Erythropoietin, Fibroblast Growth Factor 23, and Death After Kidney Transplantation

 , , , and
, , , and
Abstract
:1. Introduction
2. Methods
2.1. Patient Population
2.2. Data Collection
2.3. Laboratory Procedures
2.4. Statistical Analyses
3. Results
3.1. Baseline Characteristics
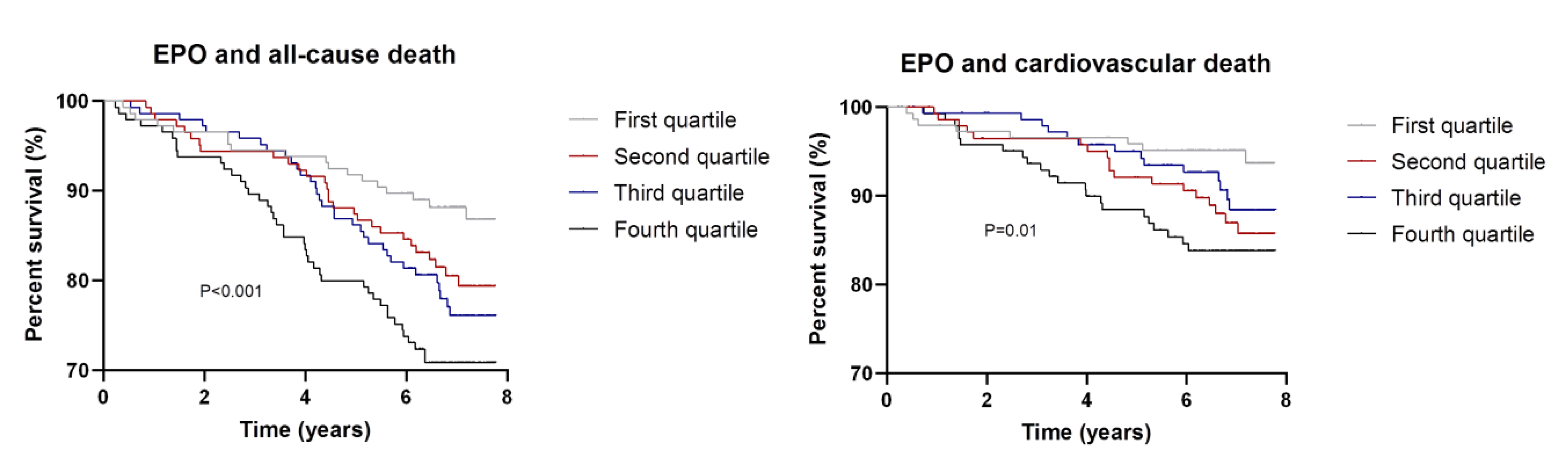
3.2. EPO, FGF23, and Death
3.3. EPO, FGF23, and Graft Failure
3.4. Percentage Change HR and Mediation Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jardine, A.; Gaston, R.S.; Fellström, B.C.; Holdaas, H. Prevention of cardiovascular disease in adult recipients of kidney transplants. Lancet 2011, 378, 1419–1427. [Google Scholar] [CrossRef]
- Sinkeler, S.J.; Zelle, D.M.; van der Heide, J.J.H.; Gans, R.; Navis, G.; Bakker, S.J.L. Endogenous plasma erythropoietin, cardiovascular mortality and all-cause mortality in renal transplant recipients. Arab. Archaeol. Epigr. 2011, 12, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Molnar, M.Z.; Tabak, A.G.; Alam, A.; Czira, M.E.; Rudas, A.; Ujszaszi, A.; Beko, G.; Novak, M.; Kalantar-Zadeh, K.; Kovesdy, C.P.; et al. Serum erythropoietin level and mortality in kidney transplant recipients. Clin. J. Am. Soc. Nephrol. 2011, 6, 2879–2886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Elzen, W.P.; Willems, J.M.; Westendorp, R.G.; de Craen, A.J.; Blauw, G.J.; Ferrucci, L.; Assendelft, W.J.; Gussekloo, J. Effect of erythropoietin levels on mortality in old age: The leiden 85-plus study. Can. Med. Assoc. J. 2010, 182, 1953–1958. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, M.A.; Burdmann, E.; Chen, C.-Y.; E Cooper, M.; de Zeeuw, D.; Eckardt, K.-U.; Ivanovich, P.; KewalRamani, R.; Levey, A.S.; Lewis, E.F.; et al. Baseline characteristics in the trial to reduce cardiovascular events with aranesp therapy (TREAT). Am. J. Kidney Dis. 2009, 54, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Szczech, L.; Tang, K.L.; Barnhart, H.; Sapp, S.; Wolfson, M.; Reddan, D.; CHOIR Investigators. Correction of anemia with epoetin alfa in chronic kidney disease. New Engl. J. Med. 2006, 355, 2085–2098. [Google Scholar] [CrossRef] [Green Version]
- Clinkenbeard, E.L.; Hanudel, M.R.; Stayrook, K.R.; Appaiah, H.N.; Farrow, E.G.; Cass, T.A.; Summers, L.J.; Ip, C.S.; Hum, J.M.; Thomas, J.C.; et al. Erythropoietin stimulates murine and human fibroblast growth factor-23, revealing novel roles for bone and bone marrow. Haematologica 2017, 102, e427–e430. [Google Scholar] [CrossRef]
- Rabadi, S.; Udo, I.; Leaf, D.E.; Waikar, S.S.; Christov, M. Acute blood loss stimulates fibroblast growth factor 23 production. Am. J. Physiol. Ren. Physiol. 2017, 314, F132–F139. [Google Scholar] [CrossRef] [Green Version]
- Hanudel, M.R.; Eisenga, M.; Rappaport, M.; Chua, K.; Qiao, B.; Jung, G.; Gabayan, V.; Gales, B.; Ramos, G.; A De Jong, M.; et al. Effects of erythropoietin on fibroblast growth factor 23 in mice and humans. Nephrol. Dial. Transpl. 2018, 34, 2057–2065. [Google Scholar] [CrossRef]
- Toro, L.; Barrientos, V.; León, P.; Rojas, M.; González, M.; González-Ibáñez, A.; Illanes, S.; Sugikawa, K.; Abarzúa, N.; Bascuñán, C.; et al. Erythropoietin induces bone marrow and plasma fibroblast growth factor 23 during acute kidney injury. Kidney Int. 2018, 93, 1131–1141. [Google Scholar] [CrossRef]
- Baia, L.C.; Humalda, J.K.; Vervloet, M.G.; Navis, G.; Bakker, S.J.; De Borst, M.H. Fibroblast growth factor 23 and cardiovascular mortality after kidney transplantation. Clin. J. Am. Soc. Nephrol. 2013, 8, 1968–1978. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Molnar, M.Z.; Amaral, A.P.; Czira, M.E.; Rudas, A.; Ujszaszi, A.; Kiss, I.; Rosivall, L.; Kosa, J.; Lakatos, P.; et al. Elevated fibroblast growth factor 23 is a risk factor for kidney transplant loss and mortality. J. Am. Soc. Nephrol. 2011, 22, 956–966. [Google Scholar] [CrossRef] [Green Version]
- Stockmann, C.; Fandrey, J. Hypoxia-Induced erythropoietin production: A paradigm for oxygen-regulated gene expression. Clin. Exp. Pharmacol. Physiol. 2006, 33, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J. Boil. Med. 2007, 80, 51–60. [Google Scholar]
- Hamrick, S.E.G.; McQuillen, P.S.; Jiang, X.; Mu, D.; Madan, A.; Ferriero, D.M. A role for hypoxia-inducible factor-1α in desferoxamine neuroprotection. Neurosci. Lett. 2005, 379, 96–100. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.; Grondin, F.; McDonald, P.P.; Richard, D.E.; Dubois, C.M. Hypoxia-enhanced expression of the proprotein convertase furin is mediated by hypoxia-inducible factor-1: Impact on the bioactivation of proproteins. J. Biol. Chem. 2005, 280, 6561–6569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, M.; White, K.E. Coupling fibroblast growth factor 23 production and cleavage. Curr. Opin. Nephrol. Hypertens. 2014, 23, 411–419. [Google Scholar] [CrossRef]
- Hanudel, M.R.; Wesseling-Perry, K.; Gales, B.; Ramos, G.; Campbell, V.; Ethridge, K.; Scotti, M.; Elashoff, D.A.; Alejos, J.; Reemtsen, B.; et al. Effects of acute kidney injury and chronic hypoxemia on fibroblast growth factor 23 levels in pediatric cardiac surgery patients. Pediatr. Nephrol. 2015, 31, 661–669. [Google Scholar] [CrossRef] [Green Version]
- de Vries, A.; Bakker, S.J.L.; van Son, W.J.; van der Heide, J.J.H.; Ploeg, R.J.; The, H.T.; de Jong, P.E.; Gans, R.O.B. Metabolic syndrome is associated with impaired long-term renal allograft function; Not all component criteria contribute equally. Arab. Archaeol. Epigr. 2004, 4, 1675–1683. [Google Scholar] [CrossRef]
- Benson, E.W.; Hardy, R.; Chaffin, C.; Robinson, C.A.; Konrad, R.J. New automated chemiluminescent assay for erythropoietin. J. Clin. Lab. Anal. 2000, 14, 271–273. [Google Scholar] [CrossRef]
- Heijboer, A.C.; Levitus, M.; Vervloet, M.; Lips, P.; Wee, P.M.T.; Dijstelbloem, H.M.; Blankenstein, M. Determination of fibroblast growth factor 23. Ann. Clin. Biochem. Int. J. Lab. Med. 2009, 46, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; Stevens, L.A.; Schmid, C.H.; Zhang, Y.L.; Castro, A.F., 3rd; Feldman, H.I.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Greene, T.; et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009, 150, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Beau, M.M.L.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Oterdoom, L.H.; de Vries, A.; van Ree, R.M.; Gansevoort, R.T.; van Son, W.J.; van der Heide, J.J.H.; Navis, G.; de Jong, P.E.; Gans, R.O.; Bakker, S.J. N-Terminal Pro-B-Type natriuretic peptide and mortality in renal transplant recipients versus the general population. Transplantation 2009, 87, 1562–1570. [Google Scholar] [CrossRef]
- Preacher, K.J.; Hayes, A.F. SPSS and SAS procedures for estimating indirect effects in simple mediation models. Behav. Res. Methods Instrum. Comput. 2004, 36, 717–731. [Google Scholar] [CrossRef] [Green Version]
- Hayes, A.F. Beyond baron and kenny: Statistical mediation analysis in the new millennium. Commun. Monogr. 2009, 76, 408–420. [Google Scholar] [CrossRef]
- White, I.R.; Wood, A.M.; Royston, P. Multiple imputation using chained equations: Issues and guidance for practice. Stat. Med. 2010, 30, 377–399. [Google Scholar] [CrossRef]
- Rubin, D.B. Multiple Imputation for Nonresponse in Surveys; Wiley: New York, NY, USA, 1987. [Google Scholar]
- Jelkmann, W. Regulation of erythropoietin production. J. Physiol. 2010, 589, 1251–1258. [Google Scholar] [CrossRef]
- Rossert, J.; Eckardt, K.-U. Erythropoietin receptors: Their role beyond erythropoiesis. Nephrol. Dial. Transpl. 2005, 20, 1025–1028. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, O.M.; Mannstadt, M.; Isakova, T.; Rauh-Hain, J.A.; Tamez, H.; Shah, A.; Smith, K.; Lee, H.; Thadhani, R.; Jüppner, H.; et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. New Engl. J. Med. 2008, 359, 584–592. [Google Scholar] [CrossRef] [Green Version]
- Isakova, T.; Xie, H.; Yang, W.; Xie, D.; Anderson, A.H.; Scialla, J.; Wahl, P.; Gutierrez, O.M.; Steigerwalt, S.; He, J.; et al. Chronic renal insufficiency cohort (CRIC) study group fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. J. Am. Med. Assoc. 2011, 305, 2432–2439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leaf, D.E.; Christov, M.; Jüppner, H.; Siew, E.; Ikizler, T.A.; Bian, A.; Chen, G.; Sabbisetti, V.S.; Bonventre, J.V.; Cai, X.; et al. Fibroblast growth factor 23 levels are elevated and associated with severe acute kidney injury and death following cardiac surgery. Kidney Int. 2016, 89, 939–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendrick, J.; Cheung, A.K.; Kaufman, J.S.; Greene, T.; Roberts, W.L.; Smits, G.; Chonchol, M.; HOST Investigators. FGF-23 associates with death, cardiovascular events, and initiation of chronic dialysis. J. Am. Soc. Nephrol. 2011, 22, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, J.M.; Isakova, T.; Cai, X.; Bayes, L.Y.; Faul, C.; Scialla, J.J.; Lash, J.P.; Chen, J.; He, J.; Navaneethan, S.; et al. CRIC study investigators inflammation and elevated levels of fibroblast growth factor 23 are independent risk factors for death in chronic kidney disease. Kidney Int. 2017, 91, 711–719. [Google Scholar] [CrossRef] [Green Version]
- Nguyen-Yamamoto, L.; Karaplis, A.C.; St–Arnaud, R.; Goltzman, D. Fibroblast growth factor 23 regulation by systemic and local osteoblast-synthesized 1,25-Dihydroxyvitamin D. J. Am. Soc. Nephrol. 2016, 28, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Kovesdy, C.P.; Quarles, L.D. Fibroblast growth factor-23: What we know, what we don’t know, and what we need to know. Nephrol. Dial. Transpl. 2013, 28, 2228–2236. [Google Scholar] [CrossRef]
- Eisenga, M.; van Londen, M.; Leaf, D.E.; Nolte, I.M.; Navis, G.; Bakker, S.J.; De Borst, M.H.; Gaillard, C.A. C-Terminal fibroblast growth factor 23, iron deficiency, and mortality in renal transplant recipients. J. Am. Soc. Nephrol. 2017, 28, 3639–3646. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.; Koch, T.A.; Bregman, D.B. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J. Bone Miner. Res. 2013, 28, 1793–1803. [Google Scholar] [CrossRef]
- David, V.; Martin, A.; Isakova, T.; Spaulding, C.; Qi, L.; Ramirez, V.; Zumbrennen-Bullough, K.B.; Sun, C.C.; Lin, H.Y.; Babitt, J.L.; et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016, 89, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Flamme, I.; Ellinghaus, P.; Urrego, D.; Krüger, T. FGF23 expression in rodents is directly induced via erythropoietin after inhibition of hypoxia inducible factor proline hydroxylase. PLoS ONE 2017, 12, e0186979. [Google Scholar] [CrossRef] [Green Version]
- Silswal, N.; Touchberry, C.; Daniel, D.R.; McCarthy, D.L.; Zhang, S.; Andresen, J.; Stubbs, J.R.; Wacker, M.J. FGF23 directly impairs endothelium-dependent vasorelaxation by increasing superoxide levels and reducing nitric oxide bioavailability. Am. J. Physiol. Metab. 2014, 307, E426–E436. [Google Scholar] [CrossRef] [Green Version]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.-C.; Sloan, A.; Isakova, T.; Gutierrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.R.; Tan, S.-J.; Holt, S.G.; Hewitson, T. FGF23 is synthesised locally by renal tubules and activates injury-primed fibroblasts. Sci. Rep. 2017, 7, 3345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Grabner, A.; Yanucil, C.; Schramm, K.; Czaya, B.; Krick, S.; Czaja, M.J.; Bartz, R.; Abraham, R.; Marco, G.S.D.; et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. 2016, 90, 985–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossaint, J.; Oehmichen, J.; van Aken, H.; Reuter, S.; Pavenstädt, H.J.; Meersch, M.; Unruh, M.L.; Zarbock, A. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J. Clin. Investig. 2016, 126, 962–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, R.; Nakada, Y.; Hu, M.C.; Kurosu, H.; Wang, L.; Nakatani, T.; Shi, M.; Eliseenkova, A.V.; Razzaque, M.S.; Moe, O.W.; et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc. Natl. Acad. Sci. USA 2009, 107, 407–412. [Google Scholar] [CrossRef] [Green Version]
- Courbebaisse, M.; Mehel, H.; Petit-Hoang, C.; Ribeil, J.-A.; Sabbah, L.; Tuloup-Minguez, V.; Bergerat, D.; Arlet, J.-B.; Stanislas, A.; Souberbielle, J.-C.; et al. Carboxy-terminal fragment of fibroblast growth factor 23 induces heart hypertrophy in sickle cell disease. Haematologica 2016, 102, e33–e35. [Google Scholar] [CrossRef] [Green Version]
- El-Maouche, D.; Dumitrescu, C.E.; Andreopoulou, P.; Gafni, R.I.; Brillante, B.A.; Bhattacharyya, N.; Fedarko, N.S.; Collins, M.T. Stability and degradation of fibroblast growth factor 23 (FGF23): The effect of time and temperature and assay type. Osteoporos. Int. 2016, 27, 2345–2353. [Google Scholar] [CrossRef]
- Koenig, W.; Sund, M.; Fröhlich, M.; Löwel, H.; Hutchinson, W.L.; Pepys, M.B. Refinement of the association of serum C-reactive protein concentration and coronary heart disease risk by correction for within-subject variation over time: The MONICA Augsburg studies, 1984 and 1987. Am. J. Epidemiol. 2003, 158, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Danesh, J.; Wheeler, J.G.; Hirschfield, G.; Eda, S.; Eiriksdottir, G.; Rumley, A.; Lowe, G.; Pepys, M.B.; Gudnason, V. C-Reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. New Engl. J. Med. 2004, 350, 1387–1397. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Quartiles of EPO (IU/L) | ||||||
|---|---|---|---|---|---|---|
| All Patients (n = 579) | Q1 (n = 146) [4.0–11.9] | Q2 (n = 143) [12.0–17.2] | Q33 (n = 145) [17.3–23.9] | Q4 (n = 145) [24.2–182.0] | P-value | |
| Age (years) | 51 ± 12 | 47 ± 13 | 50 ± 12 | 53 ± 11 | 54 ± 11 | <0.001 |
| Male sex (n, %) | 317 (55) | 92 (63) | 78 (55) | 73 (50) | 74 (51) | 0.11 |
| Body surface area, m2 | 1.87 ± 0.19 | 1.86 ± 0.19 | 1.86 ± 0.20 | 1.87 ± 0.18 | 1.88 ± 0.20 | 0.81 |
| Alcohol use (n, %) | 290 (50) | 77 (52) | 73 (51) | 64 (44) | 76 (52) | 0.35 |
| Smoking status | 0.29 | |||||
| Never smoker (n, %) | 205 (35) | 47 (32) | 49 (34) | 59 (41) | 50 (35) | |
| Former smoker (n, %) | 246 (43) | 64 (44) | 68 (48) | 59 (41) | 55 (38) | |
| Current smoker (n, %) | 126 (22) | 35 (24) | 25 (18) | 27 (19) | 39 (27) | |
| Time since Tx (yrs) | 6.0 (2.6–11.6) | 4.6 (2.1–9.2) | 5.7 (3.1–11.2) | 6.5 (3.3–12.4) | 7.0 (2.8–13.7) | 0.007 |
| Diabetes mellitus (n, %) | 102 (18) | 25 (17) | 21 (15) | 27 (19) | 29 (20) | 0.67 |
| SBP (mmHg) | 153 ± 23 | 150 ± 20 | 151 ± 21 | 153 ± 22 | 157 ± 26 | 0.05 |
| DBP (mmHg) | 90 ± 10 | 90 ± 9 | 90 ± 9 | 90 ± 10 | 90 ± 11 | 0.91 |
| Laboratory measurements | ||||||
| FGF23 (RU/mL) | 137 (94–212) | 115 (81–168) | 125 (88–184) | 138 (95–212) | 195 (115–363) | <0.001 |
| Hemoglobin (g/dL) | 13.9 ± 1.5 | 14.2 ± 1.5 | 14.0 ± 1.5 | 13.8 ± 1.5 | 13.5 ± 1.5 | 0.001 |
| Erythrocytosis (n, %)‡ | 27 (5) | 11 (8) | 8 (6) | 5 (3) | 3 (2) | 0.13 |
| MCV (fL) | 91 ± 6 | 89 ± 4 | 91 ± 6 | 92 ± 6 | 92 ± 8 | <0.001 |
| Ferritin (µg/L) | 154 (76–282) | 154 (76–320) | 164 (100–305) | 159 (89–283) | 118 (61–240) | 0.02 |
| Total cholesterol (mmol/L) | 5.6 ± 1.1 | 5.7 ± 0.9 | 5.7 ± 1.3 | 5.6 ± 1.0 | 5.6 ± 1.1 | 0.78 |
| Phosphate (mmol/L) | 1.1 ± 0.2 | 1.05 ± 0.21 | 1.07 ± 0.21 | 1.05 ± 0.19 | 1.07 ± 0.22 | 0.85 |
| Calcium (mmol/L) | 2.39 ± 0.16 | 2.39 ± 0.14 | 2.38 ± 0.16 | 2.39 ± 0.18 | 2.41 ± 0.15 | 0.51 |
| Vit. 25(OH) D, nmol/l * | 53 ± 23 | 52 ± 24 | 51 ± 21 | 57 ± 21 | 53 ± 25 | 0.33 |
| Vit. 1,25(OH)2 D, pmol/L* | 109 ± 46 | 106 ± 50 | 112 ± 46 | 107 ± 40 | 110 ± 47 | 0.80 |
| PTH (pmol/L) | 9.1 (6.0–13.4) | 8.8 (6.2–13.2) | 9.6 (5.8–13.9) | 9.2 (6.0–13.7) | 8.9 (6.0–14.0) | 0.93 |
| eGFR (ml/min/1.73m2) | 48 ± 15 | 50 ± 16 | 48 ± 14 | 47 ± 15 | 46 ± 16 | 0.16 |
| Creatinine (µmol/L) | 144 ± 52 | 145 ± 51 | 139 ± 40 | 142 ± 51 | 148 ± 62 | 0.50 |
| Proteinuria (>0.5g) (n, %) | 155 (27) | 34 (23) | 32 (22) | 39 (27) | 50 (35) | 0.07 |
| hs-CRP (mg/L) | 2.0 (0.8–4.8) | 1.4 (0.6–3.8) | 2.0 (0.7–4.1) | 2.1 (1.0–4.2) | 3.2 (1.2–7.2) | <0.001 |
| Treatment | ||||||
| ACE-i/AII-antagonists (n, %) | 190 (33) | 65 (45) | 50 (35) | 37 (26) | 38 (26) | 0.001 |
| ACE-I (n, %) | 154 (27) | 53 (36) | 37 (26) | 31 (24) | 33 (23) | |
| AII-antagonists (n,%) | 36 (6) | 12 (8) | 13 (9) | 6 (4) | 5 (3) | |
| Bèta-blocker (n, %) | 356 (62) | 88 (63) | 96 (67) | 81 (56) | 91 (63) | 0.26 |
| Ca2+ channel blockers (n, %) | 220 (39) | 56 (38) | 52 (36) | 51 (35) | 61 (42) | 0.45 |
| Diuretic use (n, %) | 250 (43) | 52 (36) | 62 (43) | 60 (41) | 76 (52) | 0.04 |
| Proliferation inhibitor (n, %) | 428 (74) | 95 (65) | 99 (69) | 115 (79) | 119 (82) | 0.002 |
| Azathioprine (n, %) | 187 (32) | 18 (12) | 42 (29) | 55 (38) | 72 (50) | |
| Mycophenolic acid (n, %) | 241 (42) | 77 (53) | 57 (40) | 60 (41) | 47 (32) | |
| Calcineurin inhibitor (n, %) | 457 (79) | 131 (90) | 120 (84) | 103 (71) | 103 (71) | <0.001 |
| Ciclosporin (n,%) | 376 (65) | 108 (78) | 101 (71) | 81 (56) | 86 (59) | |
| Tacrolimus (n, %) | 81 (14) | 23 (16) | 19 (13) | 22 (15) | 17 (12) | |
| EPO (IU/L) | ||
|---|---|---|
| All–cause death | HR (95% CI) * | P-value |
| Univariable | 1.74 (1.29–2.34) | <0.001 |
| Model 1 | 1.66 (1.16–2.36) | 0.005 |
| Model 2 | 1.72 (1.21–2.46) | 0.003 |
| Model 3 | 1.80 (1.25–2.60) | 0.002 |
| Model 4 | 1.60 (1.12–2.29) | 0.01 |
| Model 5 | 1.28 (0.87–1.88) | 0.20 |
| Cardiovascular death | HR (95% CI) * | P-value |
| Univariable | 1.70 (1.12–2.58) | 0.01 |
| Model 1 | 1.87 (1.14–3.06) | 0.01 |
| Model 2 | 1.90 (1.16–3.12) | 0.01 |
| Model 3 | 2.05 (1.22–3.44) | 0.006 |
| Model 4 | 1.87 (1.14–3.06) | 0.01 |
| Model 5 | 1.45 (0.84–2.48) | 0.18 |
| Quartiles of EPO (IU/L) | ||||
|---|---|---|---|---|
| Q1 | Q2 | Q3 | Q4 | |
| All–cause death | Ref | HR (95% CI) | HR (95% CI) | HR (95% CI) |
| Univariable | 1.00 | 1.64 (0.91–2.97) | 1.93 (1.08–3.42) | 2.62 (1.51–4.55) |
| Model 1 | 1.00 | 1.57 (0.85–2.89) | 1.47 (0.81–2.69) | 2.11 (1.15–3.86) |
| Model 2 | 1.00 | 1.54 (0.83–2.84) | 1.51 (0.83–2.75) | 2.19 (1.19–4.05) |
| Model 3 | 1.00 | 1.65 (0.88–3.11) | 1.57 (0.84–2.93) | 2.29 (1.21–4.31) |
| Model 4 | 1.00 | 1.55 (0.85–2.85) | 1.42 (0.78–2.58) | 1.99 (1.09–3.65) |
| Model 5 | 1.00 | 1.63 (0.89–3.01) | 1.41 (0.77–2.57) | 1.55 (0.82–2.91) |
| Cardiovascular Death | Ref | HR (95% CI) | HR (95% CI) | HR (95% CI) |
| Univariable | 1.00 | 2.38 (1.04–5.48) | 1.85 (0.78–4.40) | 3.08 (1.37–6.92) |
| Model 1 | 1.00 | 2.73 (1.15–6.45) | 1.69 (0.68–4.18) | 3.34 (1.36–8.20) |
| Model 2 | 1.00 | 2.58 (1.08–6.16) | 1.71 (0.69–4.25) | 3.41 (1.31–8.47) |
| Model 3 | 1.00 | 2.91 (1.17–7.23) | 1.77 (0.68–4.59) | 3.57 (1.39–9.20) |
| Model 4 | 1.00 | 2.65 (1.12–6.25) | 1.60 (0.65–3.95) | 3.10 (1.27–7.60) |
| Model 5 | 1.00 | 2.90 (1.22–6.91) | 1.65 (0.66–4.08) | 2.47 (0.97–6.31) |
| Potential Mediator | Outcome | Effect (path) * | Multivariable Model ** | |
|---|---|---|---|---|
| Coefficient (95% CI, bc) † | Proportion Mediated *** | |||
| FGF23 | All-cause death | Indirect effect (ab path) | 0.090 (0.044; 0.139) | 72% |
| Total effect (ab + c’ path) | 0.124 (−0.011; 0.255) | |||
| Unstandardized total effect ‡ | 0.120 (−0.385; 0.624) | |||
| FGF23 | Cardiovascular death | Indirect effect (ab path) | 0.065 (0.015; 0.122) | 50% |
| Total effect (ab + c’ path) | 0.129 (−0.040; 0.290) | |||
| Unstandardized total effect ‡ | 0.218 (−0.405; 0.840) | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eisenga, M.F.; De Jong, M.A.; Leaf, D.E.; Nolte, I.M.; De Borst, M.H.; Bakker, S.J.L.; Gaillard, C.A.J.M. Erythropoietin, Fibroblast Growth Factor 23, and Death After Kidney Transplantation. J. Clin. Med. 2020, 9, 1737. https://doi.org/10.3390/jcm9061737
Eisenga MF, De Jong MA, Leaf DE, Nolte IM, De Borst MH, Bakker SJL, Gaillard CAJM. Erythropoietin, Fibroblast Growth Factor 23, and Death After Kidney Transplantation. Journal of Clinical Medicine. 2020; 9(6):1737. https://doi.org/10.3390/jcm9061737
Chicago/Turabian StyleEisenga, Michele F., Maarten A. De Jong, David E. Leaf, Ilja M. Nolte, Martin H. De Borst, Stephan J. L. Bakker, and Carlo A. J. M. Gaillard. 2020. "Erythropoietin, Fibroblast Growth Factor 23, and Death After Kidney Transplantation" Journal of Clinical Medicine 9, no. 6: 1737. https://doi.org/10.3390/jcm9061737