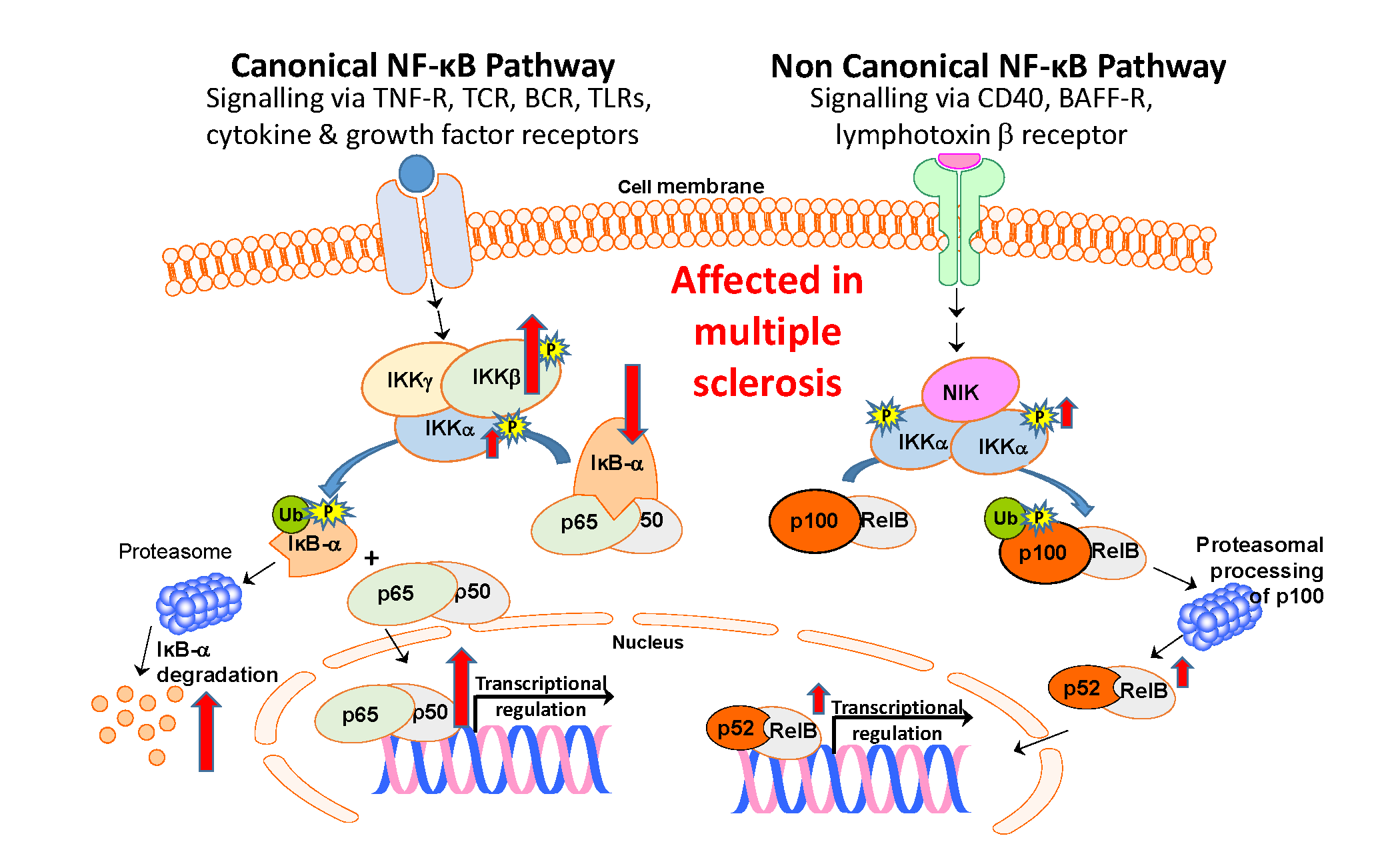
Reduced IκB-α Protein Levels in Peripheral Blood Cells of Patients with Multiple Sclerosis—A Possible Cause of Constitutive NF-κB Activation


Abstract
:
1. Introduction
2. Experimental Section
2.1. Patients and Controls
2.2. Collection of Blood, Separation of Peripheral Blood Mononuclear Cells (PBMC) and Preparation of PBMC Fractions.
2.3. Western Blotting to Detect IĸB-α and p52
2.4. Assay for NF-κB p65 DNA Binding Activity
2.5. Detection of Phosphorylated (activated) IKKα and IKKβ
2.6. Statistical Analyses
3. Results
3.1. IĸB-α Protein Levels in PBMC from MS Patients and Controls

3.2. Binding Activity of NFĸB-α p65 in PBMC from MS Patients and Controls

3.3. Activation (Phosphorylation) of IKKα and IKKβ is Increased in MS Patients vs Healthy Subjects

3.4. Increased transloCation of p52 to the Nucleus in MS Patients

4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pender, M.P.; Greer, J.M. Immunology of multiple sclerosis. Curr. Allergy Asthma Rep. 2007, 7, 285–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.A.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; Goris, A.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365. [Google Scholar] [CrossRef] [Green Version]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sorensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [Green Version]
- Baeuerle, P.A.; Baltimore, D. I-Kappa-B—A Specific Inhibitor of the Nf-Kappa-B Transcription Factor. Science 1988, 242, 540–546. [Google Scholar] [CrossRef]
- Bonetti, B.; Stegagno, C.; Cannella, B.; Rizzuto, N.; Moretto, G.; Raine, C.S. Activation of NF-kappa B and c-jun transcription factors in multiple sclerosis lesions—Implications for oligodendrocyte pathology. Am. J. Pathol. 1999, 155, 1433–1438. [Google Scholar] [CrossRef]
- Flores, N.; Duran, C.; Blasco, M.R.; Puerta, C.; Dorado, B.; Garcia-Merino, A.; Ballester, S. NFkappaB and AP-1 DNA binding activity in patients with multiple sclerosis. J. Neuroimmunol. 2003, 135, 141–147. [Google Scholar] [CrossRef]
- Yan, J.; Greer, J.M. NF-kappa B, a potential therapeutic target for the treatment of multiple sclerosis. CNS Neurol. Disord. Drug Targets 2008, 7, 536–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mc Guire, C.; Prinz, M.; Beyaert, R.; van Loo, G. Nuclear factor kappa B (NF-kappa B) in multiple sclerosis pathology. Trends Mol. Med. 2013, 19, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, S.M.; Yan, J. NF-kappa B Pathways in the Pathogenesis of Multiple Sclerosis and the Therapeutic Implications. Front. Mol. Neurosci. 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Gveric, D.; Kaltschmidt, C.; Cuzner, M.L.; Newcombe, J. Transcription factor NF-kappaB and inhibitor I kappaBalpha are localized in macrophages in active multiple sclerosis lesions. J. Neuropathol. Exp. Neurol. 1998, 57, 168–178. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Winterford, C.M.; Catts, V.S.; Pat, B.K.; Pender, M.P.; McCombe, P.A.; Greer, J.M. Increased constitutive activation of NF-kappaB p65 (RelA) in peripheral blood cells of patients with progressive multiple sclerosis. J. Neuroimmunol. 2018, 320, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Hussman, J.P.; Beecham, A.H.; Schmidt, M.; Martin, E.R.; McCauley, J.L.; Vance, J.M.; Haines, J.L.; Pericak-Vance, M.A. GWAS analysis implicates NF-kappaB-mediated induction of inflammatory T cells in multiple sclerosis. Genes Immun. 2016, 17, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Karin, M.; Delhase, M. The I kappa B kinase (IKK) and NF-kappa B: Key elements of proinflammatory signalling. Semin. Immunol. 2000, 12, 85–98. [Google Scholar] [CrossRef]
- Hu, Y.; Baud, V.; Delhase, M.; Zhang, P.; Deerinck, T.; Ellisman, M.; Johnson, R.; Karin, M. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science 1999, 284, 316–320. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, J.; Mair, F.; Greter, M.; Schmidt-Supprian, M.; Becher, B. NIK signaling in dendritic cells but not in T cells is required for the development of effector T cells and cell-mediated immune responses. J. Exp. Med. 2011, 208, 1917–1929. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Zhou, X.F.; Yu, J.; Cheng, X.; Sun, S.C. Regulation of Th17 cell differentiation and EAE induction by MAP3K NIK. Blood 2009, 113, 6603–6610. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhou, X.; Nakaya, M.; Jin, W.; Cheng, X.; Sun, S.C. T cell-intrinsic function of the noncanonical NF-kappaB pathway in the regulation of GM-CSF expression and experimental autoimmune encephalomyelitis pathogenesis. J. Immunol. 2014, 193, 422–430. [Google Scholar] [CrossRef]
- Chen, G.; Hardy, K.; Pagler, E.; Ma, L.; Lee, S.; Gerondakis, S.; Daley, S.; Shannon, M.F. The NF-kappaB transcription factor c-Rel is required for Th17 effector cell development in experimental autoimmune encephalomyelitis. J. Immunol. 2011, 187, 4483–4491. [Google Scholar] [CrossRef] [Green Version]
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Poser, C.M.; Paty, D.W.; Scheinberg, L.; McDonald, W.I.; Davis, F.A.; Ebers, G.C.; Johnson, K.P.; Sibley, W.A.; Silberberg, D.H.; Tourtellotte, W.W. New diagnostic criteria for multiple sclerosis: Guidelines for research protocols. Ann. Neurol. 1983, 13, 227–231. [Google Scholar] [CrossRef]
- Berko, D.; Tabachnick-Cherny, S.; Shental-Bechor, D.; Cascio, P.; Mioletti, S.; Levy, Y.; Admon, A.; Ziv, T.; Tirosh, B.; Goldberg, A.L.; et al. The direction of protein entry into the proteasome determines the variety of products and depends on the force needed to unfold its two termini. Mol. Cell 2012, 48, 601–611. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Granados, E.; Keenan, J.E.; Kinney, M.C.; Leo, H.; Jain, N.; Ma, C.A.; Quinones, R.; Gelfand, E.W.; Jain, A. A novel mutation in NFKBIA/IKBA results in a degradation-resistant N-truncated protein and is associated with ectodermal dysplasia with immunodeficiency. Hum. Mutat. 2008, 29, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Zhong, X.; Wu, T.; Yang, T.; Chen, G.; Xie, X.; Wei, Y.; Ye, M.; Zhou, Y.; Du, Z. Identification of a NFKBIA polymorphism associated with lower NFKBIA protein levels and poor survival outcomes in patients with glioblastoma multiforme. Int. J. Mol. Med. 2014, 34, 1233–1240. [Google Scholar] [CrossRef] [Green Version]
- Mathes, E.; O’Dea, E.L.; Hoffmann, A.; Ghosh, G. NF-kappa B dictates the degradation pathway of I kappa B alpha. EMBO J. 2008, 27, 1357–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.; Gerstberger, S.; Carlson, L.; Franzoso, G.; Siebenlist, U. Control of I-kappa-B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science 1995, 267, 1485–1488. [Google Scholar] [CrossRef]
- Krappmann, D.; Wulczyn, F.G.; Scheidereit, C. Different mechanisms control signal-induced degradation and basal turnover of the NF-kappa B inhibitor I kappa B alpha in vivo. EMBO J. 1996, 15, 6716–6726. [Google Scholar] [CrossRef] [PubMed]
- Truhlar, S.M.; Mathes, E.; Cervantes, C.F.; Ghosh, G.; Komives, E.A. Pre-folding IkappaBalpha alters control of NF-kappaB signaling. J. Mol. Biol. 2008, 380, 67–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miterski, B.; Bohringer, S.; Klein, W.; Sindern, E.; Haupts, M.; Schimrigk, S.; Epplen, J.T. Inhibitors in the NFkappaB cascade comprise prime candidate genes predisposing to multiple sclerosis, especially in selected combinations. Genes Immun. 2002, 3, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Codarri, L.; Fontana, A.; Becher, B. Cytokine networks in multiple sclerosis: Lost in translation. Curr. Opin. Neurol. 2010, 23, 205–211. [Google Scholar] [CrossRef]
- Sharief, M.K.; Hentges, R. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N. Engl. J. Med. 1991, 325, 467–472. [Google Scholar] [CrossRef]
- Maimone, D.; Guazzi, G.C.; Annunziata, P. IL-6 detection in multiple sclerosis brain. J. Neurol. Sci. 1997, 146, 59–65. [Google Scholar] [CrossRef]
- Gallo, P.; Pagni, S.; Piccinno, M.G.; Giometto, B.; Argentiero, V.; Chiusole, M.; Bozza, F.; Tavolato, B. On the role of interleukin-2 (IL-2) in multiple sclerosis (MS). IL-2-mediated endothelial cell activation. Ital. J. Neurol. Sci. 1992, 13, 65–68. [Google Scholar]
- Comabella, M.; Balashov, K.; Issazadeh, S.; Smith, D.; Weiner, H.L.; Khoury, S.J. Elevated interleukin-12 in progressive multiple sclerosis correlates with disease activity and is normalized by pulse cyclophosphamide therapy. J. Clin. Investig. 1998, 102, 671–678. [Google Scholar] [CrossRef]
- Shajarian, M.; Alsahebfosoul, F.; Etemadifar, M.; Sedaghat, N.; Shahbazi, M.; Firouzabadi, F.P.; Dezashibi, H.M. IL-23 plasma level measurement in relapsing remitting multiple sclerosis (RRMS) patients compared to healthy subjects. Immunol. Investig. 2015, 44, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Khaibullin, T.; Ivanova, V.; Martynova, E.; Cherepnev, G.; Khabirov, F.; Granatov, E.; Rizvanov, A.; Khaiboullina, S. Elevated levels of proinflammatory cytokines in cerebrospinal fluid of multiple sclerosis patients. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Szczuciński, A.; Losy, J. Chemokines and chemokine receptors in multiple sclerosis. Potential targets for new therapies. Acta Neurol. Scand. 2007, 115, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Elovaara, I.; Ukkonen, M.; Leppakynnas, M.; Lehtimaki, T.; Luomala, M.; Peltola, J.; Dastidar, P. Adhesion molecules in multiple sclerosis: Relation to subtypes of disease and methylprednisolone therapy. Arch. Neurol. 2000, 57, 546–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharief, M.K.; Douglas, M.; Noori, M.; Semra, Y.K. The expression of pro- and anti-apoptosis Bcl-2 family proteins in lymphocytes from patients with multiple sclerosis. J. Neuroimmunol. 2002, 125, 155–162. [Google Scholar] [CrossRef]
- Waiczies, S.; Weber, A.; Lunemann, J.D.; Aktas, O.; Zschenderlein, R.; Zipp, F. Elevated Bcl-X(L) levels correlate with T cell survival in multiple sclerosis. J. Neuroimmunol. 2002, 126, 213–220. [Google Scholar] [CrossRef]
- De Oliveira, G.L.; Ferreira, A.F.; Gasparotto, E.P.; Kashima, S.; Covas, D.T.; Guerreiro, C.T.; Brum, D.G.; Barreira, A.A.; Voltarelli, J.C.; Simoes, B.P.; et al. Defective expression of apoptosis-related molecules in multiple sclerosis patients is normalized early after autologous haematopoietic stem cell transplantation. Clin. Exp. Immunol. 2017, 187, 383–398. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subjects | Number | % Female | Age (Mean and Range) | EDSS (Mean and Range) |
|---|---|---|---|---|
| Whole study | ||||
| Total MS | 39 | 76.9 | 45.8 (25–76) § | 3.9 (0–6.5) |
| * RR-MS | 21 | 81.0 | 37.9 (25–51) | 2.7 (0–5.0) |
| SP-MS | 10 | 80.0 | 56.3 (48–72) § | 5.1 (2.5–6.5) |
| PP-MS | 8 | 62.5 | 53.5 (40–76) § | 4.8 (2.0–6.5) |
| Healthy controls | 32 | 90.6 | 35.5 (22-55) | N/A |
| Samples used in Figure 2 | ||||
| Total MS | 20 | 70.0 | 48.6 (30–76) § | 3.9 (1.0–6.5) |
| RR-MS | 8 | 87.5 | 39.8 (30–47) | 2.7 (1.0–3.5) |
| SP-MS | 6 | 83.3 | 54.2 (48–67) § | 4.6 (2.5–6.0) |
| PP-MS | 6 | 50.0 | 54.6 (40–76) § | 4.7 (2.0–6.5) |
| Healthy controls | 24 | 86.4 | 37.1 (22-55) | N/A |
| Samples used in Figure 3 | ||||
| Total MS | 29 | 79.3 | 44.8 (25–76) | 3.7 (0–6.5) |
| RR-MS | 18 | 77.8 | 37.8 (25–50) | 2.4 (0–3.5) |
| SP-MS | 7 | 85.7 | 56.7 (48–72) § | 5.4 (3.5–6.5) |
| PP-MS | 4 | 75.0 | 55.3 (40–76) § | 5.8 (5.0–6.5) |
| Healthy controls | 18 | 94.4 | 37.8 (23–55) | N/A |
| Samples used in Figure 4 | ||||
| Total MS | 25 | 84.0 | 43.6 (25–76) | 3.5 (0–6.5) |
| RR-MS | 18 | 77.8 | 37.4 (25–50) | 2.5 (0–3.5) |
| SP-MS | 5 | 100.0 | 60.0 (50–67) § | 5.5 (3.5–6.5) |
| PP-MS | 2 | 50.0 | 63.5 (51–76) § | 6.5 |
| Healthy controls | 9 | 88.9 | 40.0 (27–55) | N/A |
| Samples used in Figure 5 | ||||
| Total MS | 11 | 90.9 | 45.0 (25–76) | 3.5 (2.0–6.5) |
| RR-MS | 7 | 85.7 | 31.0 (25–54) | 3.5 (2.0–5.0) |
| SP-MS | 1 | 100.0 | 51.0 | 4.0 |
| PP-MS | 3 | 100.0 | 46.0 (40–76) | 5.5 (2.0–6.5) |
| Healthy controls | 7 | 100.0 | 36.7 (27–55) | N/A |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, J.; McCombe, P.A.; Pender, M.P.; Greer, J.M. Reduced IκB-α Protein Levels in Peripheral Blood Cells of Patients with Multiple Sclerosis—A Possible Cause of Constitutive NF-κB Activation. J. Clin. Med. 2020, 9, 2534. https://doi.org/10.3390/jcm9082534
Yan J, McCombe PA, Pender MP, Greer JM. Reduced IκB-α Protein Levels in Peripheral Blood Cells of Patients with Multiple Sclerosis—A Possible Cause of Constitutive NF-κB Activation. Journal of Clinical Medicine. 2020; 9(8):2534. https://doi.org/10.3390/jcm9082534
Chicago/Turabian StyleYan, Jun, Pamela A. McCombe, Michael P. Pender, and Judith M. Greer. 2020. "Reduced IκB-α Protein Levels in Peripheral Blood Cells of Patients with Multiple Sclerosis—A Possible Cause of Constitutive NF-κB Activation" Journal of Clinical Medicine 9, no. 8: 2534. https://doi.org/10.3390/jcm9082534