Effect of Isotopic Substitution on Elementary Processes in Dye-Sensitized Solar Cells: Deuterated Amino-Phenyl Acid Dyes on TiO2

Abstract
:1. Introduction
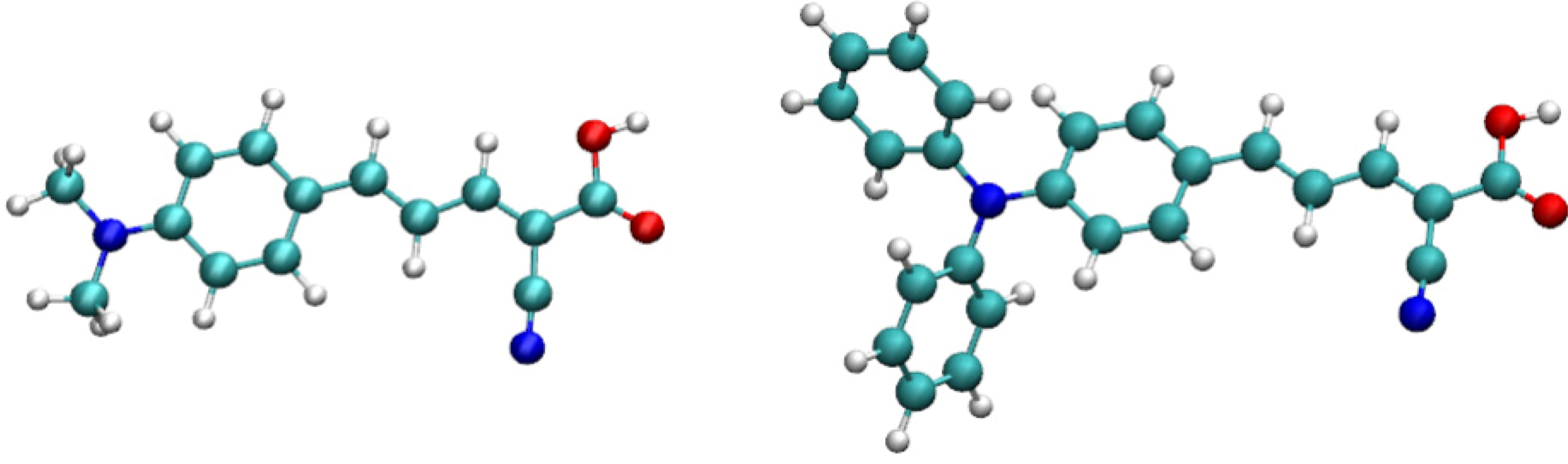
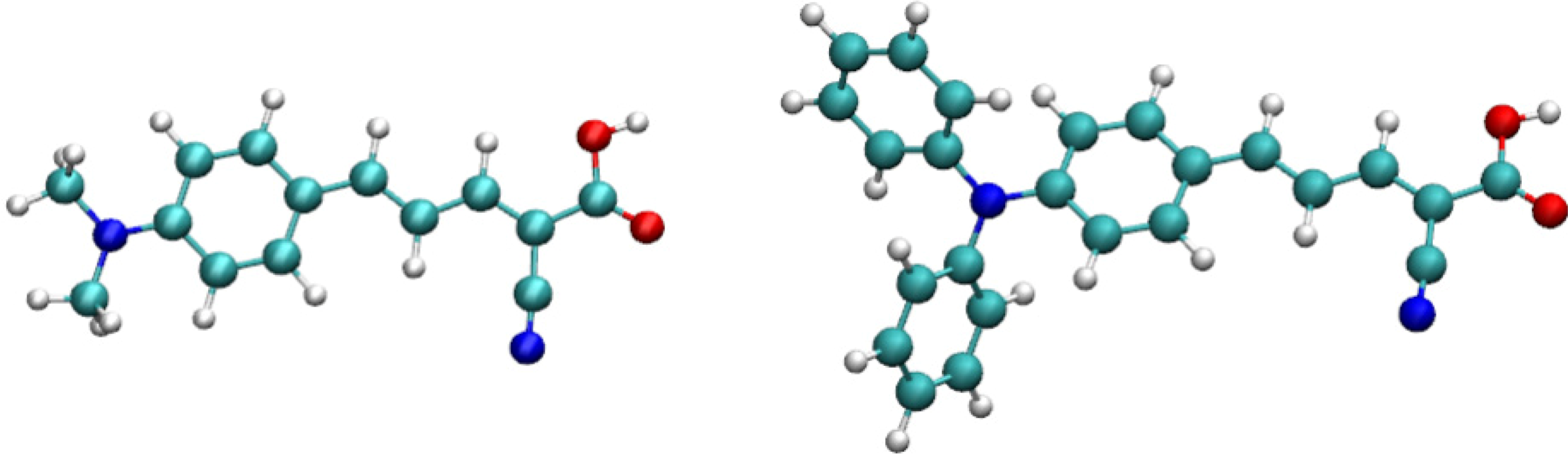
2. Theoretical and Computational Methods
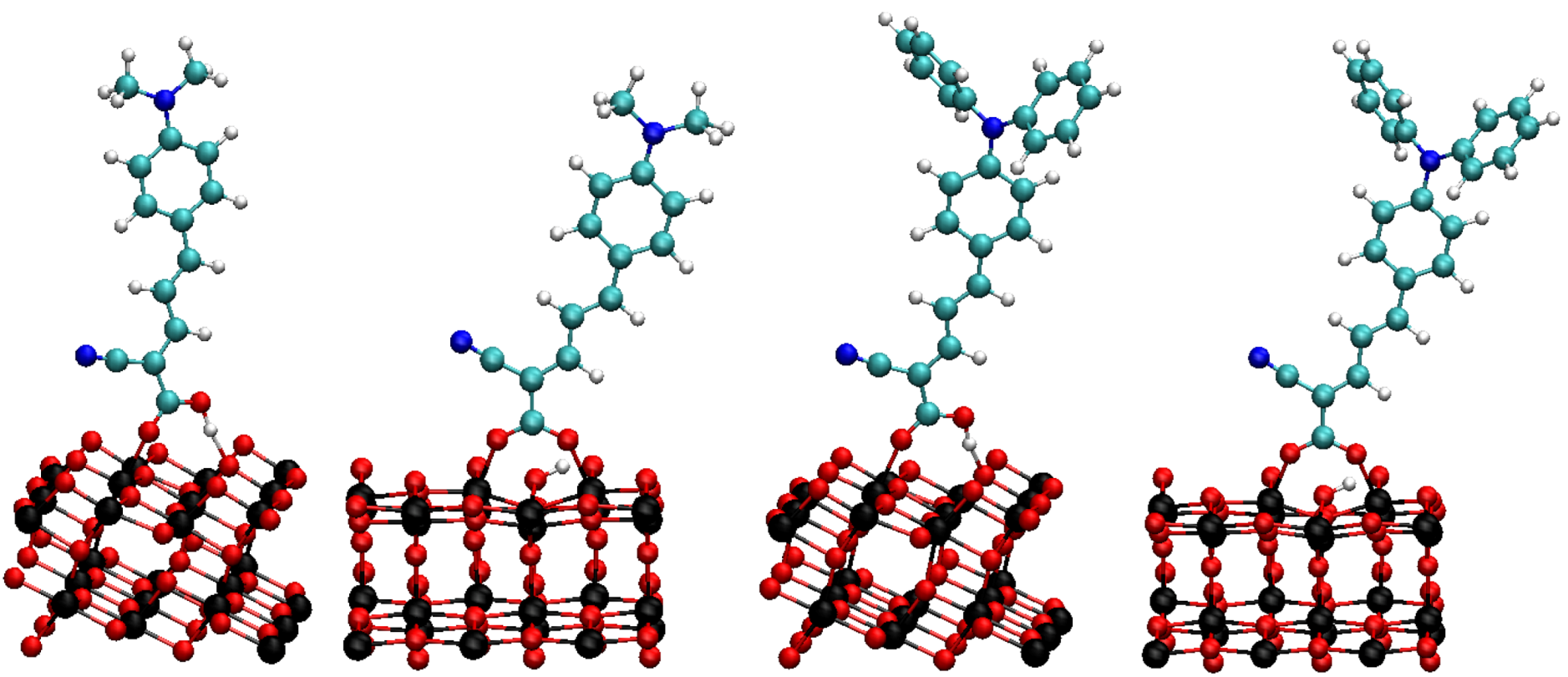

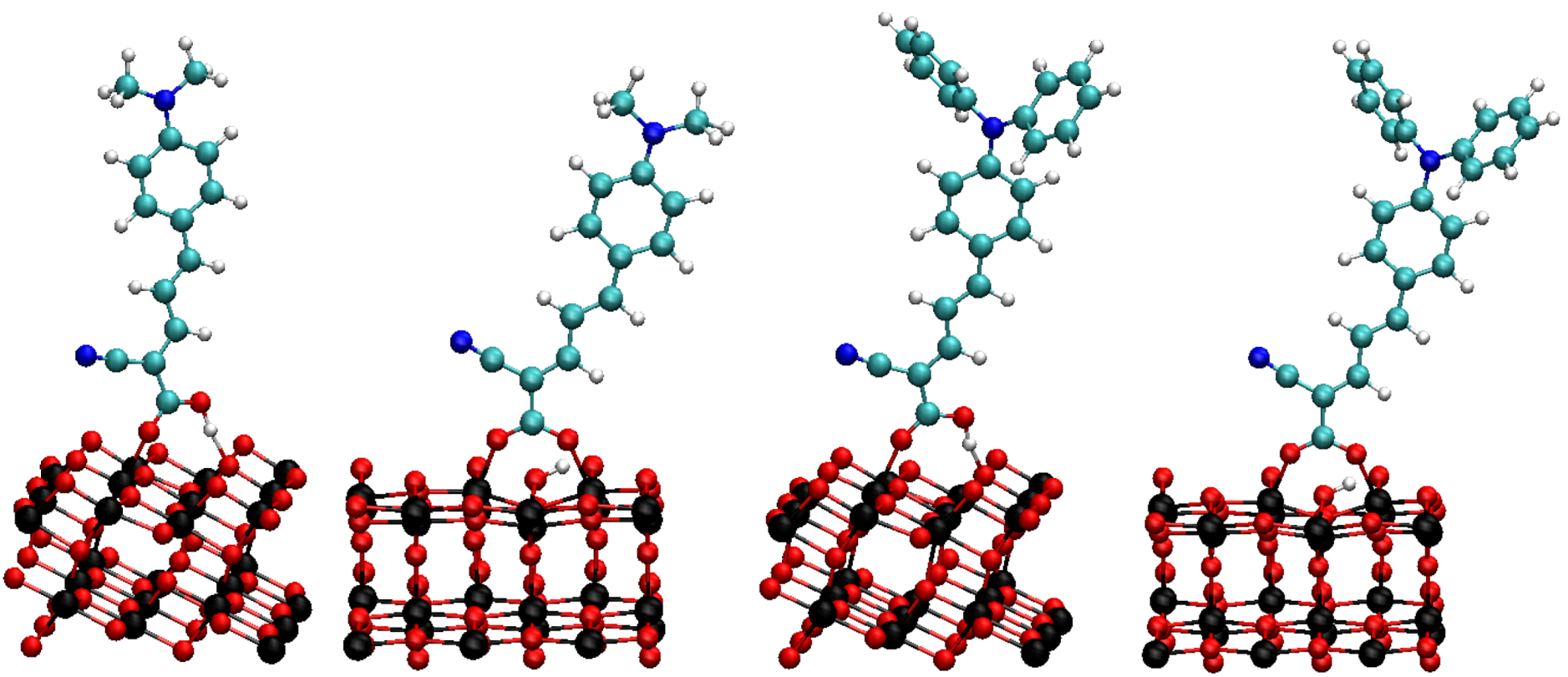
3. Results and Discussion
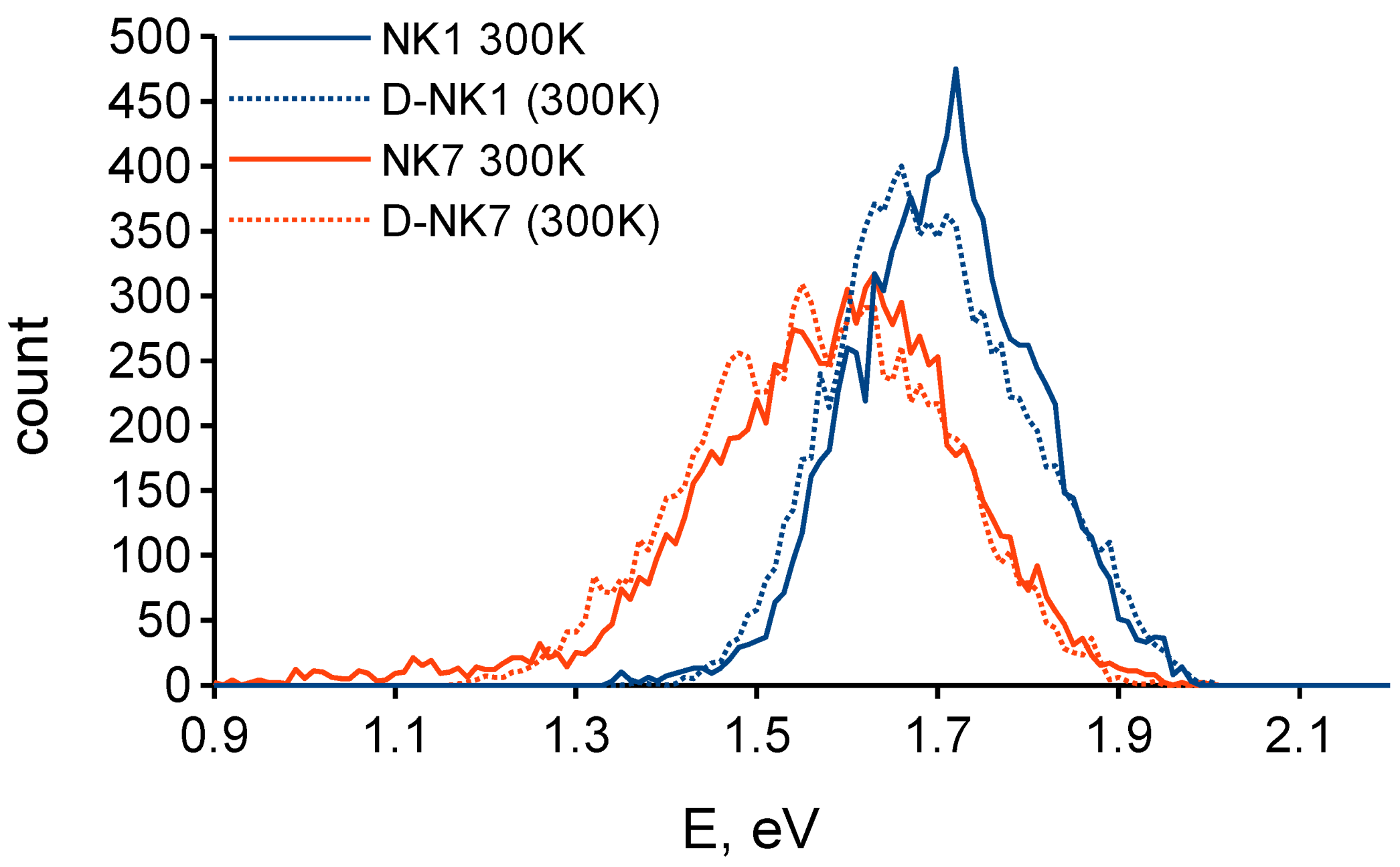
3.1. Effect of Deuteration on Absorption Spectra and Dye Regeneration
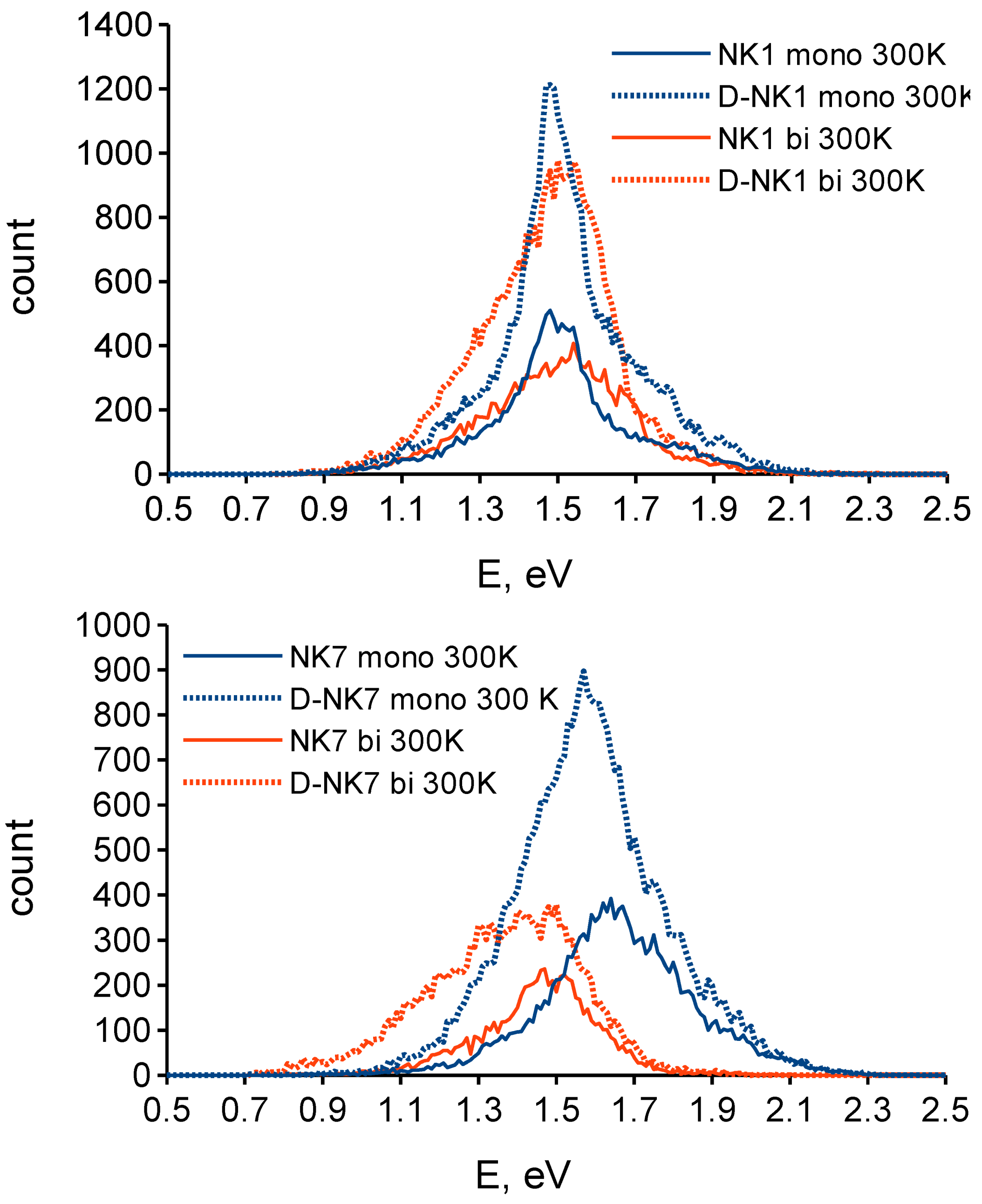

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
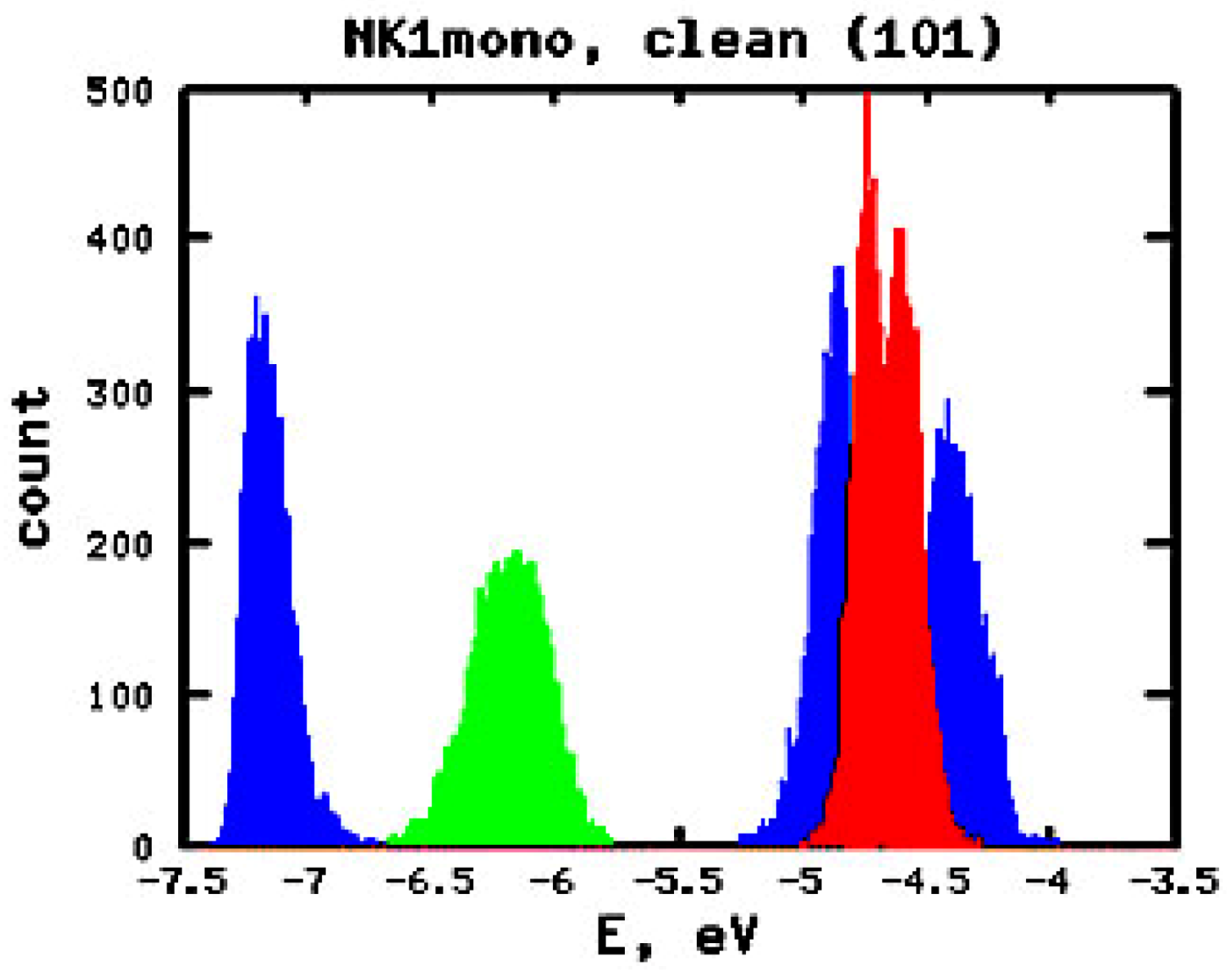
| System | Equilibrium/MD averaged (non-deuterated)/MD averaged (deuterated) | ||||
|---|---|---|---|---|---|
| HOMO | LUMO* | gap | CBM | ΔGi | |
| NK1 mono | −6.26/−6.16/−6.18 | −4.56/−4.67/−4.67 | 1.70/1.48/1.52 | −4.69/−4.89/−4.88 | 0.13/0.23/0.22 |
| NK1 bi | −6.11/−6.03/−6.06 | −4.41/−4.56/−4.58 | 1.70/1.47/1.48 | −4.84/−4.98/−5.00 | 0.43/0.42/0.42 |
| NK7 mono | −6.19/−6.23/−6.19 | −4.60/−4.58/−4.61 | 1.59/1.66/1.59 | −4.72/−4.79/−4.82 | 0.11/0.21/0.22 |
| NK7 bi | −6.10/−6.04/−6.02 | −4.57/−4.59/−4.63 | 1.53/1.45/1.39 | −4.84/−4.92/−4.94 | 0.28/0.32/0.31 |
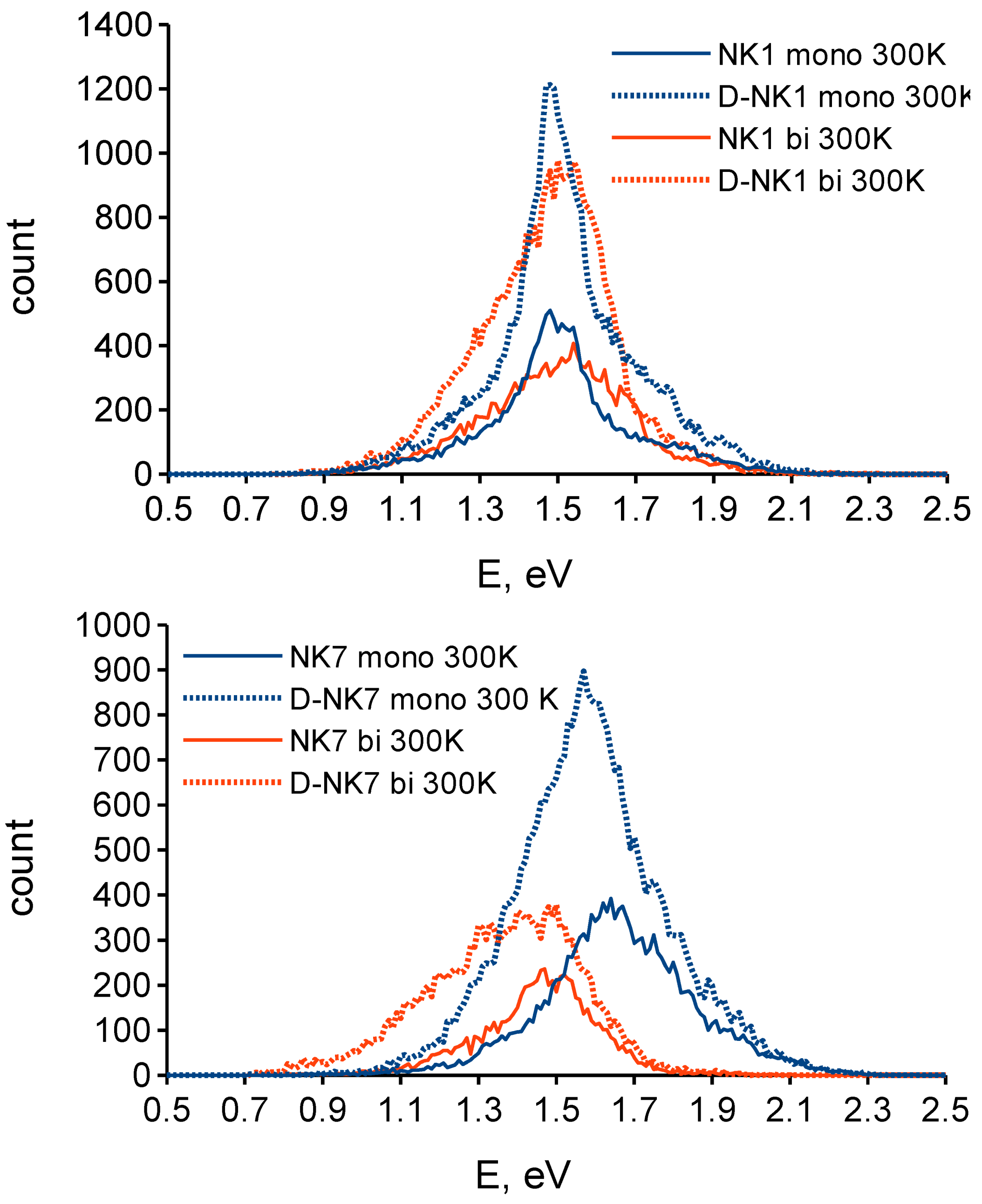
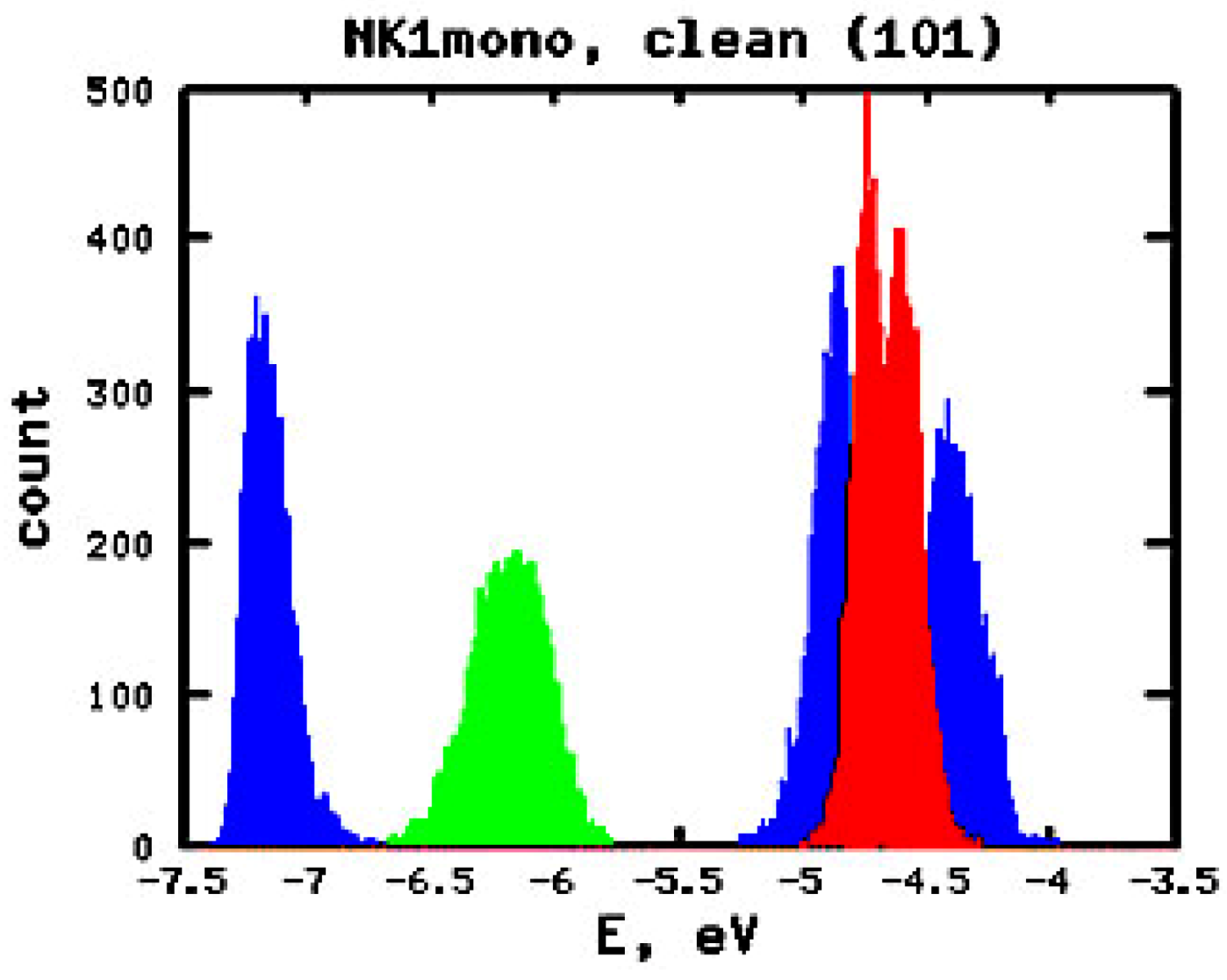
3.2. Effect of Deuteration on Injection Conditions
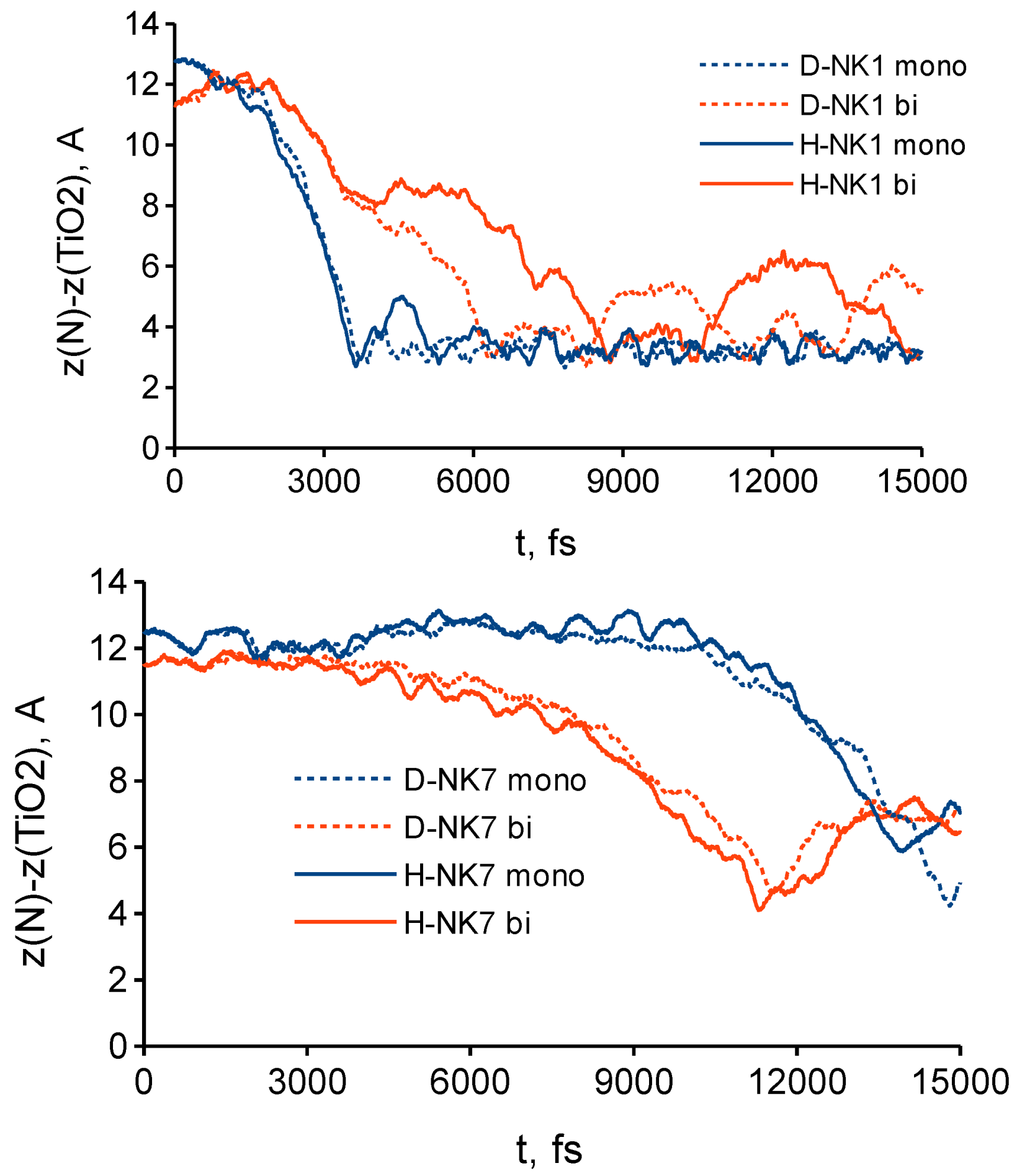
3.3. Effect of Deuteration on Orientational Motions and Recombination to the Dye Cation
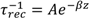

4. Conclusions
Acknowledgments
Conflict of Interest
References
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pattersson, H. Dye-sensitized solar cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef]
- Mora-Sero, I.; Bisquert, J. Breakthroughs in the development of semiconductor-sensitized solar cells. J. Phys. Chem. Lett. 2010, 1, 3046–3052. [Google Scholar] [CrossRef]
- Peter, L.M. The Grätzel cell: Where next? J. Phys. Chem. Lett. 2011, 2, 1861–1867. [Google Scholar] [CrossRef] [Green Version]
- Yella, A.; Lee, H.-W.; Tsao, H.N.; Yi, C.; Chandiran, A.K.; Nazeeruddin, M.K.; Diau, E.W.-G.; Yeh, C.-Y.; Zakeeruddin, S.M.; Grätzel, M. Porphyrin-sensitized solar cells with cobalt (II/III)–based redox electrolyte exceed 12 percent efficiency. Science 2011, 334, 629–634. [Google Scholar] [CrossRef]
- Yum, J.-H.; Baranoff, E.; Kessler, F.; Moehl, T.; Ahmad, S.; Bessho, T.; Marchioro, A.; Ghadiri, E.; Moser, J.-E.; Yi, C.; Nazeeruddin, Md.K.; Grätzel, M. A cobalt complex redox shuttle for dye-sensitized solar cells with high open-circuit potentials. Nat. Commun. 2012, 3, 631:1–631:8. [Google Scholar]
- Martsinovich, N.; Troisi, A. Theoretical studies of dye-sensitised solar cells: From electronic structure to elementary processes. Energy Environ. Sci. 2011, 4, 4473–4495. [Google Scholar] [CrossRef]
- Cong, J.; Yang, X.; Kloo, L.; Sun, L. Iodine/iodide-free redox shuttles for liquid electrolyte-based dye-sensitized solar cells. Energy Environ. Sci. 2012, 5, 9180–9194. [Google Scholar] [CrossRef]
- Wang, M.; Grätzel, C.; Zakeeruddin, S.M.; Grätzel, M. Recent developments in redox electrolytes for dye-sensitized solar cells. Energy Environ. Sci. 2012, 5, 9394–9405. [Google Scholar] [CrossRef]
- Prezhdo, O.V.; Duncan, W.R.; Prezhdo, V.V. Photoinduced electron dynamics at semiconductor interfaces: A time-domain ab initio prospective. Progr. Surf. Sci. 2009, 84, 30–68. [Google Scholar] [CrossRef]
- Duncan, W.R.; Prezhdo, O.V. Theoretical studies of photoinduced electron transfer in dye-sensitized TiO2. Annu. Rev. Phys. Chem. 2008, 58, 143–184. [Google Scholar] [CrossRef]
- Manzhos, S.; Segawa, H.; Yamashita, K. A model for recombination in Type II dye-sensitized solar cells: Catechol-thiophene dyes. Chem. Phys. Lett. 2011, 504, 230–235. [Google Scholar] [CrossRef]
- Manzhos, S.; Segawa, H.; Yamashita, K. Derivative coupling constants of NK1, NK7 dyes and their relation to excited state dynamics in solar cell applications. Chem. Phys. Lett. 2011, 501, 580–586. [Google Scholar] [CrossRef]
- Manzhos, S.; Fujisawa, J.; Segawa, H.; Yamashita, K. Isotopic substitution as a strategy to control non-adiabatic dynamics in photoelectrochemical cells: Surface complexes between TiO2 and dicyanomethylene compounds. Jpn. J. Appl. Phys. 2012, 51, 10NE03:1–10NE03:6. [Google Scholar]
- Manzhos, S.; Jono, R.; Yamashita, K.; Fujisawa, J.; Nagata, M.; Segawa, H. A study of interfacial charge transfer bands and electron recombination in the surface complexes of TCNE, TCNQ, and TCNAQ with TiO2. J. Phys. Chem. C 2011, 115, 21487–21493. [Google Scholar]
- Niu, Y.; Peng, Q.; Deng, C.; Gao, X.; Shuai, Z. Theory of excited state decays and optical apectra: Application to polyatomic molecules. J. Phys. Chem. A 2010, 114, 7817–7831. [Google Scholar]
- Manzhos, S.; Segawa, H.; Yamashita, K. Computational dye design by changing the conjugation order: Failure of LR-TDDFT to predict relative excitation energies in organic dyes differing by the position of the methine unit. Chem. Phys. Lett. 2012, 527, 51–56. [Google Scholar] [CrossRef]
- Manzhos, S.; Segawa, H.; Yamashita, K. Effect of nuclear vibrations, temperature, co-adsorbed water, and dye orientation on light absorption, charge injection and recombination conditions in organic dyes on TiO2. Phys. Chem. Chem. Phys. 2013, 15, 1141–1147. [Google Scholar] [CrossRef]
- Manzhos, S.; Segawa, H.; Yamashita, K. The effect of ligand substitution and water co-adsorption on the adsorption dynamics and energy level matching of amino-phenyl acid dyes on TiO2. Phys. Chem. Chem. Phys. 2012, 14, 1749–1755. [Google Scholar] [CrossRef]
- Manzhos, S.; Segawa, H.; Yamashita, K. Effect of nuclear vibrations, temperature, and orientation on injection and recombination conditions in amino-phenyl acid dyes on TiO2. Proc. SPIE 2012, 8438, 843814:1–843814:10. [Google Scholar]
- Lim, K.; Kim, C.; Song, J.; Yu, T.; Lim, W.; Song, K.; Wang, P.; Zu, N.; Ko, J. Enhancing the performance of organic dye-sensitized solar cells via a slight structure modification. J. Phys. Chem. C 2011, 115, 22640–22646. [Google Scholar]
- Zhang, M.-D.; Pan, H.; Ju, X.-H.; Ji, Y.-J.; Qin, L.; Zheng, H.-G.; Zhou, X.-F. Improvement of dye-sensitized solar cells' performance through introducing suitable heterocyclic groups to triarylamine dyes. Phys. Chem. Chem. Phys. 2012, 14, 2809–2815. [Google Scholar]
- Myllyperkio, P.; Manzoni, C.; Polli, D.; Cerullo, G.; Korppi-Tommola, J. Electron transfer from organic aminophenyl acid sensitizers to titanium dioxide nanoparticle films. J. Phys. Chem. C 2009, 113, 13985–13992. [Google Scholar]
- Han, L.; Islam, A.; Chen, H.; Malapaka, C.; Chiranjeevi, B.; Zhang, S.; Yang, X.; Yanagida, M. High-efficiency dye-sensitized solar cell with a novel co-adsorbent. Energy Environ. Sci. 2012, 5, 6057–6060. [Google Scholar] [CrossRef]
- Long, H.; Zhou, D.; Zhang, M.; Peng, C.; Uchida, S.; Wang, P. Probing dye-correlated interplay of energetics and kinetics in mesoscopic titania solar cells with 4-tert-butylpyridine. J. Phys. Chem. C 2011, 115, 14408–14414. [Google Scholar]
- Ren, X.; Feng, Q.; Zhou, G.; Huang, C.-H.; Wang, Z.-S. Effect of cations in coadsorbate on charge recombination and conduction band edge movement in dye-sensitized solar cells. J. Phys. Chem. C 2010, 114, 7190–7195. [Google Scholar]
- Asuduzzaman, A.M.; Schreckenbach, G. Computational studies on the interactions among redox couples, additives and TiO2: Implications for dye-sensitized solar cells. Phys. Chem. Chem. Phys. 2010, 12, 14609–14618. [Google Scholar]
- Shizu, K.; Sato, T.; Tanaka, K. Vibronic coupling density analysis for α-oligothiophene cations: A new insight for polaronic defects. Chem. Phys. 2010, 369, 108–121. [Google Scholar] [CrossRef]
- Abe, T.; Miyazawa, A.; Konno, H.; Kawanishi, Y. Deuteration isotope effect on nonradiative transition of fac-tris (2-phenylpyridinato) iridium (III) complexes. Chem. Phys. Lett. 2010, 491, 199–202. [Google Scholar]
- Tong, C.C.; Hwang, K.C. Enhancement of OLED efficiencies and high-voltage stabilities of light-emitting materials by deuteration. J. Phys. Chem. C 2007, 111, 3490–3494. [Google Scholar] [CrossRef]
- Browne, W.R.; Passaniti, P.; Gandolfi, M.T.; Ballardini, R.; Henry, W.; Guckian, A.; O'Boyle, N.; McGarvey, J.J.; Vos, J.G. Probing inter-ligand excited state interaction in homo and heteroleptic ruthenium(II) polypyridyl complexes using selective deuteriation. Inorg. Chim. Acta 2007, 360, 1183–1190. [Google Scholar] [CrossRef]
- Browne, W.R.; Vos, J.G. The effect of deuteriation on the emission lifetime of inorganic compounds. Coord. Chem. Rev. 2001, 219–221, 761–787. [Google Scholar]
- Keyes, T.E.; O’Connor, C.M.; O’Dwyer, U.; Coates, C.G.; Callaghan, P.; McGarvey, J.J.; Vos, J.G. Isotope and temperature dependence of dual emission in a mononuclear ruthenium(II) polypyridyl compound. J. Phys. Chem. A 1999, 103, 8915–8920. [Google Scholar]
- Tang, C.W.; VanSlyke, S.A. Organic electroluminescent diodes. Appl. Phys. Lett. 1987, 51, 913:1–913:3. [Google Scholar]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Dale, J.D.; Garcia, A.; Junquera, J.; Ordejon, P.; Sanchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys.: Condens. Matter. 2002, 14, 2745–2779. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhoff, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Artacho, E.; Anglada, E.; Dieguez, O.; Gale, J.D.; Garcia, A.; Junquera, J.; Martin, R.M.; Ordejon, P.; Pruneda, J.M.; Sanchez-Portal, D.; Soler, J.M. The SIESTA method: Developments and applicability. J. Phys.: Condens. Matter. 2008, 20, 064208:1–064208:6. [Google Scholar]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef]
- Lazzeri, M.; Vittadini, A.; Selloni, A. Structure and energetics of stoichiometric TiO2 anatase surfaces. Phys. Rev. B 2001, 63, 155409:1–155409:9. [Google Scholar]
- Perron, H.; Domain, C.; Roques, J.; Drot, R.; Simoni, E.; Catalette, H. Optimisation of accurate rutile TiO2 (110), (100), (101) and (001) surface models from periodic DFT calculations. Theor. Chem. Acc. 2007, 117, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Xiao, D.; Martini, L.A.; Snoeberger, R.C., III; Crabtree, R.H.; Batista, V.S. Inverse design and synthesis of acac-Coumarin anchors for robust TiO2 sensitization. J. Am. Chem. Soc. 2011, 133, 9014–9022. [Google Scholar]
- Pastore, M.; de Angelis, F. Aggregation of organic dyes on TiO2 in dye-sensitized solar cells models: An ab initio investigation. ACS Nano 2010, 4, 556–562. [Google Scholar]
- De Angelis, F. Direct vs. indirect injection mechanisms in perylene dye-sensitized solar cells: A DFT/TDDFT investigation. Chem. Phys. Lett. 2010, 493, 323–327. [Google Scholar] [CrossRef]
- De Angelis, F.; Tilocca, A.; Selloni, A. Time-Dependent DFT study of [Fe(CN)6]4- sensitization of TiO2 nanoparticles. J. Am. Chem. Soc. 2004, 126, 15024–15025. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, X.; Lu, G. Electron dynamics in dye-sensitized solar cells: Effects of surface terminations and defects. J. Phys. Chem. B 2010, 114, 17077–17083. [Google Scholar]
- Duncan, W.R.; Prezhdo, O.V. Temperature independence of the photoinduced electron injection in dye-sensitized TiO2 rationalized by ab initio time-domain density functional theory. J. Am. Chem. Soc. 2008, 130, 9756–9762. [Google Scholar] [CrossRef]
- Mosconi, E.; Yum, J.-H.; Kessler, F.; Gomez Garcia, C.J.; Zuccaccia, C.; Cinti, A.; Nazeeruddin, Md.K.; Grätzel, M.; de Angelis, F. Cobalt eectrolyte/dye interactions in dye-sensitized solar cells: a combined computational and experimental study. J. Am. Chem. Soc. 2012, 134, 19438–19453. [Google Scholar] [CrossRef]
- Daeneke, T.; Mozer, A.J.; Uemura, Y.; Makuta, S.; Fekete, M.; Tachibana, Y.; Koumura, N.; Bach, U.; Spicca, L. Dye regeneration kinetics in dye-sensitized solar cells. J. Am. Chem. Soc. 2012, 134, 16925–16928. [Google Scholar] [CrossRef]
- Ahmad, S.; Bessho, T.; Kessler, F.; Baranoff, E.; Frey, J.; Yi, C.; Grätzel, M.; Nazeeruddin, Md.K. A new generation of platinum and iodine free efficient dye-sensitized solar cells. Phys. Chem. Chem. Phys. 2012, 14, 10631–10639. [Google Scholar]
- Dos Santos, T.; Morandeira, A.; Koops, S.; Mozer, A.J.; Tsekouras, G.; Dong, Y.; Wagner, P.; Wallace, G.; Earles, J.C.; Gordon, K.C.; Officer, D.; Durrant, J.R. Injection limitations in a series of porphyrin dye-sensitized solar cells. J. Phys. Chem. C 2010, 114, 3276–3279. [Google Scholar] [CrossRef]
- Koops, S.E.; O’Regan, B.C.; Barnes, P.R.F.; Durrant, J.R. Parameters influencing the efficiency of electron injection in dye-sensitized solar cells. J. Am. Chem. Soc. 2009, 131, 4808–4818. [Google Scholar]
- Haque, S.A.; Handa, S.; Peter, K.; Palomares, E.; Thelakkat, M.; Durrant, J.R. Supermolecular control of charge transfer in dye-sensitized nanocrystalline TiO2 films: Towards a quantitative structure-function relationship. Angew. Chem. Int. Ed. 2005, 44, 5740–5744. [Google Scholar]
- Clifford, J.N.; Palomares, E.; Nazeeruddin, Md.K.; Grätzel, M.; Nelson, J.; Li, X.; Long, N.J.; Durrant, J.R. Molecular control of recombination dynamics in dye-sensitized nanocrystalline TiO2 films: Free energy vs. distance dependence. J. Am. Chem. Soc. 2004, 126, 5225–5233. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Manzhos, S.; Segawa, H.; Yamashita, K. Effect of Isotopic Substitution on Elementary Processes in Dye-Sensitized Solar Cells: Deuterated Amino-Phenyl Acid Dyes on TiO2. Computation 2013, 1, 1-15. https://doi.org/10.3390/computation1010001
Manzhos S, Segawa H, Yamashita K. Effect of Isotopic Substitution on Elementary Processes in Dye-Sensitized Solar Cells: Deuterated Amino-Phenyl Acid Dyes on TiO2. Computation. 2013; 1(1):1-15. https://doi.org/10.3390/computation1010001
Chicago/Turabian StyleManzhos, Sergei, Hiroshi Segawa, and Koichi Yamashita. 2013. "Effect of Isotopic Substitution on Elementary Processes in Dye-Sensitized Solar Cells: Deuterated Amino-Phenyl Acid Dyes on TiO2" Computation 1, no. 1: 1-15. https://doi.org/10.3390/computation1010001