Screening Readthrough Compounds to Suppress Nonsense Mutations: Possible Application to β-Thalassemia

 ,
,  ,
,
Abstract
:1. Introduction
2. Overview of the Nonsense Suppression Therapeutic Approach
3. Compounds with Nonsense Suppression Properties
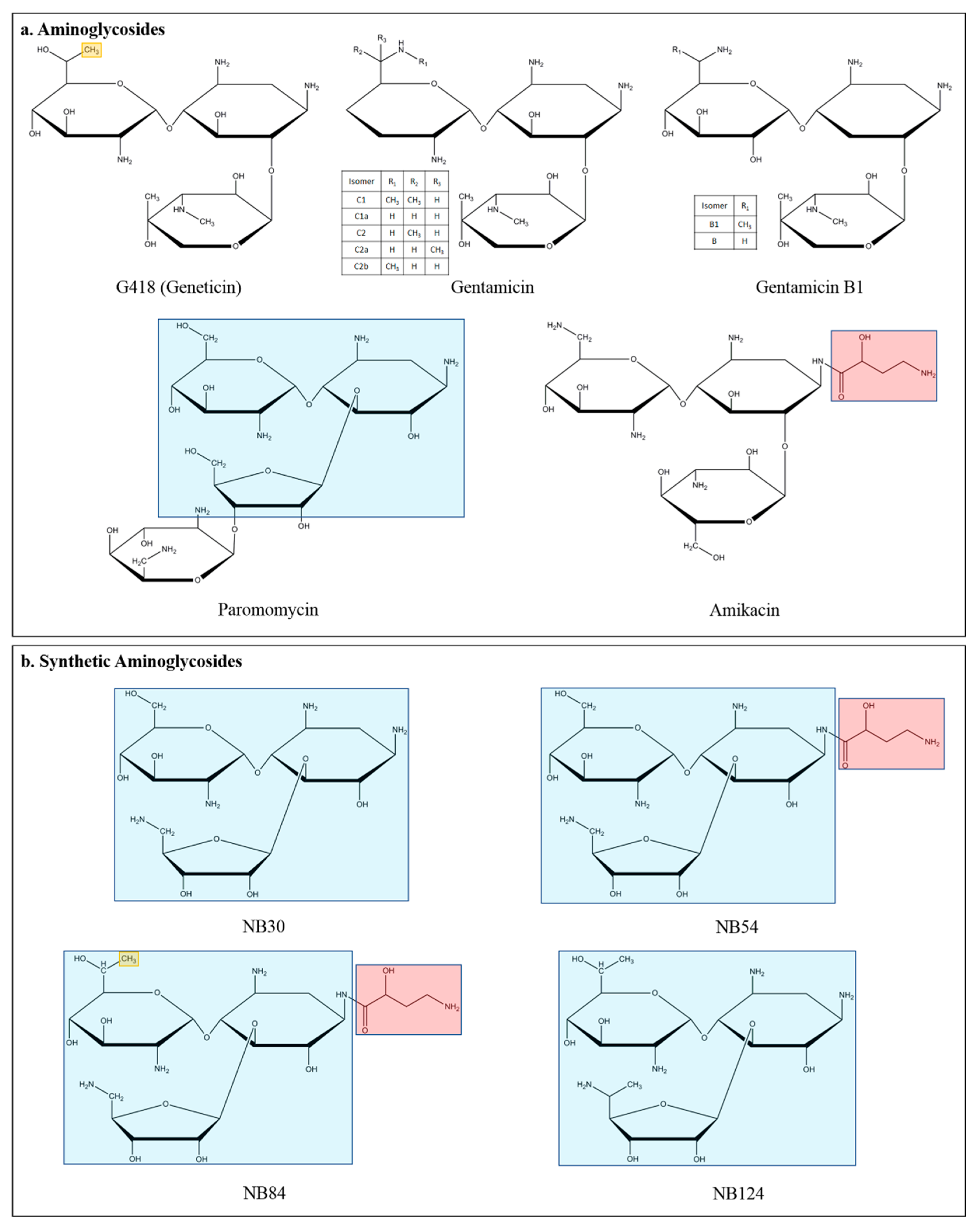
3.1. Aminoglycosides
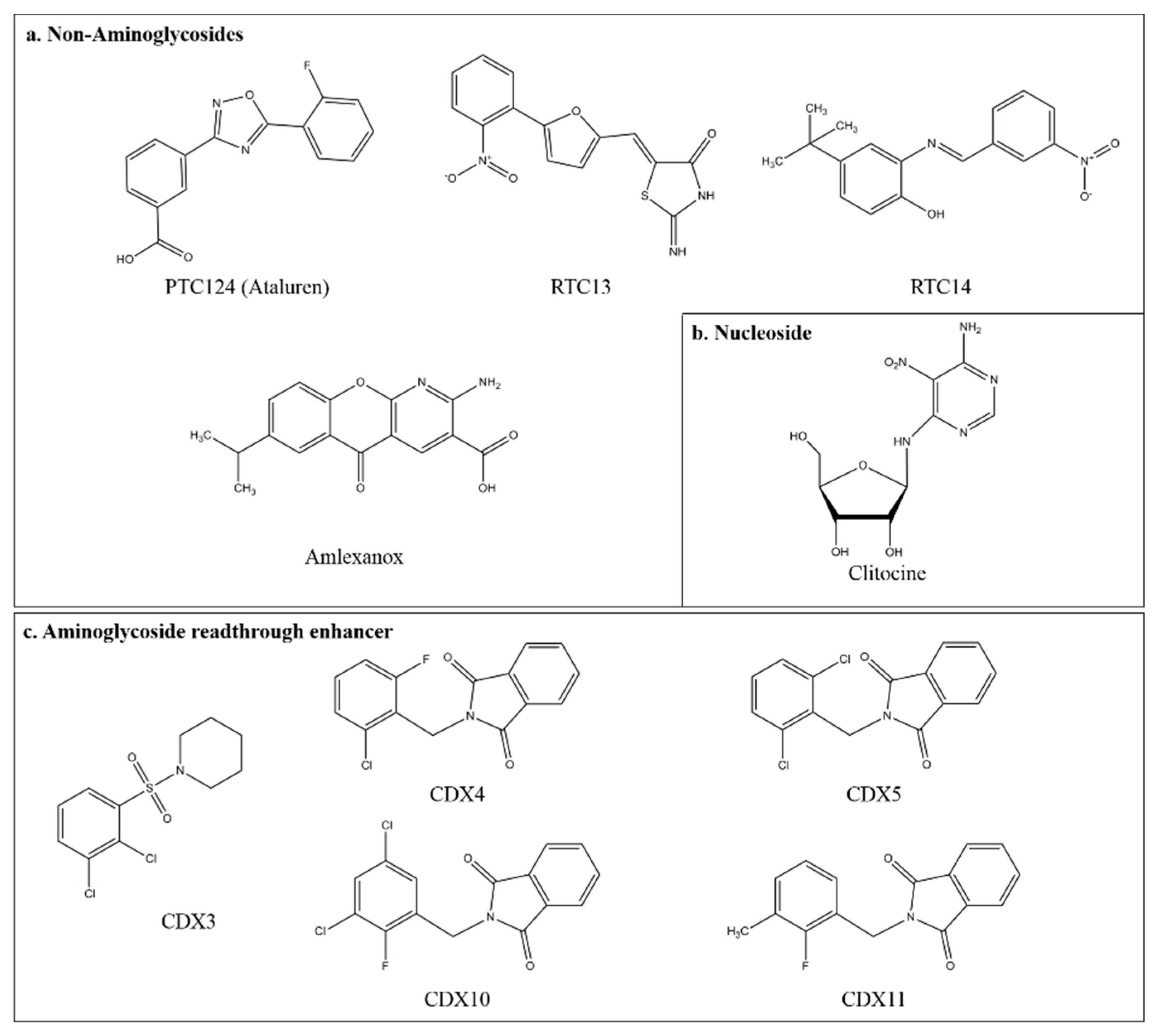
3.2. Ataluren (PTC124)
3.3. Novel Small Compounds with Nonsense Suppression Properties
4. Negative Modulation of Nonsense Mediated mRNA Decay
5. β-Thalassemia
5.1. Nonsense Mutations and Nonsense Mediated mRNA Decay in β-Thalassemia
5.2. Readthrough Approach for β-Thalassemia
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APC | adenomatous polyposis coli |
| CF | Cystic Fibrosis |
| CFTR | cystic fibrosis trans-membrane conductance regulator |
| DMD | Duchenne muscular dystrophy |
| HbA | Adult Hemoglobin (HbA) |
| HBE | human bronchial epithelial |
| HSs | hypersensitive sites |
| LCR | locus control region |
| LSD | lysosomal storage diseases |
| MPS I-H | mucopolysaccharidosis I-Hurler |
| NMD | Nonsense mRNA-mediated decay |
| PTCs | premature termination codons |
| UTRs | untranslated regions |
References
- Mort, M.; Ivanov, D.; Cooper, D.N.; Chuzhanova, N.A. A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat. 2008, 29, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Peltz, S.W.; Morsy, M.; Welch, E.M.; Jacobson, A. Ataluren as an agent for therapeutic nonsense suppression. Annu. Rev. Med. 2013, 64, 407–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shieh, P.B. Emerging strategies in the treatment of Duchenne muscular dystrophy. Neurotherapeutics 2018, 15, 840–848. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Ballabio, A.; Gieselmann, V. Lysosomal disorders: From storage to cellular damage. Biochim. Biophys. Acta 2009, 1793, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, P.D.; Raut, S.; Rivard, G.E.; Poon, M.C.; Warner, M.; McKenna, S.; Leggo, J.; Lillicrap, D. Aminoglycoside suppression of nonsense mutations in severe hemophilia. Blood 2005, 106, 3043–3048. [Google Scholar] [CrossRef]
- Gilad, S.; Bar-Shira, A.; Harnik, R.; Shkedy, D.; Ziv, Y.; Khosravi, R.; Brown, K.; Vanagaite, L.; Xu, G.; Frydman, M.; et al. Ataxia-telangiectasia: Founder effect among north African Jews. Hum. Mol. Genet. 1996, 12, 2033–2037. [Google Scholar] [CrossRef] [Green Version]
- Reiners, J.; Nagel-Wolfrum, K.; Jurgens, K.; Marker, T.; Wolfrum, U. Molecular basis of human Usher syndrome: Deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp. Eye Res. 2006, 83, 97–119. [Google Scholar] [CrossRef]
- Gwen, G.; Yanying, D.; Ming, D.; Valery, B.; Jeyakumar, K.; Trenton, R.S.; Timor, B.; David, M.B.; Kim, M.K. Long-term nonsense suppression therapy moderates MPS I-H disease progression. Mol. Genet. Metab. 2014, 111, 374–381. [Google Scholar]
- Keeling, K.M.; Xue, X.; Gunn, G.; Bedwell, D.M. Therapeutics based on stop codon readthrough. Annu. Rev. Genomics Hum. Genet. 2014, 15, 371–394. [Google Scholar] [CrossRef] [Green Version]
- Keeling, K.M.; Wang, D.; Conard, S.E.; Bedwell, D.M. Suppression of premature termination codons as a therapeutic approach. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 444–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurosaki, T.; Maquat, L.E. Nonsense mediated mRNA decay in humans at a glance. J. Cell Sci. 2016, 129, 461–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lejeune, F. Nonsense mediated mRNA decay at the crossroads of many cellular pathways. BMB Rep. 2017, 50, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brogna, S.; McLeod, T.; Petric, M. The meaning of NMD: Translate or perish. Trends Genet. 2016, 32, 395–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kervestin, S.; Jacobson, A.N. NMD: A multifaceted response to premature translational termination. Nat. Rev. Mol. Cell Biol. 2012, 13, 700–712. [Google Scholar] [CrossRef] [Green Version]
- Karousis, E.D.; Nasif, S.; Muhlemann, O. Nonsense mediated mRNA decay: Novel mechanistic insights and biological impact. WIRES RNA 2016, 7, 661–682. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Jacobson, A. Nonsense mediated mRNA decay: Degradation of defective transcripts is only part of the story. Annu. Rev. Genet. 2015, 49, 339–366. [Google Scholar] [CrossRef] [Green Version]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Bidou, L.; Allamand, V.; Rousset, J.P.; Namy, O. Sense from nonsense: Therapies for premature stop codon diseases. Trends Mol. Med. 2012, 18, 679–688. [Google Scholar] [CrossRef]
- Benhabiles, H.; Jia, J.; Lejeune, F. Nonsense Mutation Correction in Human Diseases: An Approach for Targeted Medicine, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 1–192. [Google Scholar]
- Wang, X.; Gregory-Evans, C.Y. Nonsense suppression therapies in ocular genetic diseases. Cell. Mol. Life Sci. 2015, 72, 1931–1938. [Google Scholar] [CrossRef]
- Keeling, K.M. Nonsense suppression as an approach to treat lysosomal storage diseases. Diseases 2016, 4, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, R.; Smart, M.; Tracey-White, D.; Webster, A.R.; Moosajee, M. Mechanism and evidence of nonsense suppression therapy for genetic eye disorders. Exp. Eye Res. 2017, 155, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Nagel-Wolfrum, K.; Moller, F.; Penner, I.; Baasov, T.; Wolfrum, U. Targeting nonsense mutations in diseases with Translational Read-through-Inducing Drugs (TRIDs). BioDrugs 2016, 30, 49–74. [Google Scholar] [CrossRef] [PubMed]
- Wolin, S.L.; Maquat, L.E. Cellular RNA surveillance in health and disease. Science 2019, 366, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol. Med. 2018, 24, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campofelice, A.; Lentini, L.; Di Leonardo, A.; Melfi, R.; Tutone, M.; Pace, A.; Pibiri, I. Strategies against nonsense: Oxadiazoles as Translational Readthrough-Inducing Drugs (TRIDs). Int. J. Mol. Sci. 2019, 20, 3329. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.C.; Kan, Y.W. beta 0 thalassemia, a nonsense mutation in man. Proc. Natl. Acad. Sci. USA 1979, 76, 2886–2889. [Google Scholar] [CrossRef] [Green Version]
- Maquat, L.E.; Kinniburgh, A.J.; Rachmilewitz, E.A.; Ross, J. Unstable beta-globin mRNA in mRNA-deficient beta o thalassemia. Cell 1981, 27, 543–553. [Google Scholar] [CrossRef]
- Temple, G.F.; Dozy, A.M.; Roy, K.L.; Kan, Y.W. Construction of a functional human suppressor tRNA gene: An approach to gene therapy for beta-thalassaemia. Nature 1982, 296, 537–540. [Google Scholar] [CrossRef]
- Hug, N.; Longman, D.; Caceres, J.F. Mechanism and regulation of the nonsense mediated decay pathway. Nucleic Acids Res. 2016, 44, 1483–1495. [Google Scholar] [CrossRef] [Green Version]
- Fatscher, T.; Boehm, V.; Gehring, N.H. Mechanism, factors, and physiological role of nonsense mediated mRNA decay. Cell Mol. Life Sci. 2015, 72, 4523–4544. [Google Scholar] [CrossRef] [PubMed]
- Celik, A.; Kervestin, S.; Jacobson, A. NMD: At the crossroads between translation termination and ribosome recycling. Biochimie 2015, 114, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altamura, N.; Groudinsky, O.; Dujardin, G.; Slonimski, P.P. NAM7 nuclear gene encodes a novel member of a family of helicases with a Zn-ligand motif and is involved in mitochondrial functions in Saccharomyces cerevisiae. J. Mol. Biol. 1992, 224, 575–587. [Google Scholar] [CrossRef]
- Weng, Y.; Czaplinski, K.; Peltz, S.W. Genetic and biochemical characterization of mutations in the ATPase and helicase regions of the Upf1 protein. Mol. Cell. Biol. 1996, 16, 5477–5490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, A.; Czaplinski, K.; Trifillis, P.; He, F.; Jacobson, A.; Peltz, S.W. Characterization of the biochemical properties of the human Upf1 gene product that is involved in nonsense mediated mRNA decay. RNA 2000, 6, 1226–1235. [Google Scholar] [CrossRef] [Green Version]
- Nickless, A.; Bailis, J.M.; You, Z. Control of gene expression through the nonsense mediated RNA decay pathway. Cell Biosci. 2017, 7, 26. [Google Scholar] [CrossRef]
- Celik, A.; He, F.; Jacobson, A. NMD monitors translational fidelity 24/7. Curr. Genet. 2017, 63, 1007–1010. [Google Scholar] [CrossRef] [Green Version]
- Celik, A.; Baker, R.; He, F.; Jacobson, A. High-resolution profiling of NMD targets in yeast reveals translational fidelity as a basis for substrate selection. RNA 2017, 23, 735–748. [Google Scholar] [CrossRef] [Green Version]
- Kerem, E. Pharmacologic therapy for stop mutations: How much CFTR activity is enough? Curr. Opin. Pulm. Med. 2004, 10, 547–552. [Google Scholar] [CrossRef]
- Xue, X.; Mutyam, V.; Thakerar, A.; Mobley, J.; Bridges, R.J.; Rowe, S.M.; Keeling, K.M.; Bedwell, D.M. Identification of the amino acids inserted during suppression of CFTR nonsense mutations and determination of their functional consequences. Hum. Mol. 2017, 26, 3116–3129. [Google Scholar] [CrossRef] [Green Version]
- Roy, B.; Friesen, W.J.; Tomizawa, Y.; Leszyk, J.D.; Zhuo, J.; Johnson, B.; Dakka, J.; Trotta, C.R.; Xue, X.; Mutyam, V.; et al. Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc. Natl. Acad. Sci. USA 2016, 113, 12508–12513. [Google Scholar] [CrossRef] [Green Version]
- Roy, B.; Leszyk, J.D.; Mangus, D.A.; Jacobson, A. Nonsense suppression by near-cognate tRNAs employs alternative base pairing at codon positions 1 and 3. Proc. Natl. Acad. Sci. USA 2015, 112, 3038–3043. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Ursic, D.; Davies, J. Phenotypic suppression and misreading Saccharomyces cerevisiae. Nature 1979, 277, 146–148. [Google Scholar] [CrossRef]
- Palmer, E.; Wilhelm, J.M.; Sherman, F. Phenotypic suppression of nonsense mutants in yeast by aminoglycoside antibiotics. Nature 1979, 277, 148–150. [Google Scholar] [CrossRef]
- Howard, M.; Frizzell, R.A.; Bedwell, D.M. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat. Med. 1996, 2, 467–469. [Google Scholar] [CrossRef]
- Lee, H.L.; Dougherty, J.P. Pharmaceutical therapies to recode nonsense mutations in inherited diseases. Pharmacol. Ther. 2012, 136, 227–266. [Google Scholar] [CrossRef]
- Francis, S.P.; Katz, J.; Fanning, K.D.; Harris, K.A.; Nicholas, B.D.; Lacy, M.; Pagana, J.; Agris, P.F.; Shin, J.B. A novel role of cytosolic protein synthesis inhibition in aminoglycoside ototoxicity. J. Neurosci. 2013, 33, 3079–3093. [Google Scholar] [CrossRef] [Green Version]
- Hobbie, S.N.; Akshay, S.; Kalapala, S.K.; Bruell, C.M.; Shcherbakov, D.; Bottger, E.C. Genetic analysis of interactions with eukaryotic rRNA identify the mitoribosome as target in aminoglycoside ototoxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 20888–20893. [Google Scholar] [CrossRef] [Green Version]
- Kandasamy, J.; Atia-Glikin, D.; Shulman, E.; Shapira, K.; Shavit, M.; Belakhov, V.; Baasov, T. Increased selectivity toward cytoplasmic versus mitochondrial ribosome confers improved efficiency of synthetic aminoglycosides in fixing damaged genes: A strategy for treatment of genetic diseases caused by nonsense mutations. J. Med. Chem. 2012, 55, 10630–10643. [Google Scholar] [CrossRef] [Green Version]
- Matt, T.; Ng, C.L.; Lang, K.; Sha, S.H.; Akbergenov, R.; Shcherbakov, D.; Meyer, M.; Duscha, S.; Xie, J.; Dubbaka, S.R.; et al. Dissociation of antibacterial activity and aminoglycoside ototoxicity in the 4-monosubstituted 2-deoxystreptamine apramycin. Proc. Natl. Acad. Sci. USA 2012, 109, 10984–10989. [Google Scholar] [CrossRef] [Green Version]
- Nudelman, I.; Rebibo-Sabbah, A.; Cherniavsky, M.; Belakhov, V.; Hainrichson, M.; Chen, F.; Schacht, J.; Pilch, D.S.; Ben-Yosef, T.; Baasov, T. Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J. Med. Chem. 2009, 52, 2836–2845. [Google Scholar] [CrossRef] [Green Version]
- Rowe, S.M.; Sloane, P.; Tang, L.P.; Backer, K.; Mazur, M.; Buckley-Lanier, J.; Nudelman, I.; Belakhov, V.; Bebok, Z.; Schwiebert, E.; et al. Suppression of CFTR premature termination codons and rescue of CFTR protein and function by the synthetic aminoglycoside NB54. J. Mol. Med. 2011, 89, 1149–1161. [Google Scholar] [CrossRef] [Green Version]
- Vecsler, M.; Ben Zeev, B.; Nudelman, I.; Anikster, Y.; Simon, A.J.; Amariglio, N.; Rechavi, G.; Baasov, T.; Gak, E. Ex vivo treatment with a novel synthetic aminoglycoside NB54 in primary fibroblasts from Rett syndrome patients suppresses MECP2 nonsense mutations. PLoS ONE 2011, 6, e20733. [Google Scholar] [CrossRef]
- Goldmann, T.; Overlack, N.; Moller, F.; Belakhov, V.; van Wyk, M.; Baasov, T.; Wolfrum, U.; Nagel-Wolfrum, K. A comparative evaluation of NB30, NB54 and PTC124 in translational readthrough efficacy for treatment of an USH1C nonsense mutation. EMBO Mol. Med. 2012, 4, 1186–1199. [Google Scholar] [CrossRef]
- Brendel, C.; Belakhov, V.; Werner, H.; Wegener, E.; Gartner, J.; Nudelman, I.; Baasov, T.; Huppke, P. Readthrough of nonsense mutations in Rett syndrome: Evaluation of novel aminoglycosides and generation of a new mouse model. J. Mol. Med. 2011, 89, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Belakhov, V.; Kandasamy, J.; Baasov, T.; Li, S.C.; Li, Y.T.; Bedwell, D.M.; Keeling, K.M. The designer aminoglycoside NB84 significantly reduces glycosaminoglycan accumulation associated with MPS I-H in the Idua-W392X mouse. Mol. Genet. Metab. 2012, 105, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Keeling, K.M.; Wang, D.; Dai, Y.; Murugesan, S.; Chenna, B.; Clark, J.; Belakhov, V.; Kandasamy, J.; Velu, S.E.; Baasov, T.; et al. Attenuation of nonsense mediated mRNA decay enhances in vivo nonsense suppression. PLoS ONE 2013, 8, e60478. [Google Scholar] [CrossRef] [Green Version]
- Xue, X.; Mutyam, V.; Tang, L.; Biswas, S.; Du, M.; Jackson, L.A.; Dai, Y.; Belakhov, V.; Shalev, M.; Chen, F.; et al. Synthetic aminoglycosides efficiently suppress cystic fibrosis transmembrane conductance regulator nonsense mutations and are enhanced by ivacaftor. Am. J. Respir. Cell. Mol. Biol. 2014, 50, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Bidou, L.; Bugaud, O.; Belakhov, V.; Baasov, T.; Namy, O. Characterization of new-generation aminoglycoside promoting premature termination codon readthrough in cancer cells. RNA Biol. 2017, 14, 378–388. [Google Scholar] [CrossRef] [Green Version]
- Sabbavarapu, N.M.; Shavit, M.; Degani, Y.; Smolkin, B.; Belakhov, V.; Baasov, T. Design of novel aminoglycoside derivatives with enhanced suppression of diseases-causing nonsense mutations. ACS Med. Chem. Lett. 2016, 7, 418–423. [Google Scholar] [CrossRef] [Green Version]
- Baradaran-Heravi, A.; Niesser, J.; Balgi, A.D.; Choi, K.; Zimmerman, C.; South, A.P.; Anderson, H.J.; Strynadka, N.C.; Bally, M.B.; Roberge, M. Gentamicin B1 is a minor gentamicin component with major nonsense mutation suppression activity. Proc. Natl. Acad. Sci. USA 2017, 114, 3479–3484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baradaran-Heravi, A.; Balgi, A.D.; Zimmerman, C.; Choi, K.; Shidmoossavee, F.S.; Tan, J.S.; Bergeaud, C.; Krause, A.; Flibotte, S.; Shimizu, Y.; et al. Novel small molecules potentiate premature termination codon readthrough by aminoglycosides. Nucleic Acids Res. 2016, 44, 6583–6598. [Google Scholar] [CrossRef] [PubMed]
- Mutyam, V.; Du, M.; Xue, X.; Keeling, K.M.; White, E.L.; Bostwick, J.R.; Rasmussen, L.; Liu, B.; Mazur, M.; Hong, J.S.; et al. Discovery of clinically approved agents that promote suppression of CFTR nonsense mutations. Am. J. Respir. Crit. Care Med. 2016, 194, 1092–1103. [Google Scholar] [CrossRef] [Green Version]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef]
- Du, M.; Liu, X.; Welch, E.M.; Hirawat, S.; Peltz, S.W.; Bedwell, D.M. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc. Natl. Acad. Sci. USA 2008, 105, 2064–2069. [Google Scholar] [CrossRef] [Green Version]
- Sermet-Gaudelus, I.; Boeck, K.D.; Casimir, G.J.; Vermeulen, F.; Leal, T.; Mogenet, A.; Roussel, D.; Fritsch, J.; Hanssens, L.; Hirawat, S.; et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 1262–1272. [Google Scholar] [CrossRef]
- Kerem, E.; Hirawat, S.; Armoni, S.; Yaakov, Y.; Shoseyov, D.; Cohen, M.; Nissim-Rafinia, M.; Blau, H.; Rivlin, J.; Aviram, M.; et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: A prospective phase II trial. Lancet 2008, 372, 719–727. [Google Scholar] [CrossRef]
- Wilschanski, M.; Miller, L.L.; Shoseyov, D.; Blau, H.; Rivlin, J.; Aviram, M.; Cohen, M.; Armoni, S.; Yaakov, Y.; Pugatsch, T.; et al. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur. Respir. J. 2011, 38, 59–69. [Google Scholar] [CrossRef]
- Finkel, R.S.; Flanigan, K.M.; Wong, B.; Bonnemann, C.; Sampson, J.; Sweeney, H.L.; Reha, A.; Northcutt, V.J.; Elfring, G.; Barth, J.; et al. Phase 2a study of ataluren mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS ONE 2013, 8, e81302. [Google Scholar] [CrossRef]
- McElroy, S.P.; Nomura, T.; Torrie, L.S.; Warbrick, E.; Gartner, U.; Wood, G.; McLean, W.H. A lack of premature termination codon readthrough efficacy of PTC124 (Ataluren) in a diverse array of reporter assays. PLoS Biol. 2013, 11, e1001593. [Google Scholar] [CrossRef] [Green Version]
- Auld, D.S.; Thorne, N.; Maguire, W.F.; Inglese, J. Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc. Natl. Acad. Sci. USA 2009, 106, 3585–3590. [Google Scholar] [CrossRef] [Green Version]
- Pibiri, I.; Lentini, L.; Tutone, M.; Melfi, R.; Pace, A.; Di Leonardo, A. Exploring the readthrough of nonsense mutations by non-acidic Ataluren analogues selected by ligand-based virtual screening. Eur. J. Med. Chem. 2016, 122, 429–435. [Google Scholar] [CrossRef]
- Bolze, F.; Mocek, S.; Zimmermann, A.; Klingenspor, M. Aminoglycosides, but not PTC124 (Ataluren), rescue nonsense mutations in the leptin receptor and in luciferase reporter genes. Sci. Rep. 2017, 7, 1020. [Google Scholar] [CrossRef] [Green Version]
- Kerem, E.; Konstan, M.W.; De Boeck, K.; Accurso, F.J.; Sermet-Gaudelus, I.; Wilschanski, M.; Elborn, J.S.; Melotti, P.; Bronsveld, I.; Fajac, I.; et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir. Med. 2014, 2, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Blanchet, S.; Cornu, D.; Argentini, M.; Namy, O. New insights into the incorporation of natural suppressor tRNAs at stop codons in Saccharomyces cerevisiae. Nucleic Acids Res. 2014, 42, 10061–10072. [Google Scholar] [CrossRef] [Green Version]
- Manuvakhova, M.; Keeling, K.; Bedwell, D.M. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA 2000, 6, 1044–1055. [Google Scholar] [CrossRef] [Green Version]
- Bonetti, B.; Fu, L.; Moon, J.; Bedwell, D.M. The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae. J. Mol. Biol. 1995, 251, 334–345. [Google Scholar] [CrossRef]
- Du, L.; Damoiseaux, R.; Nahas, S.; Gao, K.; Hu, H.; Pollard, J.M.; Goldstine, J.; Jung, M.E.; Henning, S.M.; Bertoni, C.; et al. Nonaminoglycoside compounds induce readthrough of nonsense mutations. J. Exp. Med. 2009, 206, 2285–2297. [Google Scholar] [CrossRef]
- Kayali, R.; Ku, J.M.; Khitrov, G.; Jung, M.E.; Prikhodko, O.; Bertoni, C. Read-through compound 13 restores dystrophin expression and improves muscle function in the mdx mouse model for Duchenne muscular dystrophy. Hum. Mol. Genet. 2012, 21, 4007–4020. [Google Scholar] [CrossRef] [Green Version]
- Zilberberg, A.; Lahav, L.; Rosin-Arbesfeld, R. Restoration of APC gene function in colorectal cancer cells by aminoglycoside- and macrolide-induced readthrough of premature termination codons. Gut 2010, 59, 496–507. [Google Scholar] [CrossRef]
- Caspi, M.; Firsow, A.; Rajkumar, R.; Skalka, N.; Moshkovitz, I.; Munitz, A.; Pasmanik-Chor, M.; Greif, H.; Megido, D.; Kariv, R.; et al. A flow cytometry-based reporter assay identifies macrolide antibiotics as nonsense mutation readthrough agents. J. Mol. Med. 2016, 94, 469–482. [Google Scholar] [CrossRef]
- Floquet, C.; Rousset, J.P.; Bidou, L. Readthrough of premature termination codons in the adenomatous polyposis coli gene restores its biological activity in human cancer cells. PLoS ONE 2011, 6, e24125. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, M.; Shiozuka, M.; Nakayama, Y.; Hara, T.; Hamada, M.; Kondo, S.; Ikeda, D.; Takahashi, Y.; Sawa, R.; Nonomura, Y.; et al. Negamycin restores dystrophin expression in skeletal and cardiac muscles of mdx mice. J. Biochem. 2003, 134, 751–758. [Google Scholar] [CrossRef]
- Gonzalez-Hilarion, S.; Beghyn, T.; Jia, J.; Debreuck, N.; Berte, G.; Mamchaoui, K.; Mouly, V.; Gruenert, D.C.; Deprez, B.; Lejeune, F. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J. Rare Dis. 2012, 7, 58. [Google Scholar] [CrossRef] [Green Version]
- Pibiri, I.; Lentini, L.; Melfi, R.; Gallucci, G.; Pace, A.; Spinello, A.; Barone, G.; Di Leonardo, A. Enhancement of premature stop codon readthrough in the CFTR gene by Ataluren (PTC124) derivatives. Eur. J. Med. Chem. 2015, 101, 236–244. [Google Scholar] [CrossRef]
- Moosajee, M.; Tracey-White, D.; Smart, M.; Weetall, M.; Torriano, S.; Kalatzis, V.; da Cruz, L.; Coffey, P.; Webster, A.R.; Welch, E. Functional rescue of REP1 following treatment with PTC124 and novel derivative PTC-414 in human choroideremia fibroblasts and the nonsense mediated zebrafish model. Hum. Mol. Genet. 2016, 25, 3416–3431. [Google Scholar] [CrossRef] [Green Version]
- Friesen, W.J.; Trotta, C.R.; Tomizawa, Y.; Zhuo, J.; Johnson, B.; Sierra, J.; Roy, B.; Weetall, M.; Hedrick, J.; Sheedy, J.; et al. The nucleoside analog clitocine is a potent and efficacious readthrough agent. RNA 2017, 23, 567–577. [Google Scholar] [CrossRef] [Green Version]
- Khajavi, M.; Inoue, K.; Lupski, J.R. Nonsense mediated mRNA decay modulates clinical outcome of genetic disease. Eur. J. Hum. Genet. 2006, 14, 1074–1081. [Google Scholar] [CrossRef] [Green Version]
- Linde, L.; Boelz, S.; Nissim-Rafinia, M.; Oren, Y.S.; Wilschanski, M.; Yaacov, Y.; Virgilis, D.; Neu-Yilik, G.; Kulozik, A.E.; Kerem, E.; et al. Nonsense mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J. Clin. Investig. 2007, 117, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Bhuvanagiri, M.; Schlitter, A.M.; Hentze, M.W.; Kulozik, A.E. NMD: RNA biology meets human genetic medicine. Biochem. J. 2010, 430, 365–377. [Google Scholar] [CrossRef] [Green Version]
- Linde, L.; Boelz, S.; Neu-Yilik, G.; Kulozik, A.E.; Kerem, B. The efficiency of nonsense mediated mRNA decay is an inherent character and varies among different cells. Eur. J. Hum. Genet. 2007, 15, 1156–1162. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.S.; Wilkinson, M.F.; Gecz, J. Nonsense mediated mRNA decay: Inter-individual variability and human disease. Neurosci. Biobehav. Rev. 2014, 46, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Lejeune, F. Triple effect of nonsense mediated mRNA decay inhibition as a therapeutic approach for cancer. Single Cell Biol. 2016, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Bhuvanagiri, M.; Lewis, J.; Putzker, K.; Becker, J.P.; Leicht, S.; Krijgsveld, J.; Batra, R.; Turnwald, B.; Jovanovic, B.; Hauer, C.; et al. 5-azacytidine inhibits nonsense mediated decay in a MYC-dependent fashion. EMBO Mol. Med. 2014, 6, 1593–1609. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Nunez, R.T.; Wallace, A.; Coyne, D.; Jansson, L.; Rush, M.; Ennajdaoui, H.; Katzman, S.; Bailey, J.; Deinhardt, K.; Sanchez-Elsner, T.; et al. Modulation of nonsense mediated decay by rapamycin. Nucleic Acids Res. 2017, 45, 3448–3459. [Google Scholar] [CrossRef] [Green Version]
- Cao, A.; Galanello, R. Beta-thalassemia. Genet. Med. 2010, 12, 61–76. [Google Scholar] [CrossRef] [Green Version]
- Galanello, R.; Origa, R. Beta-thalassemia. Orphanet J. Rare Dis. 2010, 5, 1172–1175. [Google Scholar] [CrossRef] [Green Version]
- Huisman, T.H. The structure and function of normal and abnormal haemoglobins. Baillieres Clin. Haematol. 1993, 6, 1–30. [Google Scholar] [CrossRef]
- Piras, I.; Vona, G.; Falchi, A.; Latini, V.; Ristaldi, S.; Vacca, L.; Varesi, L.; Calo, C.M. β-globin cluster haplotypes in normal individuals and β(0)39-thalassemia carriers from Sardinia, Italy. Am. J. Hum. Biol. 2005, 17, 765–772. [Google Scholar] [CrossRef]
- Trecartin, R.F.; Liebhaber, S.A.; Chang, J.C.; Lee, K.Y.; Kan, Y.W.; Furbetta, M.; Angius, A.; Cao, A. beta zero thalassemia in Sardinia is caused by a nonsense mutation. J. Clin. Investig. 1981, 68, 1012–1017. [Google Scholar] [CrossRef] [Green Version]
- Stalder, L.; Muhlemann, O. The meaning of nonsense. Trends Cell Biol. 2008, 18, 315–321. [Google Scholar] [CrossRef]
- Holbrook, J.A.; Neu-Yilik, G.; Hentze, M.W.; Kulozik, A.E. Nonsense mediated decay approaches the clinic. Nat. Genet. 2004, 36, 801–808. [Google Scholar] [CrossRef]
- Peixeiro, I.; Silva, A.L.; Romao, L. Control of human beta-globin mRNA stability and its impact on beta-thalassemia phenotype. Haematologica 2011, 96, 905–913. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, X.; Qian, Y.; Maquat, L.E. Intron function in the nonsense mediated decay of beta-globin mRNA: Indications that pre-mRNA splicing in the nucleus can influence mRNA translation in the cytoplasm. RNA 1998, 4, 801–815. [Google Scholar] [CrossRef]
- Thermann, R.; Neu-Yilik, G.; Deters, A.; Frede, U.; Wehr, K.; Hagemeier, C.; Hentze, M.W.; Kulozik, A.E. Binary specification of nonsense codons by splicing and cytoplasmic translation. EMBO J. 1998, 17, 3484–3494. [Google Scholar] [CrossRef]
- Baserga, S.J.; Benz, E.J., Jr. Nonsense mutations in the human beta-globin gene affect mRNA metabolism. Proc. Natl. Acad. Sci. USA 1988, 85, 2056–2060. [Google Scholar] [CrossRef] [Green Version]
- Hall, G.W.; Thein, S. Nonsense codon mutations in the terminal exon of the beta-globin gene are not associated with a reduction in beta-mRNA accumulation: A mechanism for the phenotype of dominant beta-thalassemia. Blood 1994, 83, 2031–2037. [Google Scholar] [CrossRef] [Green Version]
- Neu-Yilik, G.; Gehring, N.H.; Thermann, R.; Frede, U.; Hentze, M.W.; Kulozik, A.E. Splicing and 3’ end formation in the definition of nonsense mediated decay-competent human beta-globin mRNPs. EMBO J. 2001, 20, 532–540. [Google Scholar] [CrossRef] [Green Version]
- Salvatori, F.; Cantale, V.; Breveglieri, G.; Zuccato, C.; Finotti, A.; Bianchi, N.; Borgatti, M.; Feriotto, G.; Destro, F.; Canella, A.; et al. Development of K562 cell clones expressing beta-globin mRNA carrying the beta039 thalassaemia mutation for the screening of correctors of stop-codon mutations. Biotechnol. Appl. Biochem. 2009, 54, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Salvatori, F.; Breveglieri, G.; Zuccato, C.; Finotti, A.; Bianchi, N.; Borgatti, M.; Feriotto, G.; Destro, F.; Canella, A.; Brognara, E.; et al. Production of beta-globin and adult hemoglobin following G418 treatment of erythroid precursor cells from homozygous beta (0) 39 thalassemia patients. Am. J. Hematol. 2009, 84, 720–728. [Google Scholar] [CrossRef] [Green Version]
- Gardner, L.B. Hypoxic inhibition of nonsense mediated RNA decay regulates gene expression and the integrated stress response. Mol. Cell. Biol. 2008, 28, 3729–3741. [Google Scholar] [CrossRef] [Green Version]
- Koren, A.; Levin, C.; Dgany, O.; Kransnov, T.; Elhasid, R.; Zalman, L.; Palmor, H.; Tamary, H. Response to hydroxyurea therapy in beta-thalassemia. Am. J. Hematol. 2008, 83, 366–370. [Google Scholar] [CrossRef]
- Singh, G.; Rebbapragada, I.; Lykke-Andersen, J. A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense mediated mRNA decay. PLoS Biol. 2008, 6, e111. [Google Scholar] [CrossRef]
- Usuki, F.; Yamashita, A.; Kashima, I.; Higuchi, I.; Osame, M.; Ohno, S. Specific inhibition of nonsense mediated mRNA decay components, SMG-1 or Upf1, rescues the phenotype of Ullrich disease fibroblasts. Mol. Ther. 2006, 14, 351–360. [Google Scholar] [CrossRef]
- Chandrakasan, S.; Malik, P. Gene therapy for hemoglobinopathies: The state of the field and the future. Hematol. Oncol. Clin. 2014, 28, 199–216. [Google Scholar] [CrossRef] [Green Version]
- Kar, D.; Sellamuthu, K.; Kumar, S.D.; Eswarappa, S.M. Induction of Translational Readthrough across the Thalassemia-Causing Premature Stop Codon in β-Globin-Encoding mRNA. Biochemistry 2019. [Google Scholar] [CrossRef]
- Premature-Termination-Codons Readthrough Compounds. U.S. Patent 9255088, 21 March 2017. Available online: http://www.freepatentsonline.com/9255088.html (accessed on 9 February 2016).
- Readthrough Inducing Agent and Drug for Treating Genetic Disease Caused by Nonsense Mutation. U.S. Patent 9358246, 7 June 2016. Available online: http://www.freepatentsonline.com/9358246.html (accessed on 6 July 2016).
- Aminoglycosides and Uses Thereof in Treating Genetic Disorders. U.S. Patent 10398718, 3 September 2019. Available online: http://www.freepatentsonline.com/10398718.html (accessed on 9 March 2019).
- Oikonomidou, P.R.; Rivella, S. What can we learn from ineffective erythropoiesis in thalassemia? Blood Rev. 2018, 32, 130–143. [Google Scholar] [CrossRef]
- Popp, M.W.; Maquat, L.E. Leveraging rules of nonsense mediated mRNA decay for genome engineering and personalized medicine. Cell 2016, 165, 1319–1322. [Google Scholar] [CrossRef] [Green Version]
- Schweingruber, C.; Rufener, S.C.; Zund, D.; Yamashita, A.; Muhlemann, O. Nonsense mediated mRNA decay-mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochim. Biophys. Acta 2013, 1829, 612–623. [Google Scholar] [CrossRef]
- Smith, J.E.; Baker, K.E. Nonsense mediated RNA decay--a switch and dial for regulating gene expression. Bioessays 2015, 37, 612–623. [Google Scholar] [CrossRef] [Green Version]
- Nasif, S.; Contu, L.; Muhlemann, O. Beyond quality control: The role of nonsense mediated mRNA decay (NMD) in regulating gene expression. Semin. Cell Dev. Biol. 2018, 75, 78–87. [Google Scholar] [CrossRef]
- Goetz, A.E.; Wilkinson, M. Stress and the nonsense mediated RNA decay pathway. Cell Mol. Life Sci. 2017, 74, 3509–3531. [Google Scholar] [CrossRef]
- Hoek, T.A.; Khuperkar, D.; Lindeboom, R.G.; Sonneveld, S.; Verhagen, B.M.; Boersma, S.; Vermeulen, M.; Tanenbaum, M.E. Single-molecule imaging uncovers rules governing nonsense mediated mRNA decay. Mol. Cell 2019, 75, 324–339. [Google Scholar] [CrossRef]
- Lindeboom, R.G.; Vermeulen, M.; Lehner, B.; Supek, F. The impact of nonsense mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 2019, 51, 1645–1651. [Google Scholar] [CrossRef]
- Gambari, R.; Fibach, E. Medicinal chemistry of fetal hemoglobin inducers for treatment of beta-thalassemia. Curr. Med. Chem. 2007, 14, 199–212. [Google Scholar] [CrossRef]
- Baronciani, D.; Angelucci, E.; Potschger, U.; Gaziev, J.; Yesilipek, A.; Zecca, M.; Orofino, M.G.; Giardini, C.; Al-Ahmari, A.; Marktel, S.; et al. Hemopoietic stem cell transplantation in thalassemia: A report from the European Society for Blood and Bone Marrow Transplantation Hemoglobinopathy Registry, 2000–2010. Bone Marrow Transp. 2016, 51, 536–541. [Google Scholar] [CrossRef] [Green Version]
- Altamura, E.; Borgatti, M.; Finotti, A.; Gasparello, J.; Gambari, R.; Spinelli, M.; Castaldo, R.; Altamura, N. Chemical-induced readthrough at premature termination codons determined by a rapid dual-fluorescence system based on S. cerevisiae. PLoS ONE 2016, 11, e0154260. [Google Scholar] [CrossRef]
- Lentini, L.; Melfi, R.; Cancemi, P.; Pibiri, I.; Di Leonardo, A. Caffeine boosts Ataluren’s readthrough activity. Heliyon 2019, 5, e01963. [Google Scholar] [CrossRef] [Green Version]
- Jagannathan, S.; Bradley, R.K. Translational plasticity facilitates the accumulation of nonsense genetic variants in the human population. Genome Res. 2016, 26, 1639–1650. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Nonsense Mutation | Sense/Nonsense Codon | Nucleotide Substitution | Frequency | NMD Activation | Reference |
|---|---|---|---|---|---|
| β°15 | TGG/TAG | G→A | Bangladesh 10% | NO | 97 |
| β°15 | TGG/TGA | G→A | Portugal 11.79% Russia 6.45% | NO | 98 |
| β°17 | AAG/TAG | A→T | Thailand 18.56% China 14.1% | NO | 98 |
| β°22 | GAA/TAA | G→T | La Réunion (one case) | NO | 98 |
| β°26 | GAG/TAG | G→T | Thailand 0.12% | YES | 99 |
| β°35 | TAC/TAA | C→A | Thailand 1.22% | YES | 99 |
| β°37 | TGG/TAG | G→A | Afghanistan | YES | 100 |
| β°37 | TGG/TGA | G→A | Arab countries 18.6% | YES | 101 |
| β°39 | CAG/TAG | C→T | Italy 66.84%; Argentina 47.06%; Portugal 34.9%; England 34.78% | YES | 102 |
| β°43 | GAG/TAG | G→T | Singapore 0.75%; Thailand 0.37% | YES | 103 |
| β°59 | AAG/TAG | A→T | Italian-American Family | YES | 104 |
| β°61 | AAG/TAG | A→T | USA (one case) | YES | 105 |
| β°90 | GAG/TAG | G→T | Japan 13.8% | YES | 99 |
| β°112 | TGT/TGA | T→A | Slovakia (one case) | YES | 96 |
| β°121 | GAA/TAA | G→T | Czechoslovakia 11.83% England 13.04% | YES | 106 |
| β°127 | CAG/TAG | C→T | England (one case) | NO | 107 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borgatti, M.; Altamura, E.; Salvatori, F.; D’Aversa, E.; Altamura, N. Screening Readthrough Compounds to Suppress Nonsense Mutations: Possible Application to β-Thalassemia. J. Clin. Med. 2020, 9, 289. https://doi.org/10.3390/jcm9020289
Borgatti M, Altamura E, Salvatori F, D’Aversa E, Altamura N. Screening Readthrough Compounds to Suppress Nonsense Mutations: Possible Application to β-Thalassemia. Journal of Clinical Medicine. 2020; 9(2):289. https://doi.org/10.3390/jcm9020289
Chicago/Turabian StyleBorgatti, Monica, Emiliano Altamura, Francesca Salvatori, Elisabetta D’Aversa, and Nicola Altamura. 2020. "Screening Readthrough Compounds to Suppress Nonsense Mutations: Possible Application to β-Thalassemia" Journal of Clinical Medicine 9, no. 2: 289. https://doi.org/10.3390/jcm9020289