dynamics on precovered Pd surfaces: Langmuir is still right. Phys. Rev. Lett. 2007, 98, 206107. [Google Scholar] [CrossRef] [Green Version]
- Tierney, H.L.; Baber, A.E.; Kitchin, J.R.; Sykes, E.C. Hydrogen dissociation and spillover on individual isolated palladium atoms. Phys. Rev. Lett. 2009, 103, 246102. [Google Scholar] [CrossRef]
- Aich, P.; Wei, H.; Basan, B.; Kropf, A.J.; Schweitzer, N.M.; Marshall, C.L.; Miller, J.T.; Meyer, R. Single-Atom Alloy Pd–Ag Catalyst for Selective Hydrogenation of Acrolein. J. Phys. Chem. C 2015, 119, 18140–18148. [Google Scholar] [CrossRef]
- Molina, D.L.; Muir, M.; Abdel-Rahman, M.K.; Trenary, M. The influence of palladium on the hydrogenation of acetylene on Ag(111). J. Chem. Phys. 2021, 154, 184701. [Google Scholar] [CrossRef]
- Muir, M.; Trenary, M. Adsorption of CO to Characterize the Structure of a Pd/Ag(111) Single-Atom Alloy Surface. J. Phys. Chem. C 2020, 124, 14722–14729. [Google Scholar] [CrossRef]
- Hartwig, C.; Schweinar, K.; Jones, T.E.; Beeg, S.; Schmidt, F.P.; Schlögl, R.; Greiner, M. Isolated Pd atoms in a silver matrix: Spectroscopic and chemical properties. J. Chem. Phys. 2021, 154, 184703. [Google Scholar] [CrossRef]
- Liu, J.; Shan, J.; Lucci, F.R.; Cao, S.; Sykes, E.C.H.; Flytzani-Stephanopoulos, M. Palladium–gold single atom alloy catalysts for liquid phase selective hydrogenation of 1-hexyne. Catal. Sci. Tech. 2017, 7, 4276–4284. [Google Scholar] [CrossRef]
- Lucci, F.R.; Darby, M.T.; Mattera, M.F.G.; Ivimey, C.J.; Therrien, A.J.; Michaelides, A.; Stamatakis, M.; Sykes, E.C.H. Controlling Hydrogen Activation, Spillover, and Desorption with Pd–Au Single-Atom Alloys. J. Phys. Chem. Lett. 2016, 7, 480–485. [Google Scholar] [CrossRef]
- Darby, M.T.; Réocreux, R.; Sykes, E.C.H.; Michaelides, A.; Stamatakis, M. Elucidating the Stability and Reactivity of Surface Intermediates on Single-Atom Alloy Catalysts. ACS Catal. 2018, 8, 5038–5050. [Google Scholar] [CrossRef]
- Vignola, E.; Steinmann, S.N.; Vandegehuchte, B.D.; Curulla, D.; Sautet, P. C2H2-Induced Surface Restructuring of Pd-Ag Catalysts: Insights from Theoretical Modeling. J. Phys. Chem. C 2016, 120, 26320–26327. [Google Scholar] [CrossRef] [Green Version]
- Vignola, E.; Steinmann, S.N.; Le Mapihan, K.; Vandegehuchte, B.D.; Curulla, D.; Sautet, P. Acetylene Adsorptiom on Pd-Ag Alloys: Evidence for Limited Island Formation and Strong Reverse Segregation from Monte Carlo Simulations. J. Phys. Chem. C 2018, 122, 15456–15463. [Google Scholar] [CrossRef] [Green Version]
- Liu, D. DFT study of selective hydrogenation of acetylene to ethylene on Pd doping Ag nanoclusters. Appl. Surf. Sci. 2016, 386, 125–137. [Google Scholar] [CrossRef]
- Khan, N.A.; Shaikhutdinov, S.; Freund, H.-J. Acetylene and ethylene hydrogenation on alumina supported Pd-Ag model catalysts. Catal. Lett. 2006, 108, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, S.; Neyman, K.M.; Shaikhutdinov, S.; Freund, H.-J.; Illas, F. On the promoting role of Ag in selective hydrogenation reactions over Pd-Ag bimetallic catalysts: A theoretical study. J. Phys. Chem. C 2007, 111, 6852–6856. [Google Scholar] [CrossRef]
- Gao, X.; Zhou, Y.; Jing, F.; Luo, J.; Huang, Q.; Chu, W. Layered double hydroxides derived ZnO-Al2O3 supported Pd-Ag catalysts for selective hydrogenation of acetylene. Chin. J. Chem. 2017, 35, 1009–1015. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Liu, Y.; Yang, P.; Du, Y.; Feng, J.; Cao, X.; Yang, J.; Li, D. Fabrication of a PdAg mesocrystal catalyst for the partial hydrogenation of acetylene. J. Catal. 2015, 330, 61–70. [Google Scholar] [CrossRef]
- Glyzdova, D.V.; Afonasenko, T.N.; Khramov, E.V.; Leont’eva, N.N.; Prosvirin, I.P.; Bukhtiyarov, A.V.; Shlyapin, D.A. Liquid-phase acetylene hydrogenation over Ag-modified Pd/Sibunit catalysts: Effect of Pd to Ag molar ratio. Appl. Catal. A Gen. 2020, 600, 117627. [Google Scholar] [CrossRef]
- Kachala, V.V.; Khemchyan, L.L.; Kashin, A.S.; Orlov, N.V.; Grachev, A.A.; Zalesskiy, S.S.; Ananikov, V.P. Target-oriented analysis of gaseous, liquid and solid chemical systems by mass spectrometry, nuclear magnetic resonance spectroscopy and electron microscopy. Russ. Chem. Rev. 2013, 82, 648–685. [Google Scholar] [CrossRef]
- Available online: http://xpspeak.software.informer.com/4.1/ (accessed on 18 May 2008).
- Scofield, J.H. Hartree-Slater subshell photoionization cross-sections at 1254 and 1487 eV. J. Electron. Spectros. Relat. Phenom. 1976, 8, 129–137. [Google Scholar] [CrossRef]
- Červenýa, L.; Rûržička, V. Solvent and Structure Effects in Hydrogenation of Unsaturated Substances on Solid Catalysts. Adv. Catal. 1981, 30, 335–377. [Google Scholar]
- Cesarotti, E.; Ugo, R.; Kaplan, L. A discussion of the different kinds of solute-solute and solute-solvent interactions acting in homogeneous catalysis by transition metal complexes. Coord. Chem. Rev. 1982, 43, 275–298. [Google Scholar] [CrossRef]
- Bertero, N.M.; Apesteguia, C.R.; Marchi, A.J. Catalytic and kinetic study of the liquid-phase hydrogenation of acetophenone over Cu/SiO2 catalyst. Appl. Catal. A Gen. 2008, 349, 100–109. [Google Scholar] [CrossRef]
- Akpa, B.S.; D’Agostino, C.; Gladden, L.F.; Hindle, K.; Manyar, H.; McGregor, J.; Li, R.; Neurock, M.; Sinha, N.; Stitt, E.H.; et al. Solvent effects in the hydrogenation of 2-butanone. J. Catal. 2012, 289, 30–41. [Google Scholar] [CrossRef]
- Reichardt, C.; Welton, T. Solvent and Solvent Effects in Organic Chemistry; Wiley: Weinheim, Germany, 2010. [Google Scholar]
- Toukoniitty, E.; Mäki-Arvela, P.; Kuusisto, J.; Nieminen, V.; Päivarinta, J.; Hotokka, M.; Salmi, T.; Murzin, D.Y. Solvent effects in enantioselective hydrogenation of 1-phenyl-1,2-propanedione. J. Mol. Catal. A. Chem. 2003, 192, 135–151. [Google Scholar] [CrossRef]
- Markov, P.V.; Bragina, G.O.; Baeva, G.N.; Tkachenko, O.P.; Mashkovsky, I.S.; Yakushev, I.A.; Kozitsyna, N.Y.; Vargaftik, M.N.; Stakheev, A.Y. Pd–Cu catalysts from acetate complexes in liquid-phase diphenylacetylene hydrogenation. Kinet. Catal. 2015, 56, 591–597. [Google Scholar] [CrossRef]
- Ramachandran, P.A.; Chaudhari, R.V. Three-Phase Catalytic Reactors; Gordon and Breach: New York, NY, USA, 1983; p. 32. [Google Scholar]
- Weisz, P.B.; Prater, C.D. Interpretaion of Measurements in Experimental Catalysis. Adv. Catal. 1954, 6, 143–196. [Google Scholar]
- Armbrüster, M.; Behrens, M.; Cinquini, F.; Föttinger, K.; Grin, Y.; Haghofer, A.; Klötzer, B.; Knop-Gericke, A.; Lorenz, H.; Ota, A.; et al. How to Control the Selectivity of Palladium-based Catalysts in Hydrogenation Reactions: The Role of Subsurface Chemistry. ChemCatChem 2012, 4, 1048–1063. [Google Scholar] [CrossRef]
- Mashkovsky, I.S.; Markov, P.V.; Bragina, G.O.; Baeva, G.N.; Rassolov, A.V.; Yakushev, I.A.; Vargaftik, M.N.; Stakheev, A.Y. Highly-Ordered PdIn Intermetallic Nanostructures Obtained from Heterobimetallic Acetate Complex: Formation and Catalytic Properties in Diphenylacetylene Hydrogenation. Nanomaterials 2018, 8, 769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, T.; Furukawa, S. Intermetallic Compound Nanoparticles Dispersed on the Surface of Oxide Support as Active and Selective Catalysts. Mater. Trans. 2015, 56, 460–467. [Google Scholar] [CrossRef] [Green Version]
- Aduriz, H.R.; Bodnariuk, P.; Coq, B.; Figueras, F. Alumina-Supported Bimetallics of Palladium Alloyed with Germanium, Tin, Lead, or Antimony from Organometallic Precursors. I. Preparation and characterization. J. Catal. 1989, 119, 97–107. [Google Scholar] [CrossRef]
- Armbrüster, M.; Schlögl, R.; Grin, Y. Intermetallic compounds in heterogeneous catalysis—A quickly developing field. Sci. Technol. Adv. Mater. 2014, 15, 034803. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Sui, Z.J.; Zhu, Y.; Zhou, X.; Chen, D. Selective Hydrogenation of Acetylene over Pd-In/Al2O3 Catalyst: Promotional Effect of Indium and Composition-dependent Performance. ASC Catal. 2017, 7, 7835–7846. [Google Scholar] [CrossRef]
- Schauermann, S.; Hoffmann, J.; Johánek, V.; Hartmann, J.; Libuda, J.; Freund, H.-J. Catalytic Activity and Poisoning of Specific Sites on Supported Metal Nanoparticles. Angew. Chem. Int. Ed. 2002, 41, 2532–2535. [Google Scholar] [CrossRef]
- Lear, T.; Marshall, R.; Lopez-Sanchez, J.A.; Jackson, S.D.; Klapötke, T.M.; Bäumer, M.; Rupprechter, G.; Freund, H.-J.; Lennon, D. The application of infrared spectroscopy to probe the surface morthology of alumina-supported palladium catalysts. J. Chem. Phys. 2005, 123, 174706. [Google Scholar] [CrossRef] [Green Version]
- Cabilla, G.C.; Bonivardi, A.L.; Baltanás, M.A. Characterization by CO/FTIR spectroscopy of Pd/silica catalysts and its correlation with syn-gas conversion. Catal. Lett. 1998, 55, 147–156. [Google Scholar] [CrossRef]
- Wolter, K.; Seiferth, O.; Kuhlenbeck, H.; Bäumer, M.; Freund, H.-J. Infrared spectroscopic investigation of CO adsorbed on Pd aggregates deposited on an alumina model support. Surf. Sci. 1998, 399, 190–198. [Google Scholar] [CrossRef]
- McCue, A.J.; Anderson, J.A. CO induced surface segregation as a means of improving surface composition and enhancing performance of CuPd bimetallic catalysts. J. Catal. 2015, 329, 538–546. [Google Scholar] [CrossRef]
- Panafidin, M.A.; Bukhtiyarov, A.V.; Prosvirin, I.P.; Chetyrin, I.A.; Bukhtiyarov, V.I. Model Bimetallic Pd–Ag/HOPG Catalysts: An XPS and STM Study. Kinet. Catal. 2018, 59, 776–785. [Google Scholar] [CrossRef]
- Lamb, R.N.; Ngamsom, B.; Trimm, D.L.; Gong, B.; Silveston, P.L.; Praserthdam, P. Surface characterisation of Pd–Ag/Al2O3 catalysts for acetylene hydrogenation using an improved XPS procedure. Appl. Catal. A Gen. 2004, 268, 43–50. [Google Scholar] [CrossRef]
- Spee, M.P.R.; Boersma, J.; Meijer, M.D.; Slagt, M.Q.; van Koten, G.; Geus, J.W. Selective Liquid-Phase Semihydrogenation of Functionalized Acetylenes and Propargylic Alcohols with Silica-Supported Bimetallic Palladium-Copper Catalysts. J. Org. Chem. 2001, 66, 1647–1656. [Google Scholar] [CrossRef] [Green Version]
- Mashkovsky, I.S.; Markov, P.V.; Bragina, G.O.; Rassolov, A.V.; Baeva, G.N.; Stakheev, A.Y. Intermetallic Pd1-Zn1 nanoparticles in the selective liquid-phase hydrogenation of substituted alkynes. Kinet. Catal. 2017, 58, 480–491. [Google Scholar] [CrossRef]
- Marín-Astorga, N.; Alvez-Manoli, G.; Reyes, P. Stereoselective hydrogenation of phenyl alkyl acetylenes on pillared clays supported palladium catalysts. J. Mol. Catal. A Chem. 2005, 226, 81–88. [Google Scholar] [CrossRef]
- Jung, A.; Jess, A.; Schubert, T.; Schütz, W. Performance of carbon nanomaterial (nanotubes and nanofibers) supportes platinum and palladium catalysts for the hydrogenation of cinnamaldehyde and 1-octyne. Appl. Catal. A 2009, 362, 95–105. [Google Scholar] [CrossRef]
- Vilé, G.; Almora-Barrios, N.; Mitchell, S.; López, N.; Pérez-Ramírez, J. From the Lindlar Catalyst to Supported Ligand-Modified Palladium Nanoparticles: Selectivity Patterns and Accessibility Constraints in the Continious-Flow Three-Phase Hydrogenation of Acetylenic Compounds. Chem. Eur. J. 2014, 20, 5926–5937. [Google Scholar] [CrossRef]
- Chaudhari, R.V.; Jaganathan, R.; Kolhe, D.S. Effect of catalyst pretreatment on activity and selectivity of hydrogenation of phenylacetylene over palladium/carbon catalyst. Ind. Eng. Chem. Prod. Res. Dev. 1986, 25, 375–379. [Google Scholar] [CrossRef]
- Jackson, S.D.; Shaw, L.A. The liquid-phase hydrogenation of phenyl acetylene and styrene on a palladium/carbon catalyst. Appl. Catal. A 1996, 134, 91–99. [Google Scholar] [CrossRef]
- Wu, Z.; Calcio Gaudino, E.; Manzoli, M.; Martina, K.; Drobot, M.; Krtschil, U.; Cravotto, G. Selective hydrogenation of alkynes over ppm-level Pd/Boehmite/Al2O3 beads in a continuous-flow reactor. Catal. Sci. Technol. 2017, 7, 4780–4791. [Google Scholar] [CrossRef]
- Nijhuis, T.A.; van Koten, G.; Moulijn, J.A. Optimised palladium catalyst systems for the selective liquid-phase hydrogenation of functionalyzed alkynes. Appl. Catal. A Gen. 2003, 238, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Studt, F.; Abild-Pedersen, F.; Bligaard, T.; Sørensen, R.Z.; Christensen, C.H.; Nørskov, J.K. Identification of Non-Precious Metal Alloy Catalysts for Selective Hydrogenation of Acetylene. Science 2008, 320, 1320–1322. [Google Scholar] [CrossRef] [PubMed]
- Arena, F.; Cum, G.; Gallo, R.; Parmaliana, A. Palladium catalysts supported on oligomeric aramides in the liquid-phase hydrogenation of phenylacetylene. J. Mol. Catal. A: Chem. 1996, 110, 235–242. [Google Scholar] [CrossRef]
- Molero, H.; Stacchiola, D.; Tysoe, W.T. The kinetics of ethylene hydrogenation catalized by metallic palladium. Catal. Lett. 2005, 101, 145–149. [Google Scholar] [CrossRef]
- Murzin, D.Y. Selectivity in consecutive heterogeneous catalytic reactions: Case of polyatomic molecules. React. Kinet. Catal. Lett. 1996, 57, 153–158. [Google Scholar] [CrossRef]
- Murzin, D.Y. Kinetic coupling and selectivity pattern in consecutive heterogeneous catalytic reactions. React. Kinet. Catal. Lett. 1996, 58, 65–72. [Google Scholar] [CrossRef]
- Varga, M.; Molnár, Á.; Mohai, M.; Bertóti, I.; Janik-Czachor, M.; Szummer, A. Selective hydrogenation of pentynes over PdZr and PdCuZr prepared from amorphous precursors. Appl. Catal. A Gen. 2002, 234, 167–178. [Google Scholar] [CrossRef]
- Borodziński, A.; Bond, G.C. Selective Hydrogenation of Ethyne in Ethene-Rich Streams on Palladium Catalysts. Part I. Effect of Changes to the Catalyst During Reaction. Catal. Rev. Sci. Eng. 2006, 48, 91–144. [Google Scholar] [CrossRef]
- Li, X.-T.; Chen, L.; Wei, G.-F.; Shang, C.; Liu, Z.-P. Sharp increase in Catalytic Selectivity in Acetylene Semihydrogenation on Pd Achieved by a Machine Learning Simulation-Guided Experiment. ACS Catal. 2020, 10, 9694–9705. [Google Scholar] [CrossRef]
- Urmès, C.; Schweitzer, J.-M.; Cabiac, A.; Schuurman, Y. Kinetic Study of the Selective Hydrogenation of Acetylene over Supported Palladium under Tail-End Conditions. Catalysts 2019, 9, 180. [Google Scholar] [CrossRef] [Green Version]
- Haario, H. Modest 6.0-A User’s Guide; ProfMath: Helsinki, Finland, 2001. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET, m2/g | |
|---|---|---|
| initial | after catalytic test | |
| Al2O3 | 59 | - |
| Pd/Al2O3 | 57 | 56 |
| Pd1Ag3/Al2O3 | 55 | 55 |
| Parameter | Value | Error, % | Units |
|---|---|---|---|
| k1K1P1P | 1.87 | 26 | mol g−1 min−1 |
| k2K1P1P | 0.089 | 40.3 | mol g−1 min−1 |
| k3KC | 0.05 | 22.9 | mol g−1 min−1 |
| k4KT | 0.17 | 79 | mol g−1 min−1 |
| k5K1P1P | 0.063 | 50.7 | mol g−1 min−1 |
| K1P1P | 76.9 | 14.7 | l/mol |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rassolov, A.V.; Mashkovsky, I.S.; Baeva, G.N.; Bragina, G.O.; Smirnova, N.S.; Markov, P.V.; Bukhtiyarov, A.V.; Wärnå, J.; Stakheev, A.Y.; Murzin, D.Y. Liquid-Phase Hydrogenation of 1-Phenyl-1-propyne on the Pd1Ag3/Al2O3 Single-Atom Alloy Catalyst: Kinetic Modeling and the Reaction Mechanism. Nanomaterials 2021, 11, 3286. https://doi.org/10.3390/nano11123286
Rassolov AV, Mashkovsky IS, Baeva GN, Bragina GO, Smirnova NS, Markov PV, Bukhtiyarov AV, Wärnå J, Stakheev AY, Murzin DY. Liquid-Phase Hydrogenation of 1-Phenyl-1-propyne on the Pd1Ag3/Al2O3 Single-Atom Alloy Catalyst: Kinetic Modeling and the Reaction Mechanism. Nanomaterials. 2021; 11(12):3286. https://doi.org/10.3390/nano11123286
Chicago/Turabian StyleRassolov, Alexander V., Igor S. Mashkovsky, Galina N. Baeva, Galina O. Bragina, Nadezhda S. Smirnova, Pavel V. Markov, Andrey V. Bukhtiyarov, Johan Wärnå, Alexander Yu. Stakheev, and Dmitry Yu. Murzin. 2021. "Liquid-Phase Hydrogenation of 1-Phenyl-1-propyne on the Pd1Ag3/Al2O3 Single-Atom Alloy Catalyst: Kinetic Modeling and the Reaction Mechanism" Nanomaterials 11, no. 12: 3286. https://doi.org/10.3390/nano11123286