Ophthalmological Findings in Mucopolysaccharidoses

Abstract
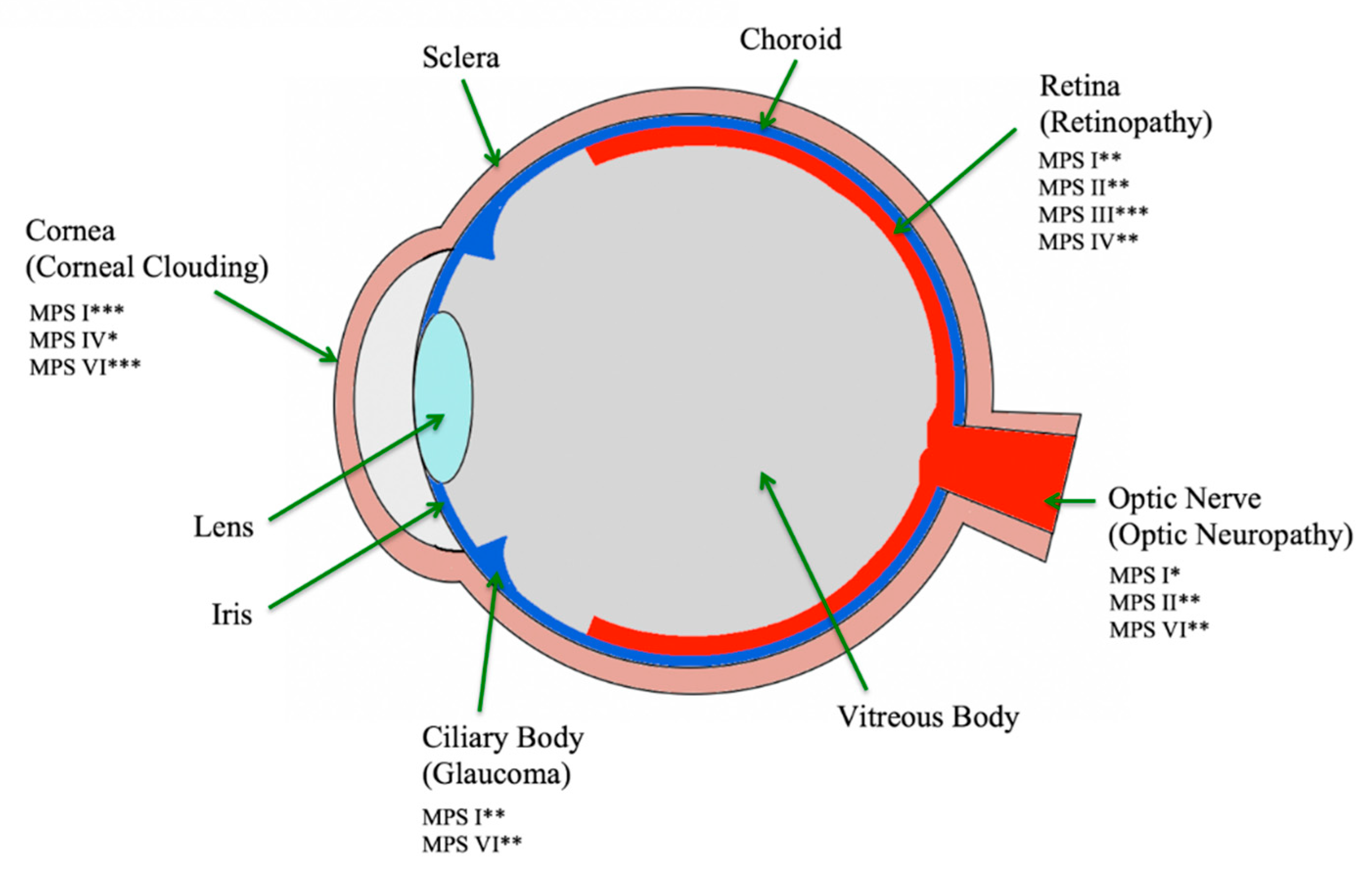
1. Introduction
2. Methods
3. MPS I
3.1. Natural History of MPS I
3.2. Ocular Issues in MPS I
4. MPS II
4.1. Natural History of MPS II
4.2. Ocular Issues in MPS II
5. MPS III
5.1. Natural History of MPS III
5.2. Ocular Issues in MPS III
6. MPS IV
6.1. Natural History of MPS IV
6.2. Ocular Issues in MPS IV
7. MPS VI
7.1. Natural History of MPS VI
7.2. Ocular Issues in MPS VI
8. MPS VII
8.1. Natural History of MPS VII
8.2. Ocular Issues in MPS VII
9. MPS IX
9.1. Natural History of MPS IX
9.2. Ocular Issues in MPS IX
10. Ophthalmological Management and Intervention
10.1. Refractive Errors
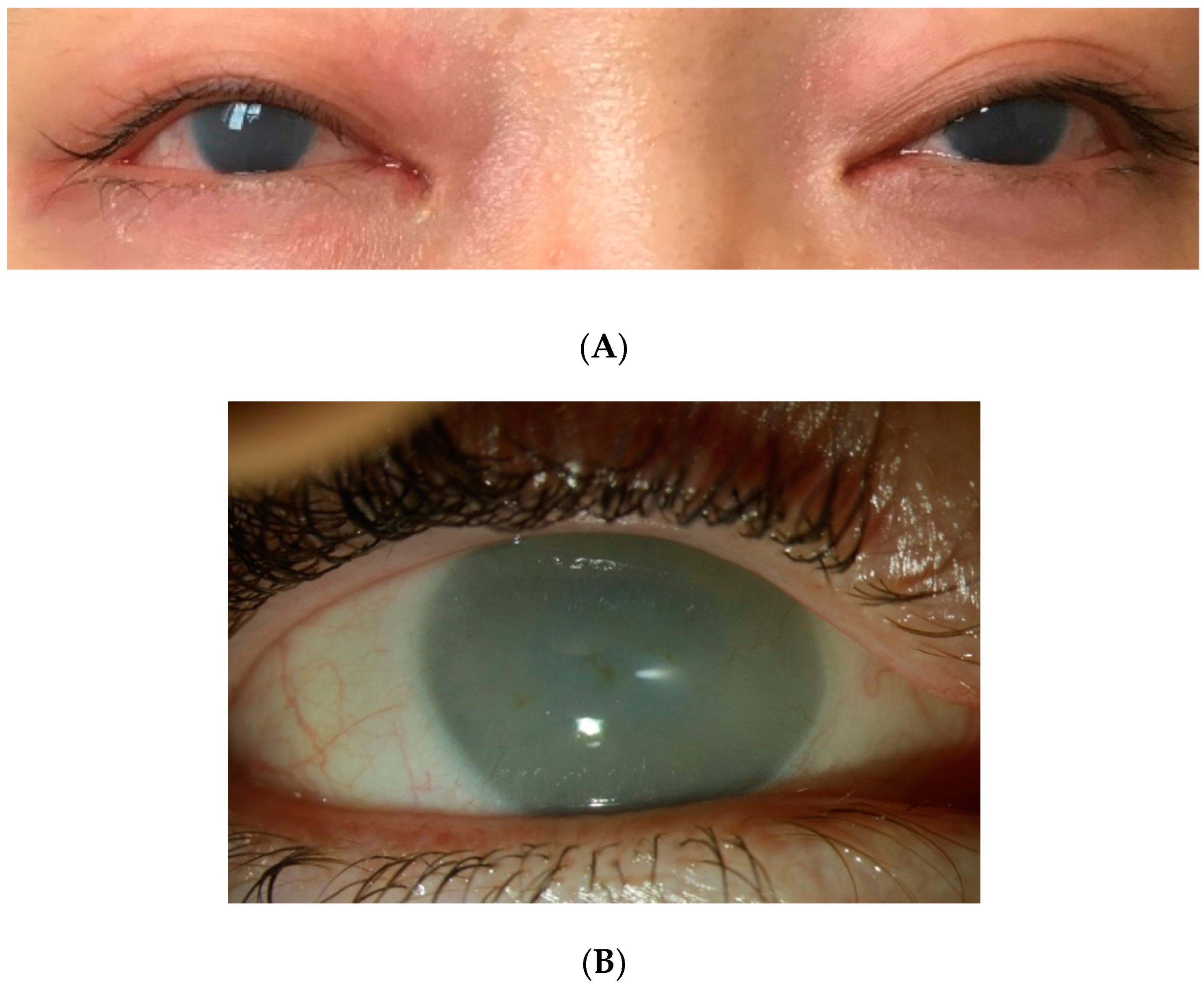
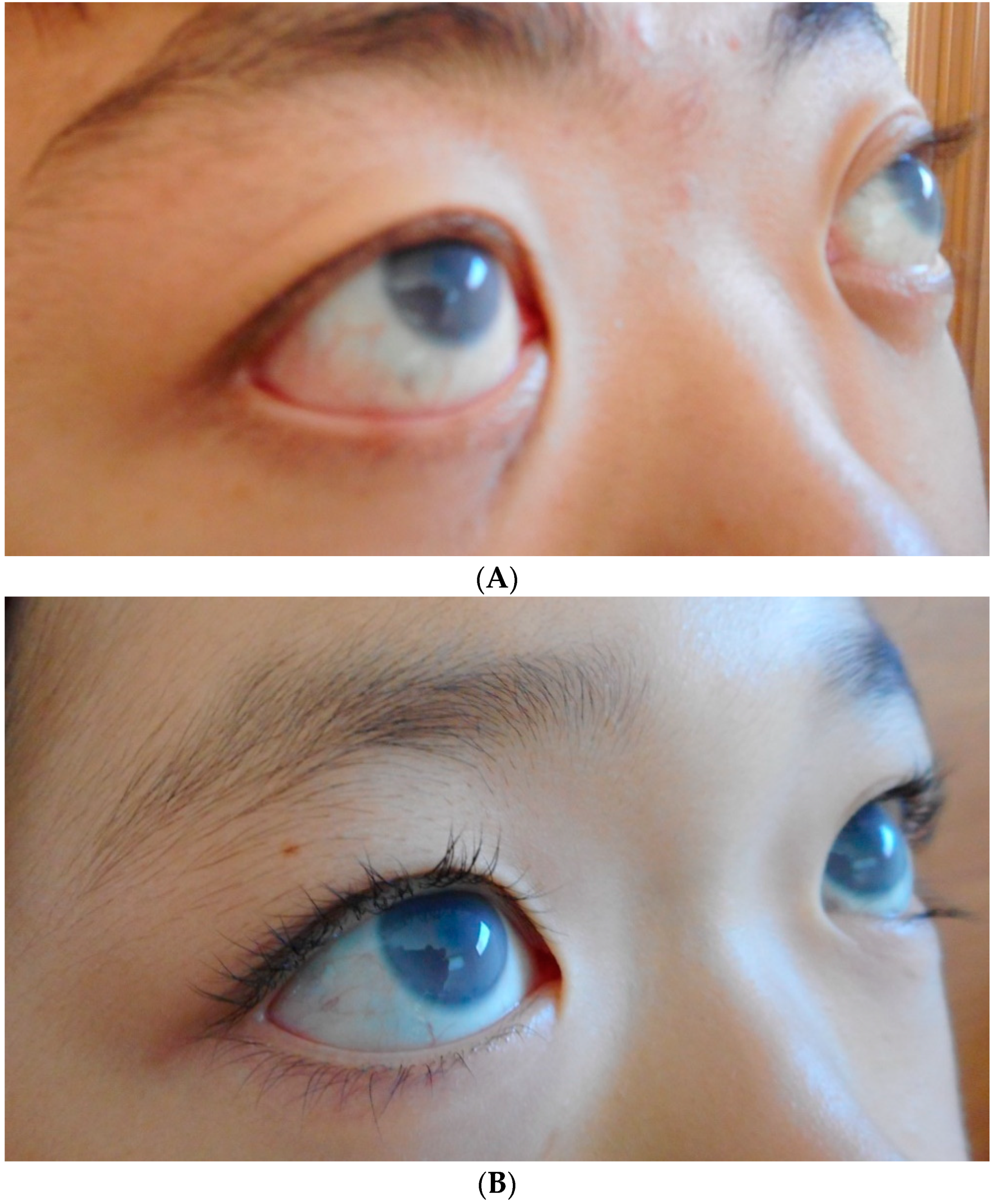
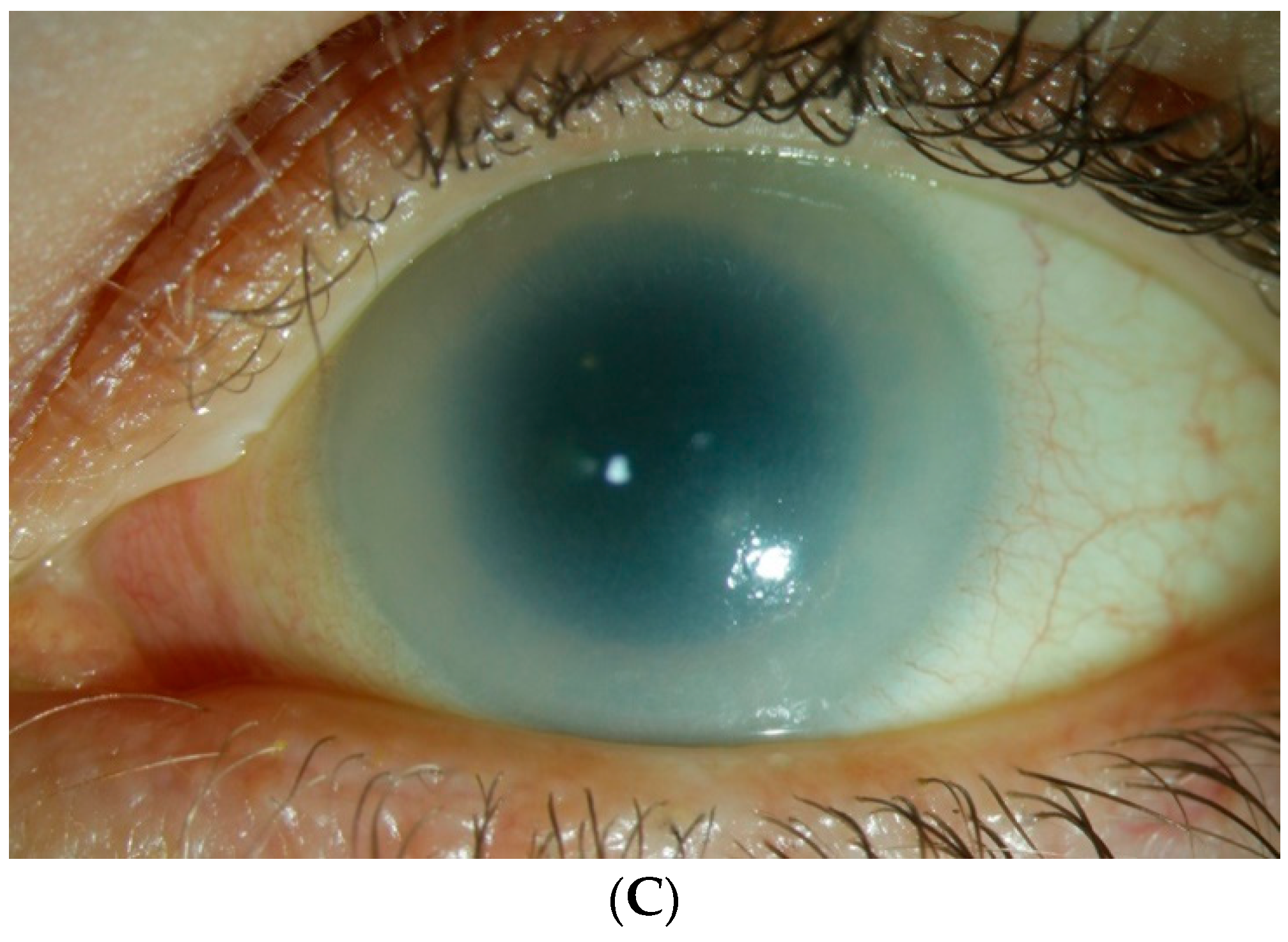
10.2. Corneal Clouding
10.3. Keratoconjunctivitis Sicca
10.4. Glaucoma
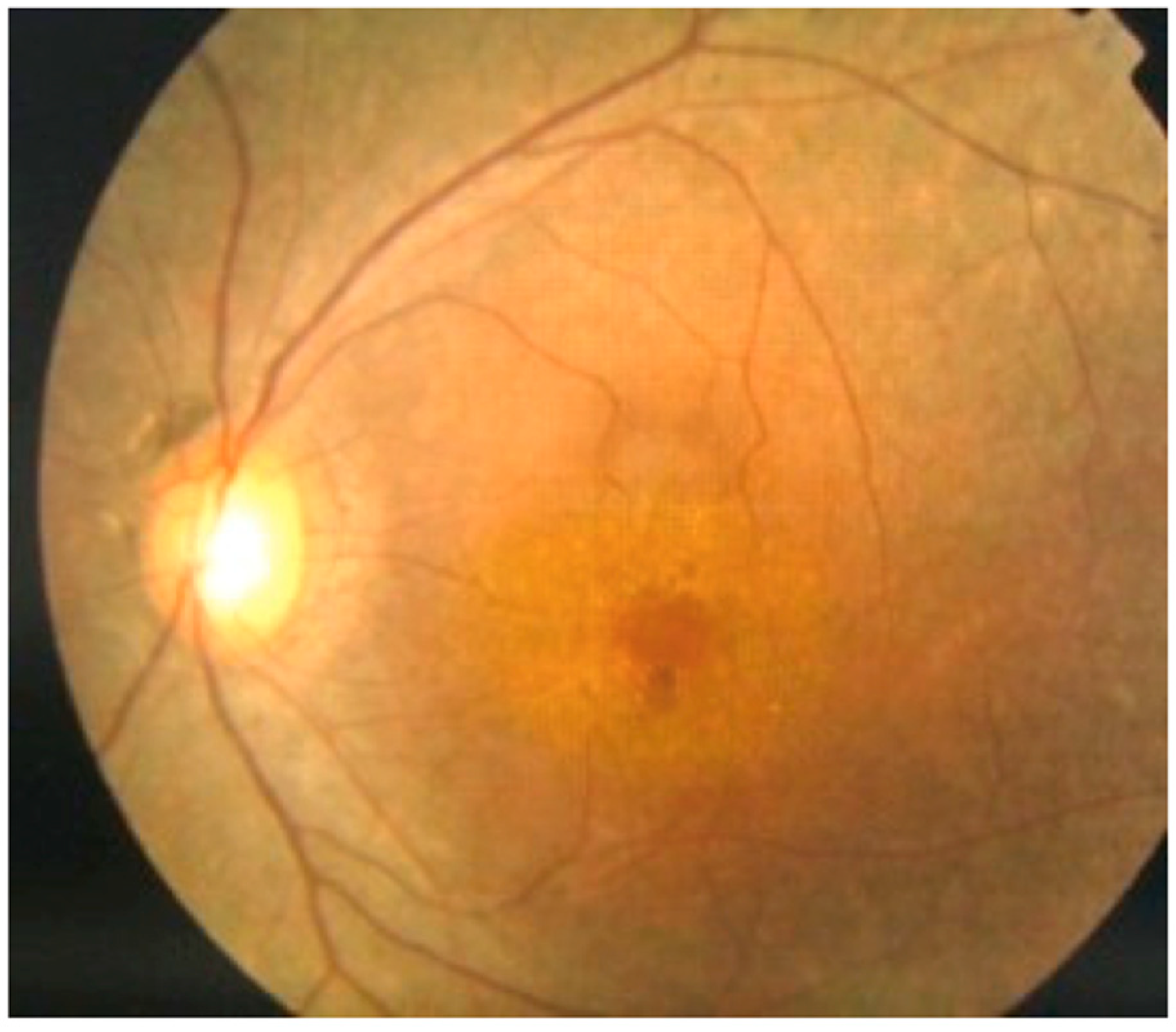
10.5. Retinopathy
10.6. Anesthesia Issues
11. Hematopoietic Stem Cell Transplant and Enzyme Replacement Therapy
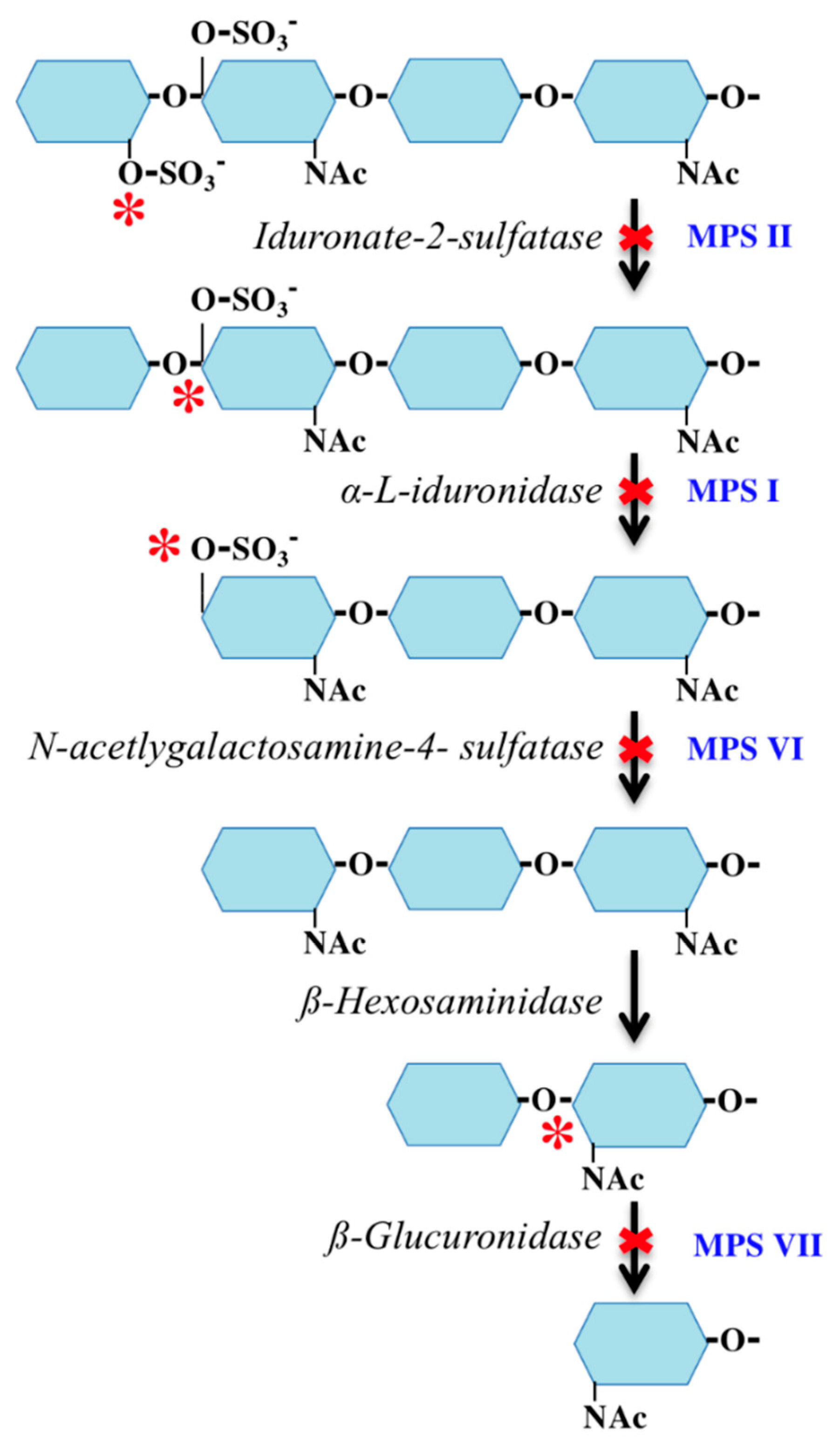
12. Corneal Clouding Theories
13. Lack of Corneal Clouding in MPS II
14. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| MPS | mucopolysaccharidosis |
| GAG | glycosaminoglycans |
| HSCT | hematopoietic stem cell transplantation |
| ERT | enzyme replacement therapy |
| DS | dermatan sulfate |
| HS | heparan sulfate |
| IOP | intraocular pressure |
| GALNS | N-acetylgalactosamine-6-sulfate sulfatase |
| KS | keratan sulfate |
| C6S | chondroitin-6-sulfate |
| C4S | chondroitin-4-sulfate |
| DALK | deep anterior lamellar keratoplasty |
| PK | penetrating keratoplasty |
| UMSC | umbilical mesenchymal stem cell |
References
- Ashworth, J.L.; Biswas, S.; Wraith, E.; Lloyd, I.C. Mucopolysaccharidoses and the eye. Surv. Ophthalmol. 2006, 51, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Fenzl, C.R.; Teramoto, K.; Moshirfar, M. Ocular manifestations and management recommendations of lysosomal storage disorders I: Mucopolysaccharidoses. Clin. Ophthalmol. 2015, 9, 1633–1644. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S. Mucopolysaccharidoses Update (2 Volume Set); Nova Medicine & Health: New York, NY, USA, 2018; 2 volumes. [Google Scholar]
- Ferrari, S.; Ponzin, D.; Ashworth, J.L.; Fahnehjelm, K.T.; Summers, C.G.; Harmatz, P.R.; Scarpa, M. Diagnosis and management of ophthalmological features in patients with mucopolysaccharidosis. Br. J. Ophthalmol. 2011, 95, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, A.; Bruwer, Z.; Al-Thihli, K. An update on ocular involvement in mucopolysaccharidoses. Curr. Opin. Ophthalmol. 2013, 24, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Summers, C.G.; Ashworth, J.L. Ocular manifestations as key features for diagnosing mucopolysaccharidoses. Rheumatology 2011, 50, v34–v40. [Google Scholar] [CrossRef] [PubMed]
- Beesley, C.E.; Meaney, C.A.; Greenland, G.; Adams, V.; Vellodi, A.; Young, E.P.; Winchester, B.G. Mutational analysis of 85 mucopolysaccharidosis type I families: Frequency of known mutations, identification of 17 novel mutations and in vitro expression of missense mutations. Hum. Genet. 2001, 109, 503–511. [Google Scholar] [CrossRef]
- Beck, M.; Arn, P.; Giugliani, R.; Muenzer, J.; Okuyama, T.; Taylor, J.; Fallet, S. The natural history of MPS I: Global perspectives from the MPS I Registry. Genet. Med. 2014, 16, 759–765. [Google Scholar] [CrossRef]
- Del Longo, A.; Piozzi, E.; Schweizer, F. Ocular features in mucopolysaccharidosis: Diagnosis and treatment. Ital. J. Pediatr. 2018, 44, 125. [Google Scholar] [CrossRef]
- Fahnehjelm, K.T.; Ashworth, J.L.; Pitz, S.; Olsson, M.; Tornquist, A.L.; Lindahl, P.; Summers, C.G. Clinical guidelines for diagnosing and managing ocular manifestations in children with mucopolysaccharidosis. Acta Ophthalmol. 2012, 90, 595–602. [Google Scholar] [CrossRef]
- Fahnehjelm, K.T.; Chen, E.; Winiarski, J. Corneal hysteresis in mucopolysaccharidosis I and VI. Acta Ophthalmol. 2012, 90, 445–448. [Google Scholar] [CrossRef]
- Collins, M.L.; Traboulsi, E.I.; Maumenee, I.H. Optic nerve head swelling and optic atrophy in the systemic mucopolysaccharidoses. Ophthalmology 1990, 97, 1445–1449. [Google Scholar] [CrossRef]
- Ashworth, J.L.; Biswas, S.; Wraith, E.; Lloyd, I.C. The ocular features of the mucopolysaccharidoses. Eye 2006, 20, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Fahnehjelm, K.T.; Tornquist, A.L.; Winiarski, J. Ocular axial length and corneal refraction in children with mucopolysaccharidosis (MPS I-Hurler). Acta Ophthalmol. 2012, 90, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, R.G.; Brzezinska, R.; Schulze-Frenking, G.; Pitz, S. Sonographic ocular findings in patients with mucopolysaccharidoses I, II and VI. Pediatr. Radiol. 2008, 38, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Sornalingam, K.; Javed, A.; Aslam, T.; Sergouniotis, P.; Jones, S.; Ghosh, A.; Ashworth, J. Variability in the ocular phenotype in mucopolysaccharidosis. Br. J. Ophthalmol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Fahnehjelm, K.T.; Olsson, M.; Chen, E.; Hengstler, J.; Naess, K.; Winiarski, J. Children with Mucopolysaccharidosis Risk Progressive Visual Dysfunction Despite Haematopoietic Stem Cell Transplants. Available online: https://mpssociety.org/learn/treatments/ert/ (accessed on 12 August 2019).
- Martin, R.; Beck, M.; Eng, C.; Giugliani, R.; Harmatz, P.; Munoz, V.; Muenzer, J. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics 2008, 121, e377–e386. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E.; Scarpa, M.; Beck, M.; Bodamer, O.A.; De Meirleir, L.; Guffon, N.; Meldgaard Lund, A.; Malm, G.; Van der Ploeg, A.T.; Zeman, J. Mucopolysaccharidosis type II (Hunter syndrome): A clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur. J. Pediatr. 2008, 167, 267–277. [Google Scholar] [CrossRef]
- Gadve, S.S.; Sarma, D.; Saikia, U.K. Short stature with umbilical hernia - Not always due to cretinism: A report of two cases. Indian J. Endocrinol. Metab. 2012, 16, 453–456. [Google Scholar] [CrossRef]
- Salvucci, I.D.M.; Finzi, S.; Oyamada, M.K.; Kim, C.A.; Pimentel, S.L.G. Multimodal image analysis of the retina in Hunter syndrome (mucopolysaccharidosis type II): Case report. Ophthalmic Genet. 2018, 39, 103–107. [Google Scholar] [CrossRef]
- Andrade, F.; Aldamiz-Echevarria, L.; Llarena, M.; Couce, M.L. Sanfilippo syndrome: Overall review. Pediatr. Int. 2015, 57, 331–338. [Google Scholar] [CrossRef]
- Wilkin, J.; Kerr, N.C.; Byrd, K.W.; Ward, J.C.; Iannaccone, A. Characterization of a Case of Pigmentary Retinopathy in Sanfilippo Syndrome Type IIIA Associated with Compound Heterozygous Mutations in the SGSH Gene. Ophthalmic Genet. 2016, 37, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Del Canho, H.; van den Bergh, F.A.; Duran, M.; Hennekam, R.C.; Groniger, A.M.; Poorthuis, B.J. Type D Sanfilippo disease in an 8-year-old boy; a rare cause of mental retardation. Ned. Tijdschr. Geneeskd. 1993, 137, 969–972. [Google Scholar] [PubMed]
- Sawamoto, K.; Suzuki, Y.; Mackenzie, W.G.; Theroux, M.C.; Pizarro, C.; Yabe, H.; Orii, K.E.; Mason, R.W.; Orii, T.; Tomatsu, S. Current therapies for Morquio A syndrome and their clinical outcomes. Expert Opin. Orphan Drugs 2016, 4, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Hendriksz, C.J.; Berger, K.I.; Giugliani, R.; Harmatz, P.; Kampmann, C.; Mackenzie, W.G.; Raiman, J.; Villarreal, M.S.; Savarirayan, R. International guidelines for the management and treatment of Morquio A syndrome. Am. J. Med. Genet. A 2015, 167A, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Kasmann-Kellner, B.; Weindler, J.; Pfau, B.; Ruprecht, K.W. Ocular changes in mucopolysaccharidosis IV A (Morquio A syndrome) and long-term results of perforating keratoplasty. Ophthalmologica 1999, 213, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, R.; Lampe, C.; Guffon, N.; Ketteridge, D.; Leao-Teles, E.; Wraith, J.E.; Jones, S.A.; Piscia-Nichols, C.; Lin, P.; Quartel, A.; et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome)—10-year follow-up of patients who previously participated in an MPS VI Survey Study. Am. J. Med. Genet. A 2014, 164A, 1953–1964. [Google Scholar] [CrossRef]
- Stürrmer, J. Type VI-A mucopolysaccharidosis (Maroteaux-Lamy disease). Clinico-pathologic case report. Klin. Monatsblätter Augenheilkd. 1989, 194, 273–281. [Google Scholar] [CrossRef]
- Montano, A.M.; Lock-Hock, N.; Steiner, R.D.; Graham, B.H.; Szlago, M.; Greenstein, R.; Pineda, M.; Gonzalez-Meneses, A.; Coker, M.; Bartholomew, D.; et al. Clinical course of sly syndrome (mucopolysaccharidosis type VII). J. Med. Genet. 2016, 53, 403–418. [Google Scholar] [CrossRef]
- Coulson-Thomas, V.J.; Caterson, B.; Kao, W.W. Transplantation of human umbilical mesenchymal stem cells cures the corneal defects of mucopolysaccharidosis VII mice. Stem. Cells 2013, 31, 2116–2126. [Google Scholar] [CrossRef]
- Bergwerk, K.E.; Falk, R.E.; Glasgow, B.J.; Rabinowitz, Y.S. Corneal transplantation in a patient with mucopolysaccharidosis type VII (Sly disease). Ophthalmic Genet. 2000, 21, 17–20. [Google Scholar] [CrossRef]
- Natowicz, M.R.; Short, M.P.; Wang, Y.; Dickersin, G.R.; Gebhardt, M.C.; Rosenthal, D.I.; Sims, K.B.; Rosenberg, A.E. Clinical and biochemical manifestations of hyaluronidase deficiency. N Engl J Med 1996, 335, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Imundo, L.; Leduc, C.A.; Guha, S.; Brown, M.; Perino, G.; Gushulak, L.; Triggs-Raine, B.; Chung, W.K. A complete deficiency of Hyaluronoglucosaminidase 1 (HYAL1) presenting as familial juvenile idiopathic arthritis. J Inherit Metab Dis 2011, 34, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Alroy, J.; Haskins, M.; Birk, D.E. Altered corneal stromal matrix organization is associated with mucopolysaccharidosis I, III and VI. Exp. Eye Res. 1999, 68, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Montano, A.M.; Oikawa, H.; Smith, M.; Barrera, L.; Chinen, Y.; Thacker, M.M.; Mackenzie, W.G.; Suzuki, Y.; Orii, T. Mucopolysaccharidosis type IVA (Morquio A disease): Clinical review and current treatment. Curr. Pharm. Biotechnol. 2011, 12, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Couprie, J.; Denis, P.; Guffon, N.; Reynes, N.; Masset, H.; Beby, F. Ocular manifestations in patients affected by Morquio syndrome (MPS IV). J. Fr. Ophtalmol. 2010, 33, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Javed, A.; Aslam, T.; Jones, S.A.; Ashworth, J. Objective Quantification of Changes in Corneal Clouding Over Time in Patients with Mucopolysaccharidosis. Investig. Ophthalmol. Vis. Sci. 2017, 58, 954–958. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Elflein, H.M.; Hofherr, T.; Berisha-Ramadani, F.; Weyer, V.; Lampe, C.; Beck, M.; Pitz, S. Measuring corneal clouding in patients suffering from mucopolysaccharidosis with the Pentacam densitometry programme. Br. J. Ophthalmol. 2013, 97, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Ohden, K.L.; Pitz, S.; Ashworth, J.; Magalhaes, A.; Marinho, D.R.; Lindahl, P.; Tear Fahnehjelm, K.; Summers, C.G. Outcomes of keratoplasty in the mucopolysaccharidoses: An international perspective. Br. J. Ophthalmol. 2017, 101, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Harding, S.A.; Nischal, K.K.; Upponi-Patil, A.; Fowler, D.J. Indications and outcomes of deep anterior lamellar keratoplasty in children. Ophthalmology 2010, 117, 2191–2195. [Google Scholar] [CrossRef]
- Pinello, L.; Busin, M.; Fontana, L.; Dua, H.S. Application of (lamellar) keratoplasty and limbal stem cell transplantation for corneal clouding in the mucopolysaccharidoses—A review. Clin. Exp. Ophthalmol. 2010, 38, 52–62. [Google Scholar] [CrossRef]
- Fahnehjelm, K.T.; Tornquist, A.L.; Winiarski, J. Dry-eye syndrome after allogeneic stem-cell transplantation in children. Acta Ophthalmol. 2008, 86, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Javed, A.; Aslam, T.; Ashworth, J. Use of new imaging in detecting and monitoring ocular manifestations of the mucopolysaccharidoses. Acta Ophthalmol. 2016, 94, e676–e682. [Google Scholar] [CrossRef] [PubMed]
- Wasielica-Poslednik, J.; Butsch, C.; Lampe, C.; Elflein, H.; Lamparter, J.; Weyer, V.; Pitz, S. Comparison of Rebound Tonometry, Perkins Applanation Tonometry and Ocular Response Analyser in Mucopolysaccharidosis Patients. PLoS ONE 2015, 10, e0133586. [Google Scholar] [CrossRef] [PubMed]
- Wasielica-Poslednik, J.; Politino, G.; Schmidtmann, I.; Lorenz, K.; Bell, K.; Pfeiffer, N.; Pitz, S. Influence of Corneal Opacity on Intraocular Pressure Assessment in Patients with Lysosomal Storage Diseases. PLoS ONE 2017, 12, e0168698. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, P.C.; Dietlein, T.S.; Krieglstein, G.K. Microendoscopic trabecular surgery in glaucoma management. Ophthalmology 1999, 106, 538–544. [Google Scholar] [CrossRef]
- Clark, B.M.; Sprung, J.; Weingarten, T.N.; Warner, M.E. Anesthesia for patients with mucopolysaccharidoses: Comprehensive review of the literature with emphasis on airway management. Bosn. J. Basic Med. Sci. 2018, 18, 1–7. [Google Scholar] [CrossRef]
- Hobbs, J.R.; Hugh-Jones, K.; Barrett, A.J.; Byrom, N.; Chambers, D.; Henry, K.; James, D.C.; Lucas, C.F.; Rogers, T.R.; Benson, P.F.; et al. Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet 1981, 2, 709–712. [Google Scholar] [CrossRef]
- Aldenhoven, M.; Wynn, R.F.; Orchard, P.J.; O’Meara, A.; Veys, P.; Fischer, A.; Valayannopoulos, V.; Neven, B.; Rovelli, A.; Prasad, V.K.; et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: An international multicenter study. Blood 2015, 125, 2164–2172. [Google Scholar] [CrossRef]
- Fahnehjelm, K.; Tornquist, A.L.; Malm, G.; Winiarski, J. Ocular findings in four children with mucopolysaccharidosis I-Hurler (MPS I-H) treated early with haematopoietic stem cell transplantation. Acta Ophthalmol. Scand. 2006, 84, 781–785. [Google Scholar] [CrossRef]
- Gullingsrud, E.O.; Krivit, W.; Summers, C.G. Ocular abnormalities in the mucopolysaccharidoses after bone marrow transplantation. Longer follow-up. Ophthalmology 1998, 105, 1099–1105. [Google Scholar] [CrossRef]
- Summers, C.G.; Fahnehjelm, K.T.; Pitz, S.; Guffon, N.; Koseoglu, S.T.; Harmatz, P.; Scarpa, M. Systemic therapies for mucopolysaccharidosis: Ocular changes following haematopoietic stem cell transplantation or enzyme replacement therapy—A review. Clin. Exp. Ophthalmol. 2010, 38, 34–42. [Google Scholar] [CrossRef]
- Boelens, J.J.; Wynn, R.F.; O’Meara, A.; Veys, P.; Bertrand, Y.; Souillet, G.; Wraith, J.E.; Fischer, A.; Cavazzana-Calvo, M.; Sykora, K.W.; et al. Outcomes of hematopoietic stem cell transplantation for Hurler’s syndrome in Europe: A risk factor analysis for graft failure. Bone Marrow Transplant. 2007, 40, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E.; Clarke, L.A.; Beck, M.; Kolodny, E.H.; Pastores, G.M.; Muenzer, J.; Rapoport, D.M.; Berger, K.I.; Swiedler, S.J.; Kakkis, E.D.; et al. Enzyme replacement therapy for mucopolysaccharidosis I: A randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-l-iduronidase (laronidase). J. Pediatr. 2004, 144, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Sifuentes, M.; Doroshow, R.; Hoft, R.; Mason, G.; Walot, I.; Diament, M.; Okazaki, S.; Huff, K.; Cox, G.F.; Swiedler, S.J.; et al. A follow-up study of MPS I patients treated with laronidase enzyme replacement therapy for 6 years. Mol. Genet. Metab. 2007, 90, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Newkirk, K.M.; Atkins, R.M.; Dickson, P.I.; Rohrbach, B.W.; McEntee, M.F. Ocular lesions in canine mucopolysaccharidosis I and response to enzyme replacement therapy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5130–5135. [Google Scholar] [CrossRef]
- Tolar, J.; Grewal, S.S.; Bjoraker, K.J.; Whitley, C.B.; Shapiro, E.G.; Charnas, L.; Orchard, P.J. Combination of enzyme replacement and hematopoietic stem cell transplantation as therapy for Hurler syndrome. Bone Marrow Transplant. 2008, 41, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Wynn, R.F.; Mercer, J.; Page, J.; Carr, T.F.; Jones, S.; Wraith, J.E. Use of enzyme replacement therapy (Laronidase) before hematopoietic stem cell transplantation for mucopolysaccharidosis I: Experience in 18 patients. J. Pediatr. 2009, 154, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Michelacci, Y.M. Collagens and proteoglycans of the corneal extracellular matrix. Braz. J. Med. Biol. Res. 2003, 36, 1037–1046. [Google Scholar] [CrossRef]
- Mizumoto, S.; Kosho, T.; Yamada, S.; Sugahara, K. Pathophysiological Significance of Dermatan Sulfate Proteoglycans Revealed by Human Genetic Disorders. Pharmaceuticals 2017, 10. [Google Scholar] [CrossRef]
- Yuan, C.; Bothun, E.D.; Hardten, D.R.; Tolar, J.; McLoon, L.K. A novel explanation of corneal clouding in a bone marrow transplant-treated patient with Hurler syndrome. Exp. Eye Res. 2016, 148, 83–89. [Google Scholar] [CrossRef]
- Pacella, E.; Pacella, F.; De Paolis, G.; Parisella, F.R.; Turchetti, P.; Anello, G.; Cavallotti, C. Glycosaminoglycans in the human cornea: Age-related changes. Ophthalmol. Eye Dis. 2015, 7, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.; Davies, Y.; Nieduszynski, I.A.; Lawrence, F.; Quantock, A.J.; Bonshek, R.; Fullwood, N.J. Ultrastructural localization of sulfated and unsulfated keratan sulfate in normal and macular corneal dystrophy type I. Glycobiology 2000, 10, 305–312. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MPS Type (Eponym) | Enzyme Deficiency | Accumulated GAG | Inheritance | Incidence | Age of Onset (Years) | |||
|---|---|---|---|---|---|---|---|---|
| Corneal Clouding | Glaucoma | Retinopathy | Optic Neuropathy | |||||
| MPS IH (Hurler) | α-l-iduronidase | HS, DS | AR | Attenuated: 1 in 115,000 to 500,000 live births | 1.1 | 1 | 10 | 17 |
| MPS IHS (Hurler-Scheie) | α-l-iduronidase | HS, DS | AR | 4.4 | ||||
| Hurler IS (Scheie) | α-l-iduronidase | HS, DS | AR | Severe: 1 in 100,000 | 10.5 | |||
| MPS II (Hunter) | Iduronate-2-sulfatase | HS, DS | XR | 1 in 100,000 to 170,000 male births | N/A | 7.5 | <21 | 33 |
| MPS IIIA (Sanfilippo A) | Heparan-N-sulfatase | HS | AR | 1 in 70,000 live births | N/A | N/A | 5 | N/A |
| MPS IIIB (Sanfilippo B) | α-N-acetylglucosaminidase | HS | AR | N/A | N/A | N/A | N/A | |
| MPS IIIC (Sanfilippo C) | α-glucosaminide acetyltransferase | HS | AR | N/A | N/A | N/A | N/A | |
| MPS IIID (Sanfilippo D) | N-acetylglucosamine-6-sulfatase | HS | AR | 8 | N/A | N/A | N/A | |
| MPS IVA (Morquio A) | N-acetylgalactosamine-6-sulfatase | KS, C6S | AR | 1 in 76,000 to 640,000 | 11 | 7.8 | 12 | N/A |
| MPS IVB (Morquio B) | β-galactosidase | KS | ||||||
| MPS VI (Maroteaux-Lamy) | N-acetylgalactosamine-4-sulfatase | DS, C4S | AR | 1 in 250,000 to 600,000 live births | 7 | 3 | N/A | 26 |
| MPS VII (Sly) | β-glucuronidase | DS, C4S, C6S, HS | AR | 1 in 250,000 live births | 15 | N/A | N/A | N/A |
| MPS IX (Natowicz) | Hyaluronidase | Hyaluronan | AR | Unknown | N/A | N/A | N/A | N/A |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomatsu, S.; Pitz, S.; Hampel, U. Ophthalmological Findings in Mucopolysaccharidoses. J. Clin. Med. 2019, 8, 1467. https://doi.org/10.3390/jcm8091467
Tomatsu S, Pitz S, Hampel U. Ophthalmological Findings in Mucopolysaccharidoses. Journal of Clinical Medicine. 2019; 8(9):1467. https://doi.org/10.3390/jcm8091467
Chicago/Turabian StyleTomatsu, Shizuka, Susanne Pitz, and Ulrike Hampel. 2019. "Ophthalmological Findings in Mucopolysaccharidoses" Journal of Clinical Medicine 8, no. 9: 1467. https://doi.org/10.3390/jcm8091467
APA StyleTomatsu, S., Pitz, S., & Hampel, U. (2019). Ophthalmological Findings in Mucopolysaccharidoses. Journal of Clinical Medicine, 8(9), 1467. https://doi.org/10.3390/jcm8091467