Single-Step Photochemical Formation of Near-Infrared-Absorbing Gold Nanomosaic within PNIPAm Microgels: Candidates for Photothermal Drug Delivery


Abstract
:
1. Introduction
1.1. Overview of Anisotropic AuNPs Usage
1.2. Toxicity Challenges
1.3. Nontoxic Tuning Methods
1.4. Photochemical Formation of AuNPs
1.5. Stabilization of AuNPs within PNIPAm Hydrogels/Microgels
1.6. Formation of AuNPs from Au(I) Precursor
2. Materials and Methods
2.1. Physical Measurements
2.2. Syntheses of Gold(I) Precursor
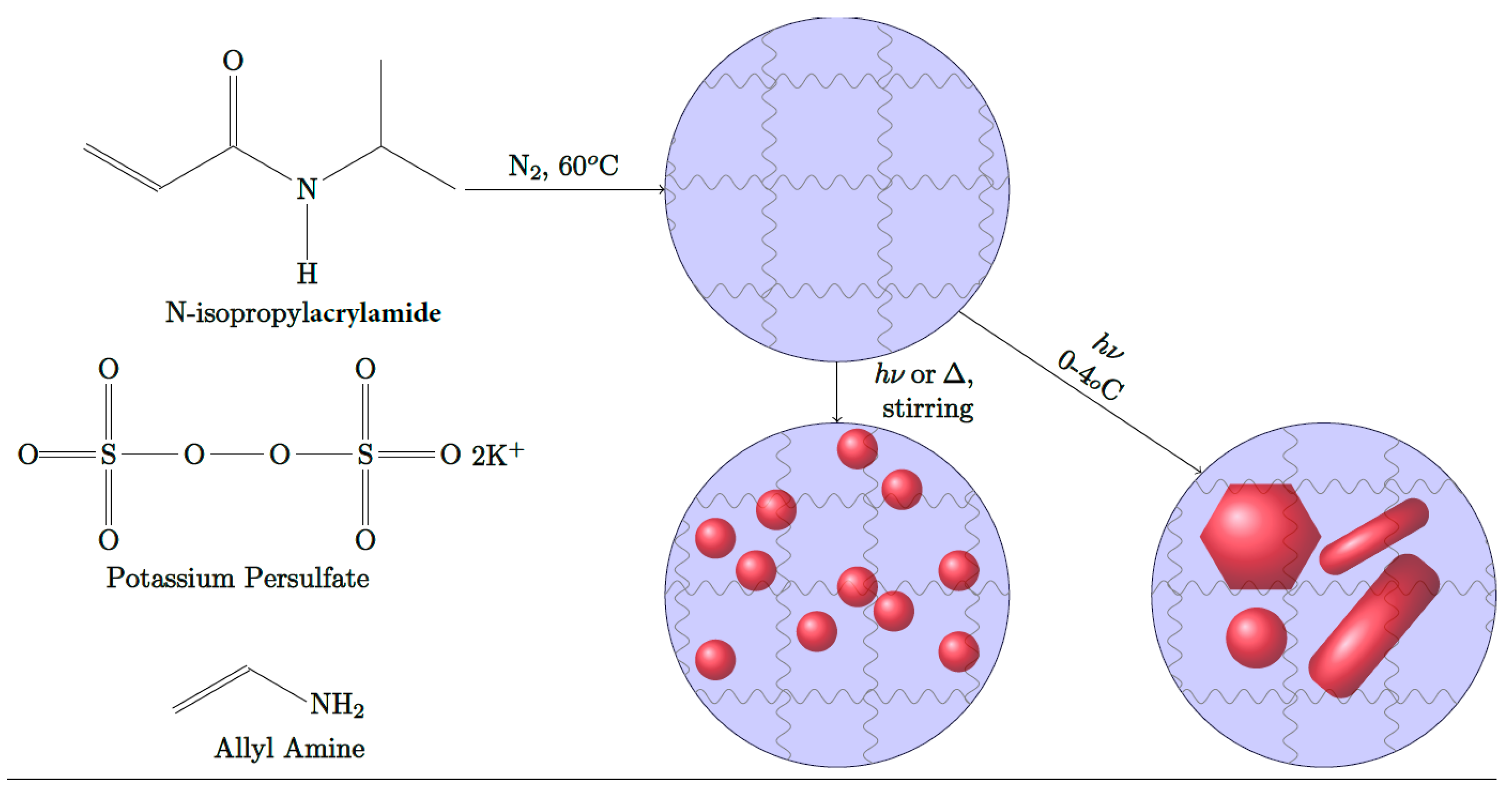
2.3. Syntheses of PNIPAm Microgels
2.4. Syntheses of Spherical AuNPs within PNIPAm Microgels
2.5. Synthesis of Spherical AuNPs within Different Media
2.6. Synthesis of Anisotropic AuNPs (NIRAuNM) within PNIPAm-Co-allylamine Microgels
2.7. Preparation of Samples for Electron Microscopy
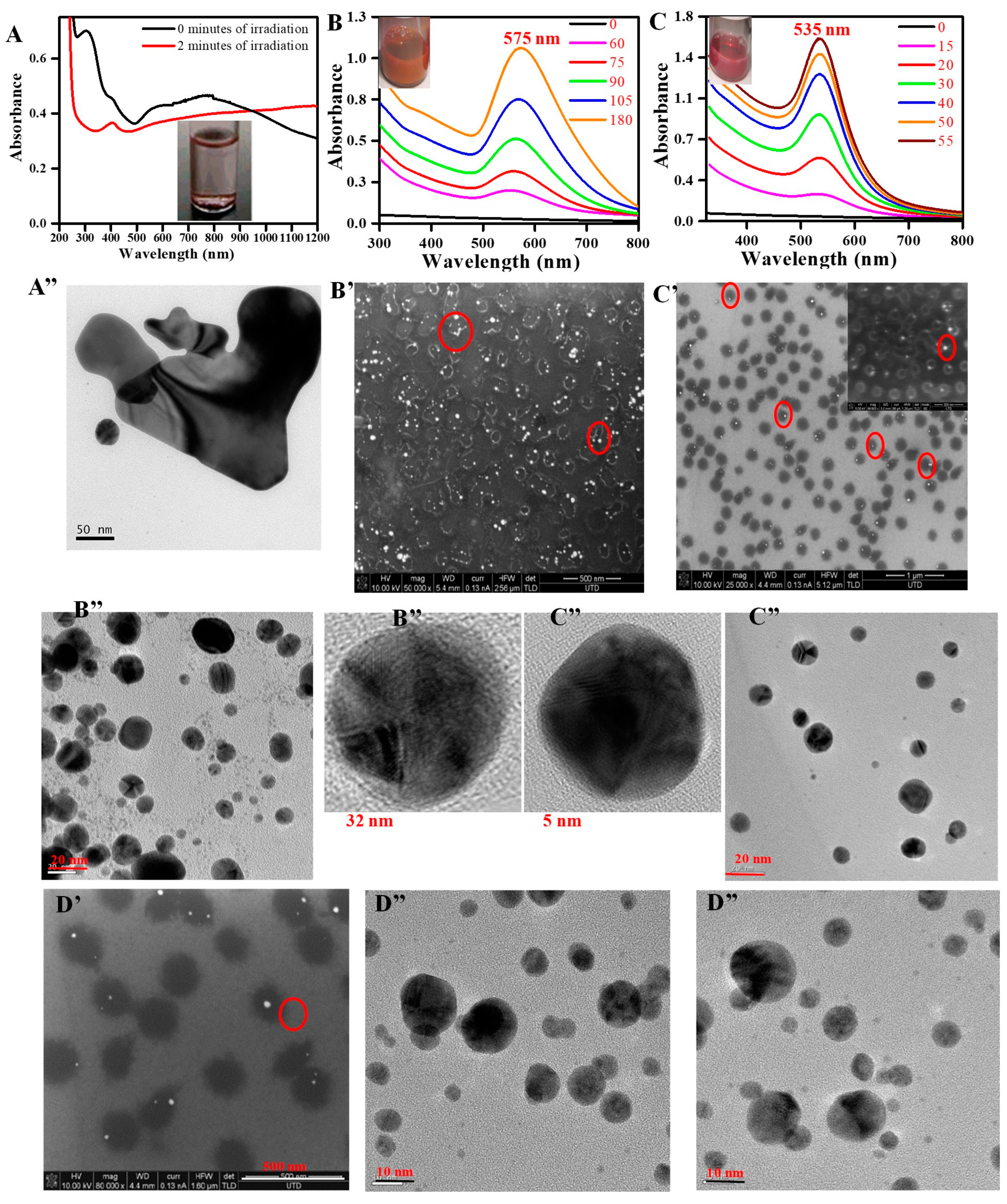
3. Results and Discussion
3.1. Potential of Au(I) Precursor as an Alternative Precursor to Au(III) Salts for Making AuNPs
3.2. In Situ Formation of Spherical Gold Nanoparticles within PNIPAm Microgels
3.3. Hybrid Crystalline PNIPAm Microgels Containing Gold Nanoparticles
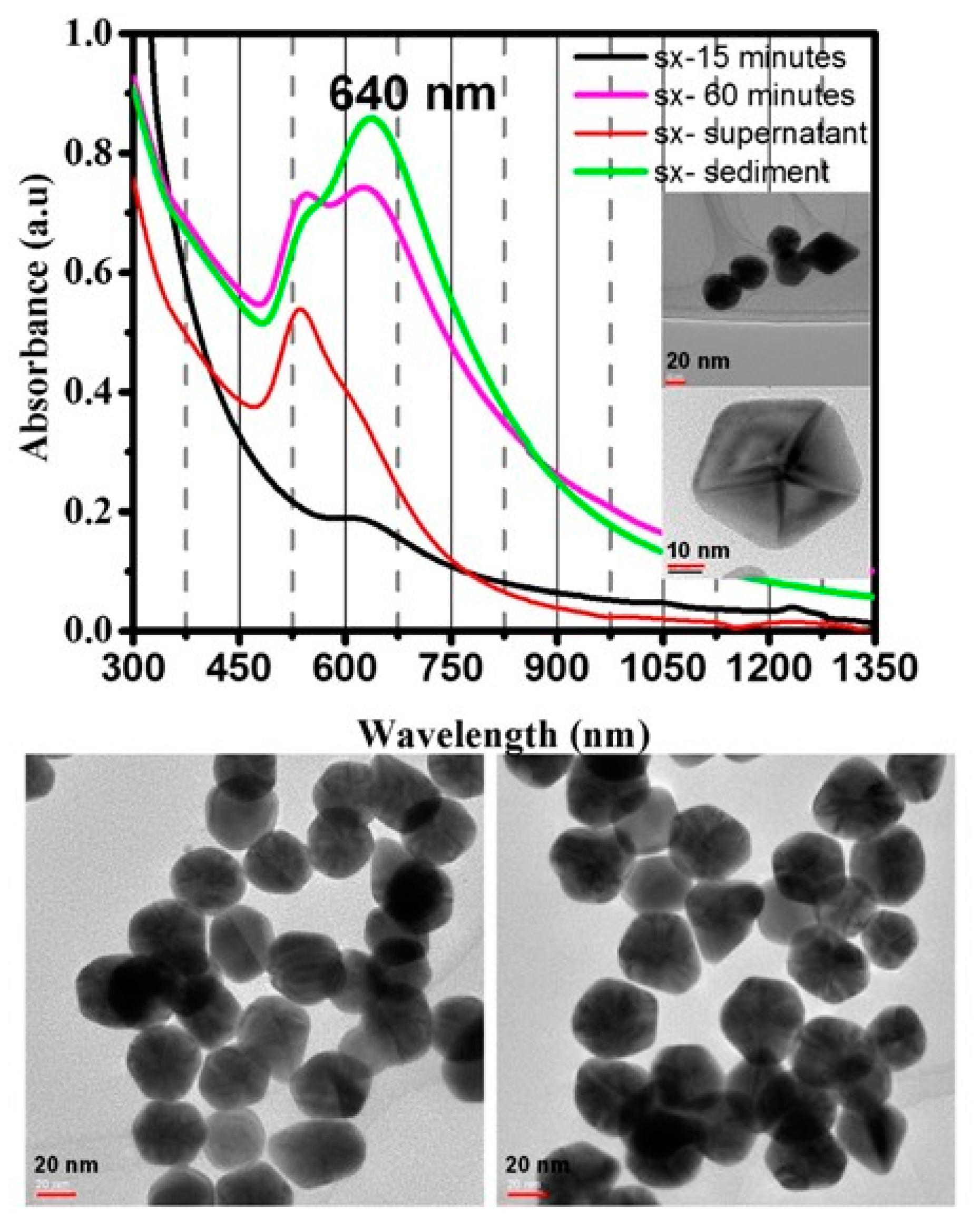
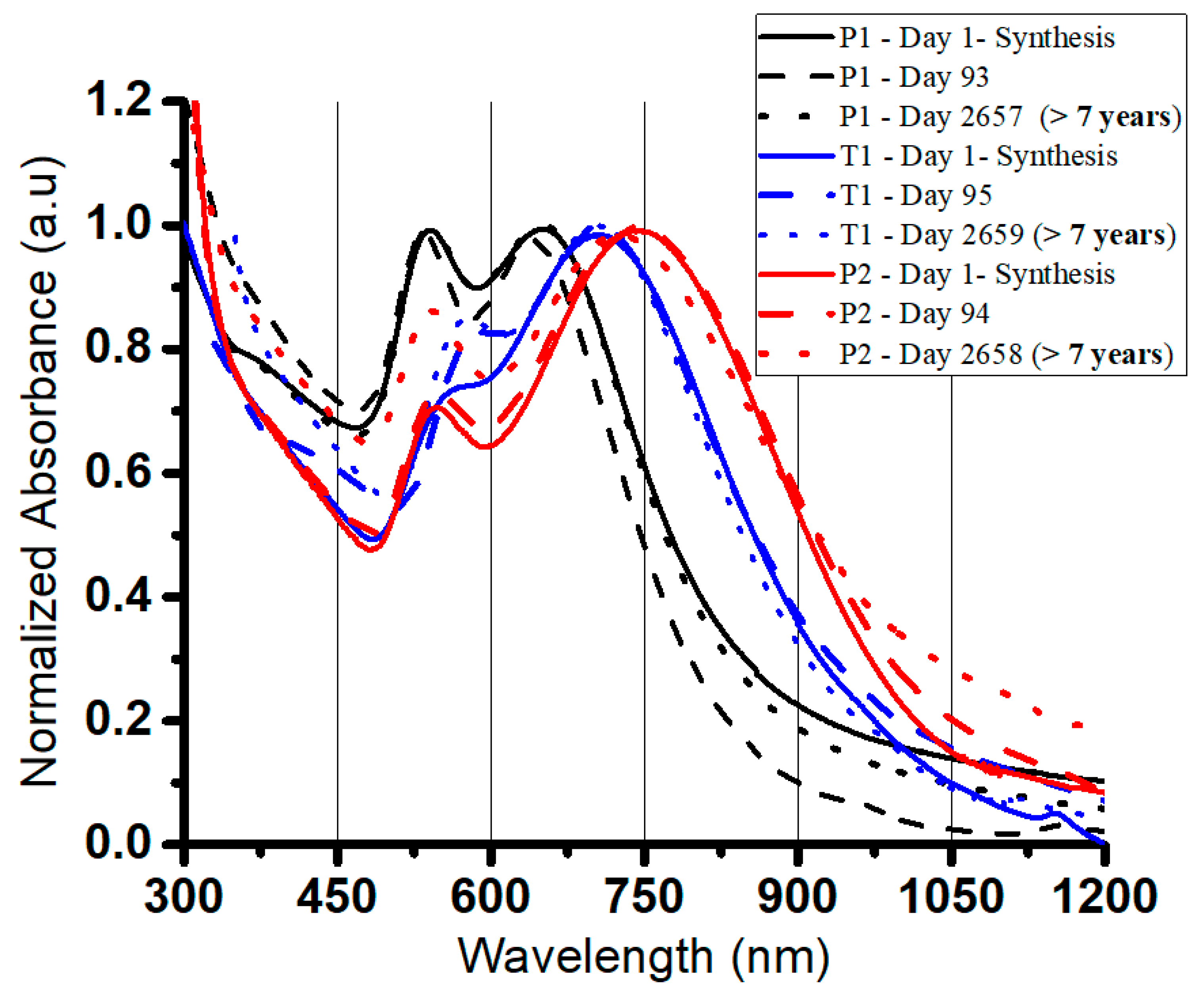
3.4. In Situ Formation of NIR-Absorbing Anisotropic Gold Nanomosaic (NIRAuNM) within PNIPAm-Co-Allylamine Microgels
3.5. Demonstrating Broad Applications by Altering the Polymer Stabilizer
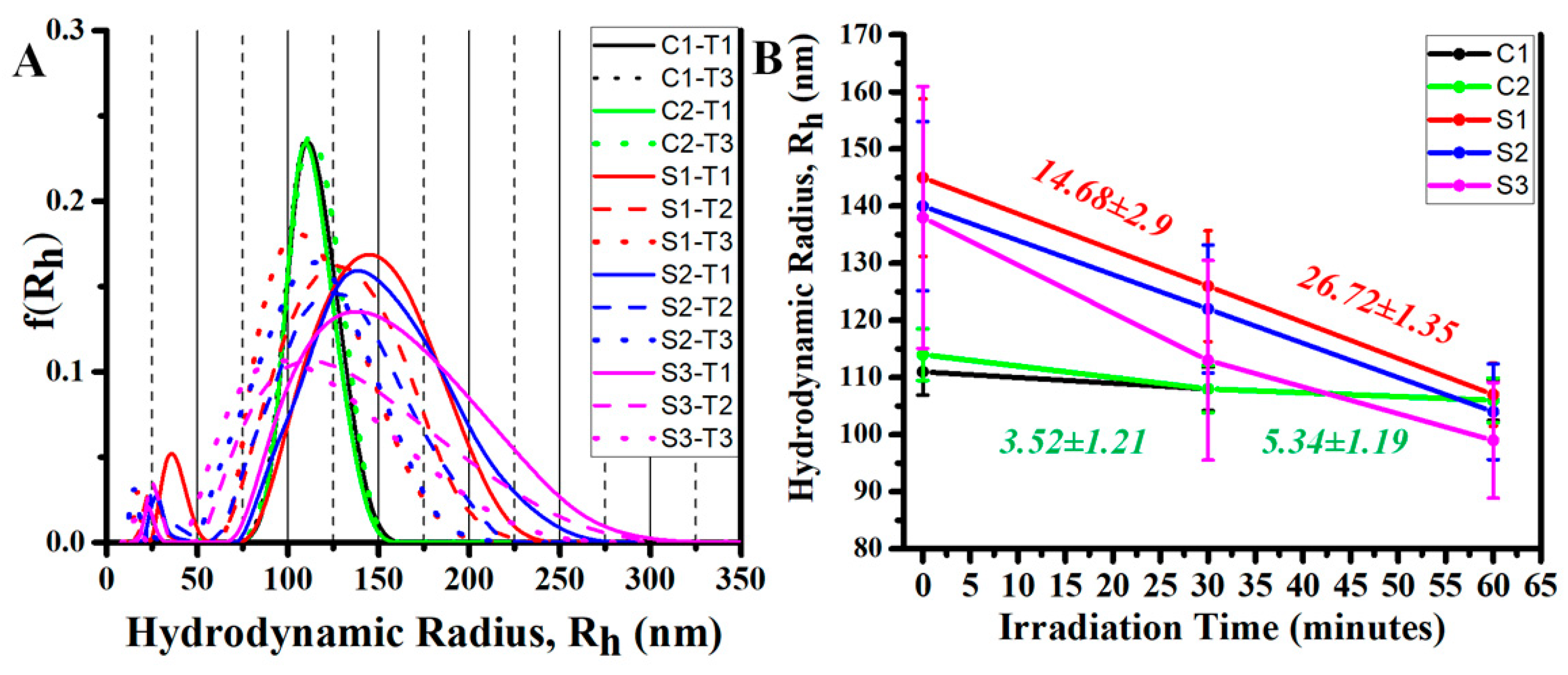
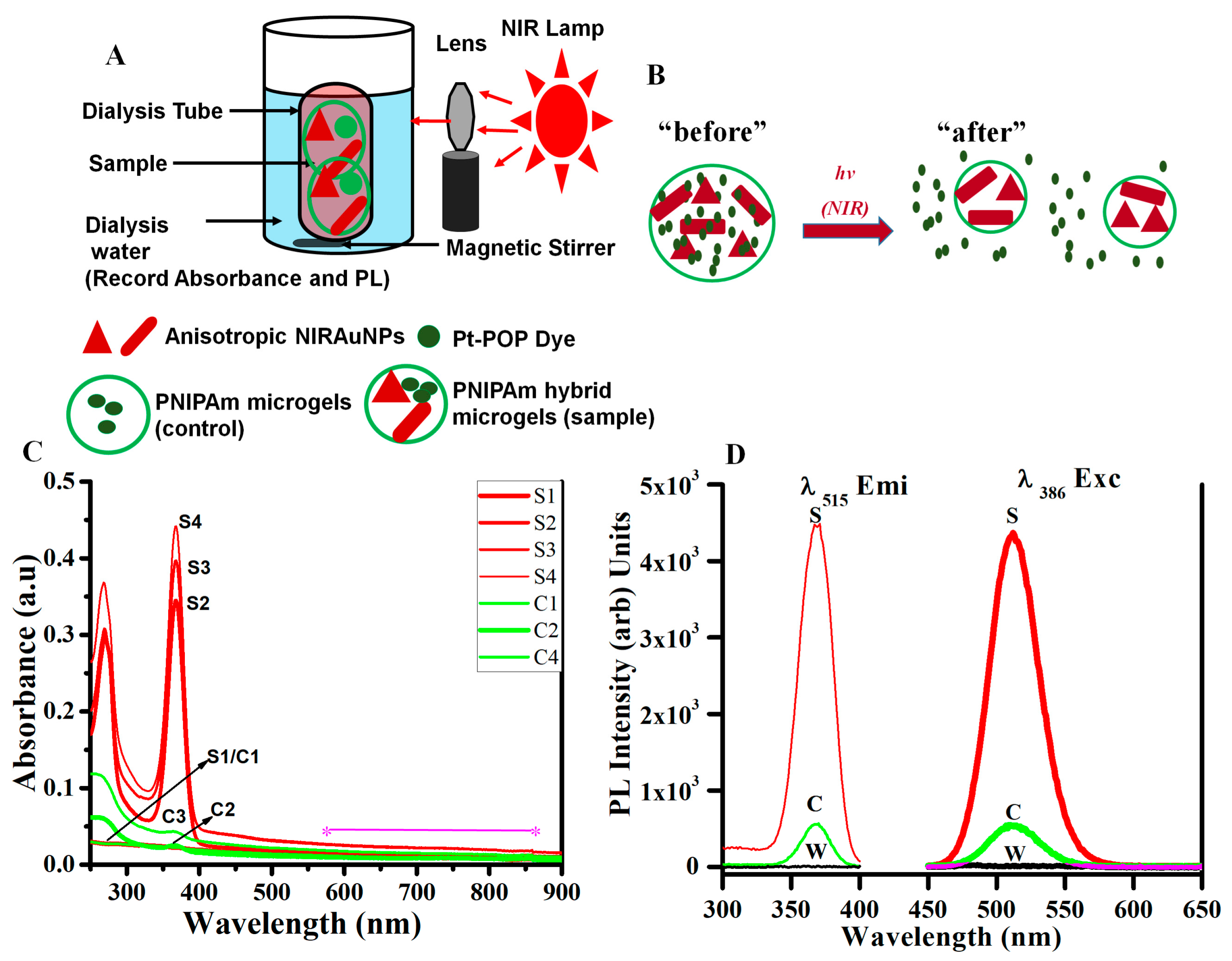
3.6. Photothermally Driven Release of a Model Dye from NIRAuNM Containing PNIPAm–Co-Allylamine (Hybrid) Microgels
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nayak, S.; Lyon, L.A. Soft Nanotechnology with Soft Nanoparticles. Ang. Chem. Int. 2005, 44, 7686–7708. [Google Scholar] [CrossRef]
- Chai, Q.; Jiao, Y.; Yu, X. Hydrogels for Biomedical Applications: Their Characteristics and the Mechanism Behind Them. Gels 2017, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Gan, D.; Lyon, L.A.; El-Sayed, A.M. Temperature-Jump Investigations of the Kinetics of Hydrogel Nanoparticle Volume Phase Transitions. J. Am. Chem. Soc. 2001, 123, 11284–11289. [Google Scholar] [CrossRef]
- Hu, Z.; Huang, G. A New Route to Crsytalline Hydrogels, Guided by a Phase Diagram. Ang. Chem. Int. 2003, 42, 4799–4802. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Choi, W.J.; Kim, S.; Nizamoglu, S.; Yun, H.S. Light-Guiding Hydrogels for Cell-based Sensing and Optgenetic Synthesis In Vivo. Nat. Photonics 2013, 7, 987–994. [Google Scholar] [CrossRef]
- Jiang, X.; Xiong, D.; An, Y.; Zheng, P.; Zhang, W.; Shi, L. Thermoresponsive Hydrogel of Poly(Glycidyl Methacrylate-Co-N-Isopropylacrylamide) as a Nanoreactor of Gold Nanoparticles. J. Polym. Sci. A Polym. Chem. 2007, 45, 2812–2819. [Google Scholar] [CrossRef]
- Li, Y.; Hong, X.M.; Collard, D.M.; El-Sayed, M.A. Suzuki Cross-Coupling Reactions Catalyzed by Palladium Nanoparticles in Aqueous Solution. Org. Lett. 2000, 2, 2385–2388. [Google Scholar] [CrossRef]
- Caló, E.; Khutoryanskiy, V.V. Biomedical Applications of Hydrogels: A Review of Patents and Commercial Products. Eur. Polym. J. 2015, 65, 252–267. [Google Scholar] [CrossRef] [Green Version]
- Marquez, G.; Wang, L.V.; Wang, C.; Hu, Z. Development of Tissue-Simulating Optical Phantoms: Poly- N -Isopropylacrylamide Solution Entrapped inside a Hydrogel. Phys. Med. Biol. 1999, 44, 309–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Link, S.; El-Sayed, M.A. Shape and Size Dependence of Radiative, Non-Radiative and Photothermal Properties of Gold Nanocrystals. Int. Rev. Phys. Chem. 2000, 19, 409–453. [Google Scholar] [CrossRef]
- Perezjuste, J.; Pastorizasantos, I.; Lizmarzan, L.; Mulvaney, P. Gold Nanorods: Synthesis, Characterization and Applications. Coord. Chem. Rev. 2005, 249, 1870–1901. [Google Scholar] [CrossRef]
- Pissuwan, D.; Valenzuela, S.M.; Cortie, M.B. Prospects for Gold Nanorod Particles in Diagnostic and Therapeutic Applications. Biotechnol. Genet. Eng. Rev. 2008, 25, 93–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissleder, R. A Clearer Vision for in Vivo Imaging. Nat. Biotechnol 2001, 19, 316–317. [Google Scholar] [CrossRef] [PubMed]
- Pissuwan, D.; Valenzuela, S.M.; Killingsworth, M.C.; Xu, X.; Cortie, M.B. Targeted Destruction of Murine Macrophage Cells with Bioconjugated Gold Nanorods. J. Nanopart Res. 2007, 9, 1109–1124. [Google Scholar] [CrossRef]
- Huang, X.; Jain, P.K.; El-Sayed, I.H.; El-Sayed, M.A. Determination of the Minimum Temperature Required for Selective Photothermal Destruction of Cancer Cells with the Use of Immunotargeted Gold Nanoparticles. Photochem Photobiol 2006, 82, 412. [Google Scholar] [CrossRef] [PubMed]
- Dreaden, E.C.; Mackey, M.A.; Huang, X.; Kang, B.; El-Sayed, M.A. Beating Cancer in Multiple Ways Using Nanogold. Chem. Soc. Rev. 2011, 40, 3391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.-Y.; Chu, H.-C.; Kuo, T.-J.; Kuo, C.-L.; Huang, M.H. Seed-Mediated Synthesis of High Aspect Ratio Gold Nanorods with Nitric Acid. Chem. Mater. 2005, 17, 6447–6451. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, F.; Guo, Y.; Chen, R.; Shen, Y.; Guo, A.; Liu, J.; Zhang, X.; Guo, S. Controlled Synthesis of Monodispersed Gold Nanorods with Different Aspect Ratios in the Presence of Aromatic Derivatives. J. Nanoparticle Res. 2014, 16, 2806. [Google Scholar] [CrossRef] [Green Version]
- Dreaden, E.C.; Alkilany, A.M.; Huang, X.; Murphy, C.J.; El-Sayed, M.A. The Golden Age: Gold Nanoparticles for Biomedicine. Chem. Soc. Rev. 2012, 41, 2740–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.-S.; Shih, C.-W.; Chen, C.-D.; Lai, W.-C.; Wang, C.R.C. The Shape Transition of Gold. Nanorods Langmuir 1999, 15, 701–709. [Google Scholar] [CrossRef]
- Alkilany, A.M.; Murphy, C.J. Toxicity and Cellular Uptake of Gold Nanoparticles: What We Have Learned so Far? J. Nanopart Res. 2010, 12, 2313–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isomaa, B.; Reuter, J.; Djupsund, B.M. The Subacute and Chronic Toxicity of Cetyltrimethylammonium Bromide (CTAB), a Cationic Surfactant, in the Rat. Arch. Toxicol. 1976, 35, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhao, P.; Astruc, D. Anisotropic Gold Nanoparticles: Synthesis, Properties, Applications, and Toxicity. Angew. Chem. Int. Ed. 2014, 53, 1756–1789. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Sharma, A.; Glomm, W. The Influence of Differently Shaped Gold Nanoparticles Functionalized with NIPAM-Based Hydrogels on the Release of Cytochrome C. Gels 2017, 3, 42. [Google Scholar] [CrossRef] [Green Version]
- Rostro-Kohanloo, B.C.; Bickford, L.R.; Payne, C.M.; Day, E.S.; Anderson, L.J.E.; Zhong, M.; Lee, S.; Mayer, K.M.; Zal, T.; Adam, L.; et al. The Stabilization and Targeting of Surfactant-Synthesized Gold Nanorods. Nanotechnology 2009, 20, 434005. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.-S.; Iqbal, M.; Lee, T.; Kim, Y.H.; Tae, G. Removal of Cetyltrimethylammonium Bromide to Enhance the Biocompatibility of Au Nanorods Synthesized by a Modified Seed Mediated Growth Process. J. Nanosci. Nanotechnol. 2008, 8, 4670–4674. [Google Scholar] [CrossRef]
- Todor, I.; Szabo, L.; Marisca, O.T.; Chis, V.; Leopold, N. Gold nanoparticle assemblies of controllable size obtained by hydroxylamine reduction at room temperature. J. Nanopart. Res. 2014, 16, 2740. [Google Scholar] [CrossRef]
- Shankar, S.S.; Rai, A.; Ankamwar, B.; Singh, A.; Ahmad, A.; Sastry, M. Biological Synthesis of Triangular Gold Nanoprisms. Nat. Mater. 2004, 3, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.B.; Sakthivel, N. Phytosynthesis of Gold Nanoparticles Using Leaf Extract of Coleus Amboinicus Lour. Mater. Charact. 2010, 61, 1232–1238. [Google Scholar] [CrossRef]
- Kitching, M.; Ramani, M.; Marsili, E. Fungal Biosynthesis of Gold Nanoparticles: Mechanism and Scale up: Fungal Biosynthesis of AuNPs. Microb. Biotechnol. 2015, 8, 904–917. [Google Scholar] [CrossRef]
- Fazal, S.; Jayasree, A.; Sasidharan, S.; Koyakutty, M.; Nair, S.V.; Menon, D. Green Synthesis of Anisotropic Gold Nanoparticles for Photothermal Therapy of Cancer. Acs Appl. Mater. Interfaces 2014, 6, 8080–8089. [Google Scholar] [CrossRef]
- Dong, S.; Tang, C.; Zhou, H.; Zhao, H. Photochemical Synthesis of Gold Nanoparticles by the Sunlight Radiation Using a Seeding Approach. Gold Bull. 2004, 37, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Eustis, S.; Hsu, H.-Y.; El-Sayed, M.A. Gold Nanoparticle Formation from Photochemical Reduction of Au 3+ by Continuous Excitation in Colloidal Solutions. A Proposed Molecular Mechanism. J. Phys. Chem. B 2005, 109, 4811–4815. [Google Scholar] [CrossRef] [PubMed]
- Pal, A. Photochemical Synthesis of Gold Nanoparticles via Controlled Nucleation Using a Bioactive Molecule. Mater. Lett. 2004, 58, 529–534. [Google Scholar] [CrossRef]
- Kim, F.; Song, J.H.; Yang, P. Photochemical Synthesis of Gold Nanorods. J. Am. Chem. Soc. 2002, 124, 14316–14317. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Shen, Y.; Xie, A.; Qiu, L.; Zhang, Q.; Zhang, S. Photoinduced Synthesis of Anisotropic Gold Nanoparticles in Room-Temperature Ionic Liquid. J. Phys. Chem. C 2007, 111, 7629–7633. [Google Scholar] [CrossRef]
- Zhai, Y.; DuChene, J.S.; Wang, Y.-C.; Qiu, J.; Johnston-Peck, A.C.; You, B.; Guo, W.; DiCiaccio, B.; Qian, K.; Zhao, E.W.; et al. Polyvinylpyrrolidone-Induced Anisotropic Growth of Gold Nanoprisms in Plasmon-Driven Synthesis. Nat. Mater. 2016, 15, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Narain, R. Rapid Synthesis of Gold Nanorods Using a One-Step Photochemical Strategy. Langmuir 2010, 26, 18392–18399. [Google Scholar] [CrossRef]
- Abdelrasoul, G.N.; Cingolani, R.; Diaspro, A.; Athanassiou, A.; Pignatelli, F. Photochemical Synthesis: Effect of UV Irradiation on Gold Nanorods Morphology. J. Photochem. Photobiol. A Chem. 2014, 275, 7–11. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, K.; Guan, Y.; Zhang, Y. Assembling of Gold Nanorods on P(NIPAM-AAPBA) Microgels: A Large Shift in the Plasmon Band and Colorimetric Glucose Sensing. RSC Adv. 2012, 2, 4768–4776. [Google Scholar] [CrossRef]
- Zhang, H.; Guo, S.; Fu, S.; Zhao, Y. A Near-Infrared Light-Responsive Hybrid Hydrogel Based on UCST Triblock Copolymer and Gold Nanorods. Polymer 2017, 9, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras-Caceres, R.; Pastoriza-Santos, I.; Perez-Juste, J.; Fernandez-Barbero, A.; Liz-Marzan, L. Au@pNIPAM Thermosensitive Nanostructures: Control Over Shell Cross-linking, Overall Dimensions, and Core Growth. Adv. Funct. Mat. 2009, 19, 3070–3076. [Google Scholar] [CrossRef]
- Kawano, T.; Niidome, Y.; Mori, T.; Katayama, Y.; Niidome, T. PNIPAM Gel-Coated Gold Nanorods for Targeted Delivery Responding to a Near-Infrared Laser. Bioconjugate Chem. 2009, 20, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Qian, Y.; Gao, Q.; Dong, J.; Qian, W. In Situ Controllable Preparation of Gold Nanorods in Thermo-Responsive Hydrogels and Their Application in Surface Enhanced Raman Scattering. J. Mater. Chem. 2010, 20, 8711. [Google Scholar] [CrossRef]
- Fernández-López, C.; Polavarapu, L.; Solís, D.M.; Taboada, J.M.; Obelleiro, F.; Contreras-Cáceres, R.; Pastoriza-Santos, I.; Pérez-Juste, J. Gold Nanorod–PNIPAM Hybrids with Reversible Plasmon Coupling: Synthesis, Modeling, and SERS Properties. Acs Appl. Mater. Interfaces 2015, 7, 12530–12538. [Google Scholar] [CrossRef] [PubMed]
- Vogler, A.; Quett, C.; Kunkely, H. Photochemistry of Azide Complexes of Gold, Silver, Platinum, and Palladium. Generation of the Metallic State. Ber. Bunsengesphys. Chem. 1988, 92, 1486–1492. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-U.; Cha, S.-H.; Shin, K.; Jho, J.Y.; Lee, J.-C. Synthesis of Gold Nanoparticles from Gold(I)-Alkanethiolate Complexes with Supramolecular Structures through Electron Beam Irradiation in TEM. J. Am. Chem. Soc. 2005, 127, 9962–9963. [Google Scholar] [CrossRef]
- Lu, X.; Tuan, H.-Y.; Korgel, B.A.; Xia, Y. Facile Synthesis of Gold Nanoparticles with Narrow Size Distribution by Using AuCl or AuBr as the Precursor. Chem. Eur. J. 2008, 14, 1584–1591. [Google Scholar] [CrossRef]
- Elbjeirami, O.; Omary, M.A. Photochemistry of Neutral Isonitrile Gold(I) Complexes: Modulation of Photoreactivity by Aurophilicity and π-Acceptance Ability. J. Am. Chem. Soc. 2007, 129, 11384–11393. [Google Scholar] [CrossRef]
- Marpu, S.B. Biocompatible Hybrid Nanomaterials Involving Polymers and Hydrogels Interfaced with Phosphorescent Complexes and Toxin-Free Metallic Nanoparticles for Biomedical Applications. Ph.D. Thesis, Denton, TX, USA, August 2011. [Google Scholar]
- Gao, J.; Frisken, B.J. Influence of Reaction Conditions on the Synthesis of Self-Cross-Linked N -Isopropylacrylamide Microgels. Langmuir 2003, 19, 5217–5222. [Google Scholar] [CrossRef]
- Xia, X.; Hu, Z.; Marquez, M. Physically Bonded Nanoparticle Networks: A Novel Drug Delivery System. J. Control. Release 2005, 103, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Hu, Z.; Zhou, B.; Cheng, Z.; Wu, J.; Marquez, M. Melting Kinetics of Thermally Responsive Microgel Crystals. Macromolecules 2007, 40, 9544–9548. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, G.; Marquez, M.; Hu, Z. The Formation of Crystalline Hydrogel Films by Self-Crosslinking Microgels. Soft Matter 2009, 5, 820. [Google Scholar] [CrossRef]
- WHO | JECFA. Available online: https://apps.who.int/food-additives-contaminants-jecfa-database/chemical.aspx?chemID=1966 (accessed on 19 June 2020).
- Tong, L.; Zhao, Y.; Huff, T.B.; Hansen, M.N.; Wei, A.; Cheng, J.-X. Gold Nanorods Mediate Tumor Cell Death by Compromising Membrane Integrity. Adv. Mater. 2007, 19, 3136–3141. [Google Scholar] [CrossRef]
- Huang, X.; Neretina, S.; El-Sayed, M.A. Gold Nanorods: From Synthesis and Properties to Biological and Biomedical Applications. Adv. Mater. 2009, 21, 4880–4910. [Google Scholar] [CrossRef]
- Energy Dispersive X-Ray Periodic Table, Hitachi. Available online: https://www.bruker.com/fileadmin/user_upload/8-PDF-Docs/X-rayDiffraction_ElementalAnalysis/HH-XRF/Misc/Periodic_Table_and_X-ray_Energies.pdf (accessed on 18 January 2020).
- Shan, J.; Tenhu, H. Recent Advances in Polymer Protected Gold Nanoparticles: Synthesis, Properties and Applications. Chem. Commun. 2007, 4580–4598. [Google Scholar] [CrossRef]
- Saxena, S.K. Polyvinyl Alcohol, Chemical and Technical Assessment. FAO. 2004. Available online: http://www.fao.org/fileadmin/templates/agns/pdf/jecfa/cta/61/PVA.pdf (accessed on 19 June 2010).
- Brody, J.R.; Kern, S.E. History and Principles of Conductive Media for Standard DNA Electrophoresis. Anal. Biochem. 2004, 333, 1–13. [Google Scholar] [CrossRef]
- Pharm, K. Hydroxypropyl Cellulose; Hercules Incorporated: Wilmington, DE, USA, 2004; p. 494. Available online: https://www.stobec.com/DATA/PRODUIT/1557~v~data_8524.pdf (accessed on 21 June 2010).
- Housni, A.; Ahmed, M.; Liu, S.; Narain, R. Monodisperse Protein Stabilized Gold Nanoparticles vis a Simple Photochemical Process. J. Phys. Chem. C 2008, 112, 12282–12290. [Google Scholar] [CrossRef]
- Jans, H.; Jans, K.; Lagae, L.; Borghs, G.; Maes, G.; Huo, Q. Poly(acryliac acid)-stabilized Colloidal Gold Nanoparticles: Synthesis and Properties. Nanotechnology 2010, 21, 455702. [Google Scholar] [CrossRef]
- Khanna, P.; Gokhale, R.; Subbarao, V.; Vishwanath, A.K.; Das, B.; Satyanarayana, C. PVA Stabilized Gold Nanoparticles by use of Unexplored Albeit Conventional Reducing Agent. Mater. Chem. Phys. 2005, 92, 229–233. [Google Scholar] [CrossRef]
- Kuo, C.-H.; Chiang, T.-F.; Chen, L.-J.; Huang, M.H. Synthesis of Highly Faceted Pentagonal and Hexagonal-Shaped Gold Nanoparticles with Controlled Sized by Sodium Dodecyl Sulfate. Langmuir 2004, 20, 7820–7824. [Google Scholar] [CrossRef] [PubMed]
- Pucci, A.; Bernabò, M.; Elvati, P.; Meza, L.I.; Galembeck, F.; Leite, C.A.D.P.; Tirelli, N.; Ruggeri, G. Photoinduced Formation of Gold Nanoparticles into Vinyl Alcohol Based Polymers. J. Mater. Chem. 2006, 16, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- Pal, A.; Esumi, K.; Pal, T. Preparation of Nanosized Gold Particles in a Biopolymer Using UV Photoactivation. J. Colloid Interface Sci. 2005, 288, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Mordoukhovski, L.; Kumacheva, E. Sequestering Gold Nanorods by Polymer Microgels. Adv. Mater. 2008, 20, 2371–2375. [Google Scholar] [CrossRef]
- Gorelikov, I.; Field, L.M.; Kumacheva, E. Hybrid Microgels Photoresponsive in the Near-Infrared Spectral Range. J. Am. Chem. Soc. 2004, 126, 15938–15939. [Google Scholar] [CrossRef] [PubMed]
- Shiotani, A.; Mori, T.; Niidome, T.; Niidome, Y.; Katayama, Y. Stable Incorporation of Gold Nanorods into N-Isopropylacrylamide Hydrogels and Their Rapid Shrinkage Induced by a Near-Infrared Laser Irradiation. Langmuir 2007, 23, 4012–4018. [Google Scholar] [CrossRef]
- Hirsch, R.L.; Gobin, M.A.; Lowery, R.A.; Tam, F.; Drezek, A.R.; Halas, J.N.; West, L.J. Metal Nanoshells. Ann. Biomed. Eng. 2006, 34, 15–22. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stabilizing Media | Experimental Condition | SPR λmax (nm) | FWHM * (nm) | FWHM * (cm−1) | TEM davg (nm) | PQY * |
|---|---|---|---|---|---|---|
| PNIPAm-co-allylamine microgels | Ambient light | 575 | 676 | 5825 | 21.0 ± 6.0 | NA |
| PNIPAm-co-allylamine microgels | Photoirradiation | 535 | 591 | 4584 | 12.1 ± 4.0 | 0.07 |
| PNIPAm-co-acrylic acid microgels | Photoirradiation | 533 | 595 | 4932 | 10.6 ± 3.7 | 0.1 |
| Stabilizing Media | Experimental Condition (Time, Minutes) | SPR ʎmax * (nm) | FWHM * (nm) | FWHM * (cm−1) | TEM davg (nm) |
|---|---|---|---|---|---|
| Alginic acid (ALA) | Photoirradiation (15 min) | 521 | 96 | 3680 | 4.1 ± 1.6 |
| Chitosan (CSN) | Photoirradiation (45 min) | 522 | 102 | 3883 | 19.8 ± 1.6 |
| Polyacrylic acid (PAA) | Photoirradiation (15 min) | 527 | 130 | 4864 | 6.5 ± 2.2 |
| Sodium dodecyl sulfate (SDS) | Heating (10 min) | 535 | 122 | 4222 | Nonspherical, fused 20 ± 4 |
| Polyvinyl alcohol (PVA) | Heating (20 min) | 538 | 166 | 5772 | Nonspherical, 25 ± 5 |
| Hydroxypropyl cellulose (HPC) | Photoirradiation (30 min) | 547 | 210 | 6764 | Nonspherical, fused, 30 ± 5 |
| Bovine serum albumin (BSA) | Photoirradiation (90 min) | 580 | 178 | 5142 | Nonspherical, 35 ± 5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marpu, S.B.; Kamras, B.L.; MirzaNasiri, N.; Elbjeirami, O.; Simmons, D.P.; Hu, Z.; Omary, M.A. Single-Step Photochemical Formation of Near-Infrared-Absorbing Gold Nanomosaic within PNIPAm Microgels: Candidates for Photothermal Drug Delivery. Nanomaterials 2020, 10, 1251. https://doi.org/10.3390/nano10071251
Marpu SB, Kamras BL, MirzaNasiri N, Elbjeirami O, Simmons DP, Hu Z, Omary MA. Single-Step Photochemical Formation of Near-Infrared-Absorbing Gold Nanomosaic within PNIPAm Microgels: Candidates for Photothermal Drug Delivery. Nanomaterials. 2020; 10(7):1251. https://doi.org/10.3390/nano10071251
Chicago/Turabian StyleMarpu, Sreekar B., Brian Leon Kamras, Nooshin MirzaNasiri, Oussama Elbjeirami, Denise Perry Simmons, Zhibing Hu, and Mohammad A. Omary. 2020. "Single-Step Photochemical Formation of Near-Infrared-Absorbing Gold Nanomosaic within PNIPAm Microgels: Candidates for Photothermal Drug Delivery" Nanomaterials 10, no. 7: 1251. https://doi.org/10.3390/nano10071251