Intervening with the Nitric Oxide Pathway to Alleviate Pulmonary Hypertension in Pulmonary Vein Stenosis

 , and
, and
Abstract
:1. Introduction
2. Methods
2.1. Animal Experimentation
2.2. Outline of This Study
2.3. Pulmonary Vein Banding
2.4. Chronic Instrumentation
2.5. Experimental Protocols
2.6. Blood Gas Measurements
2.7. Data Analysis
2.8. Sacrifice
2.9. Wire-Myograph Experiments
2.10. eNOS Protein Expression
2.11. Statistical Analysis
3. Results
3.1. Induction of Pulmonary Hypertension
3.2. Response to Exercise
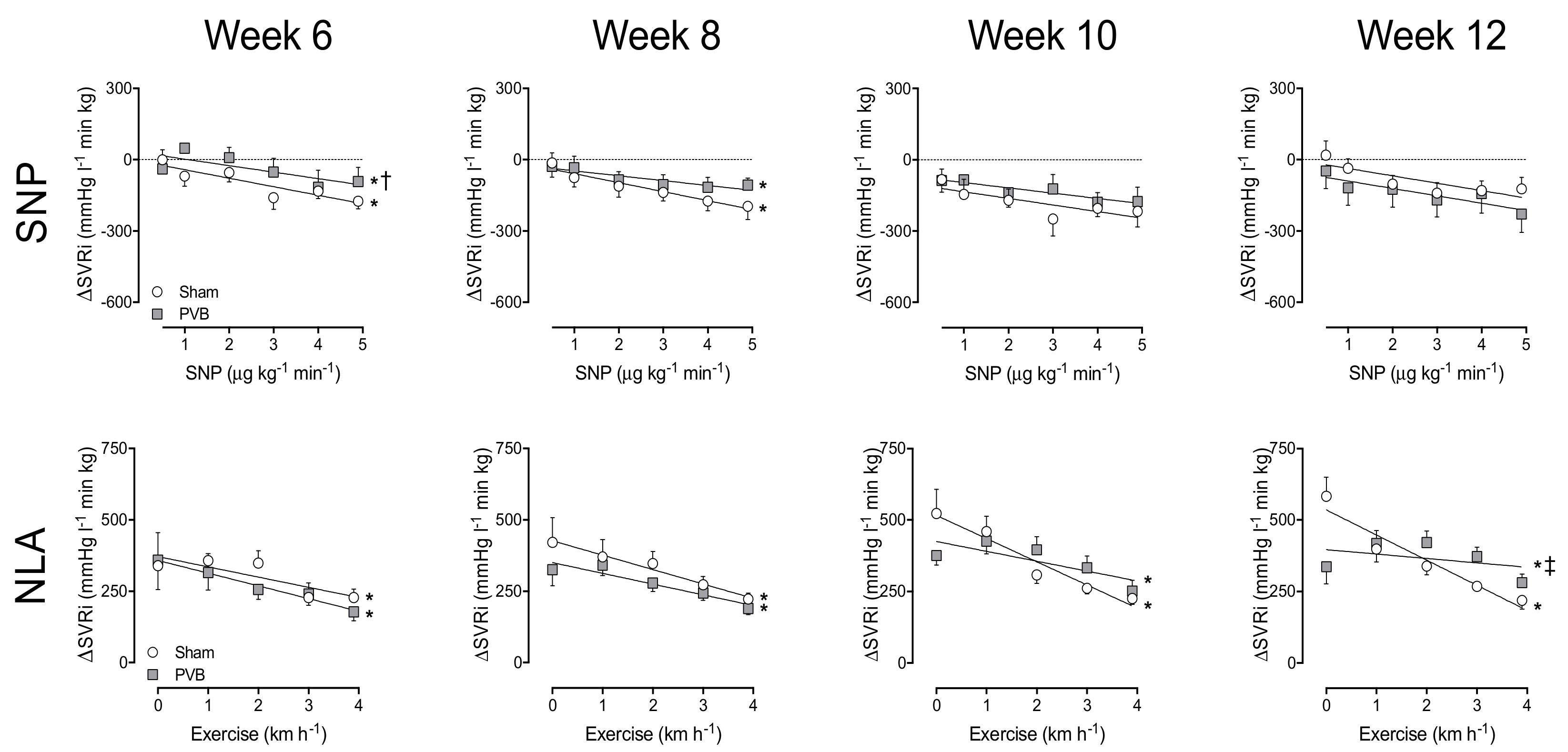
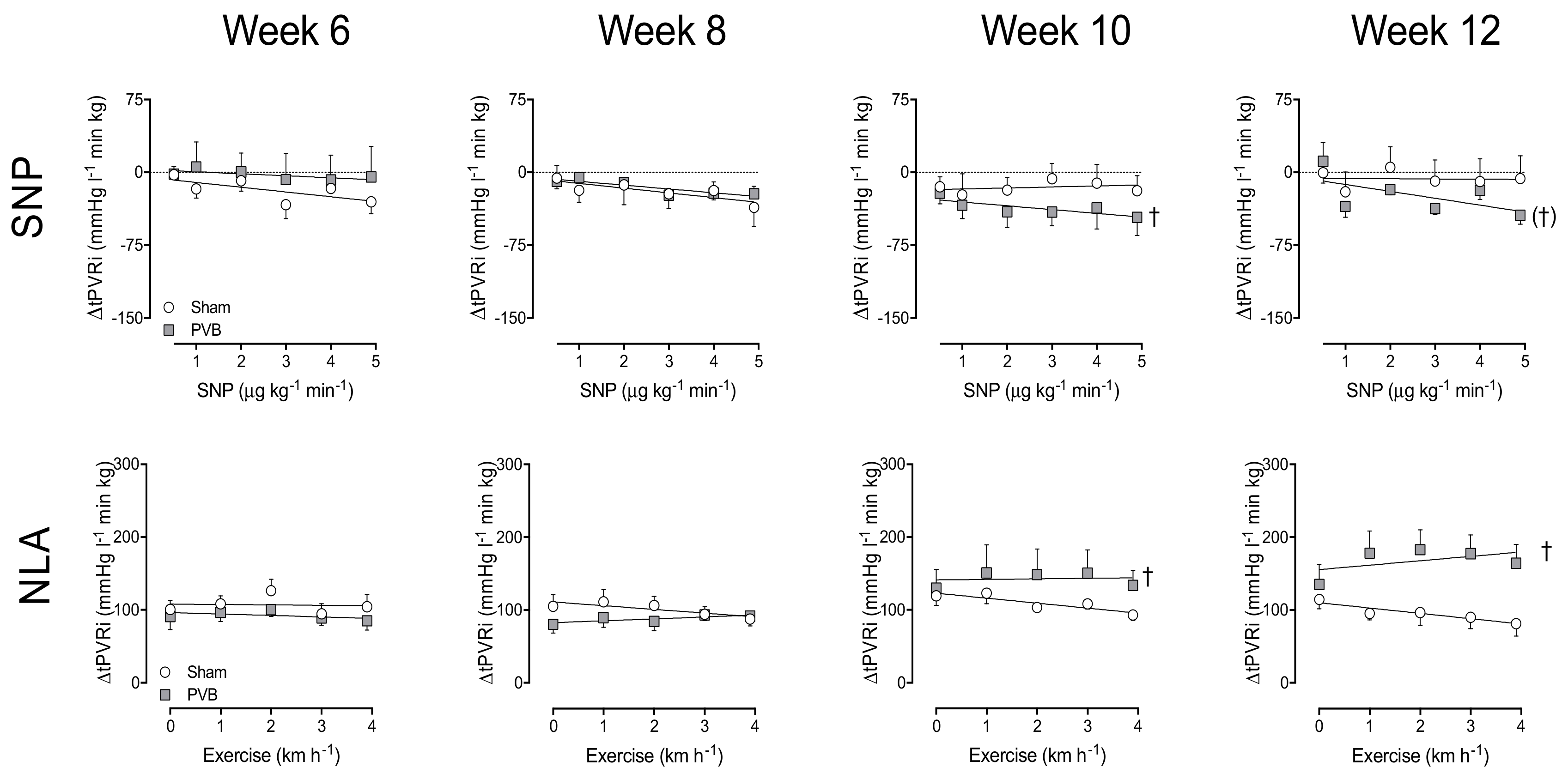
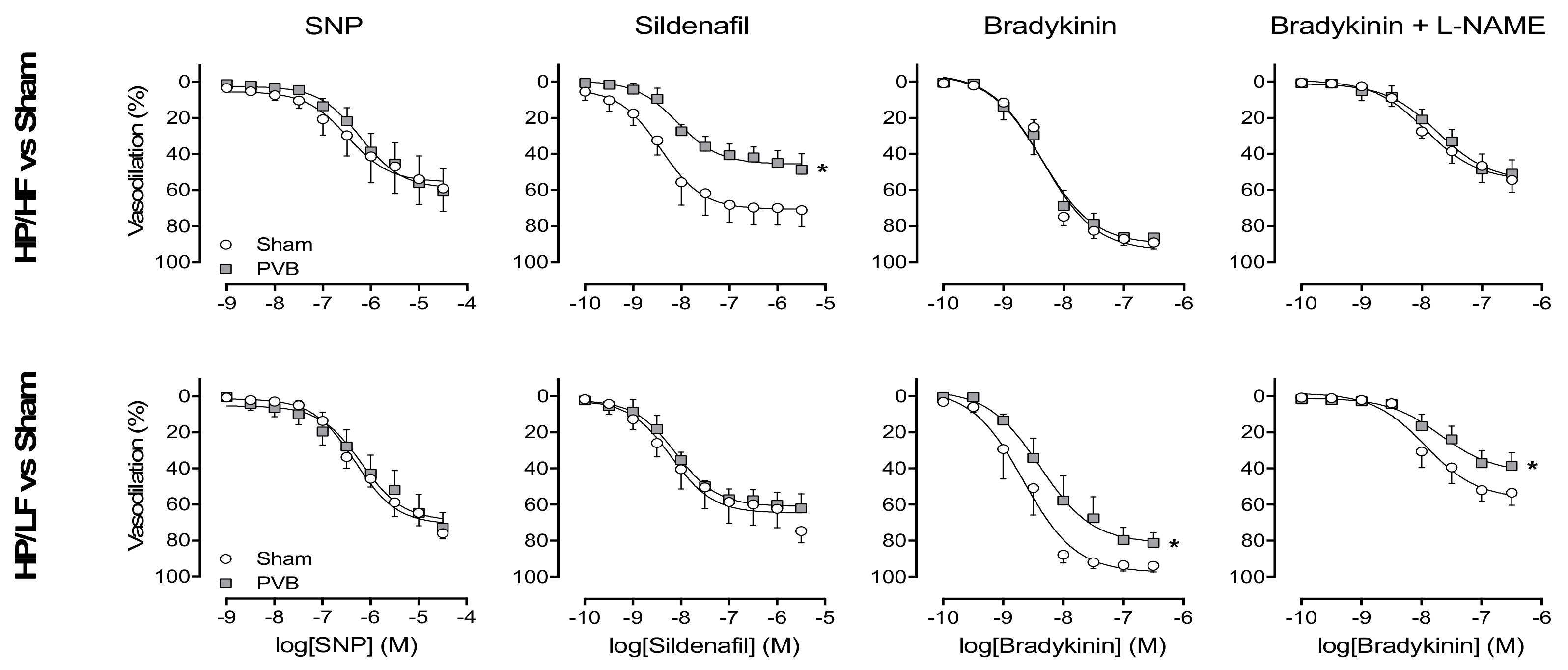
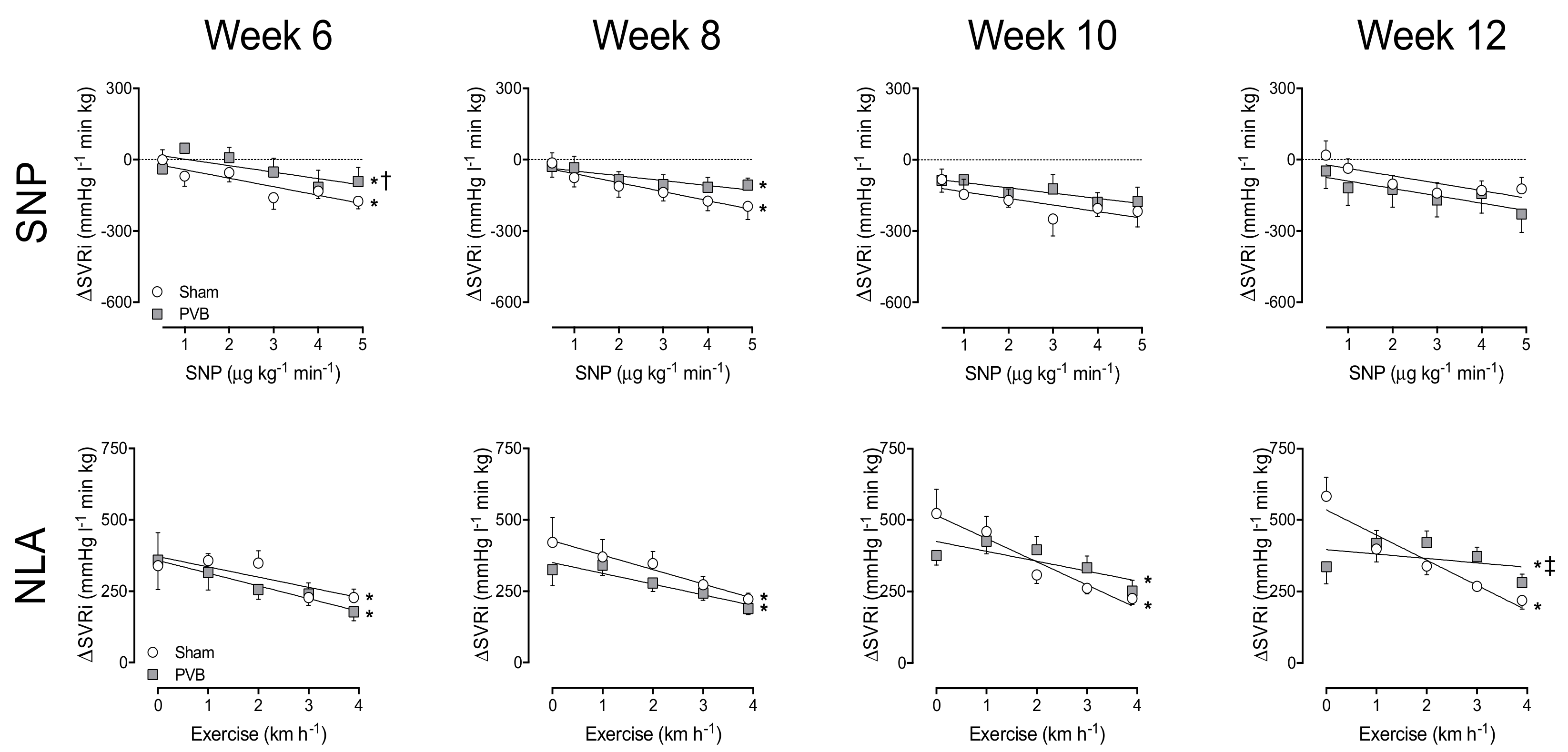
3.3. Nitric Oxide Production and Sensitivity
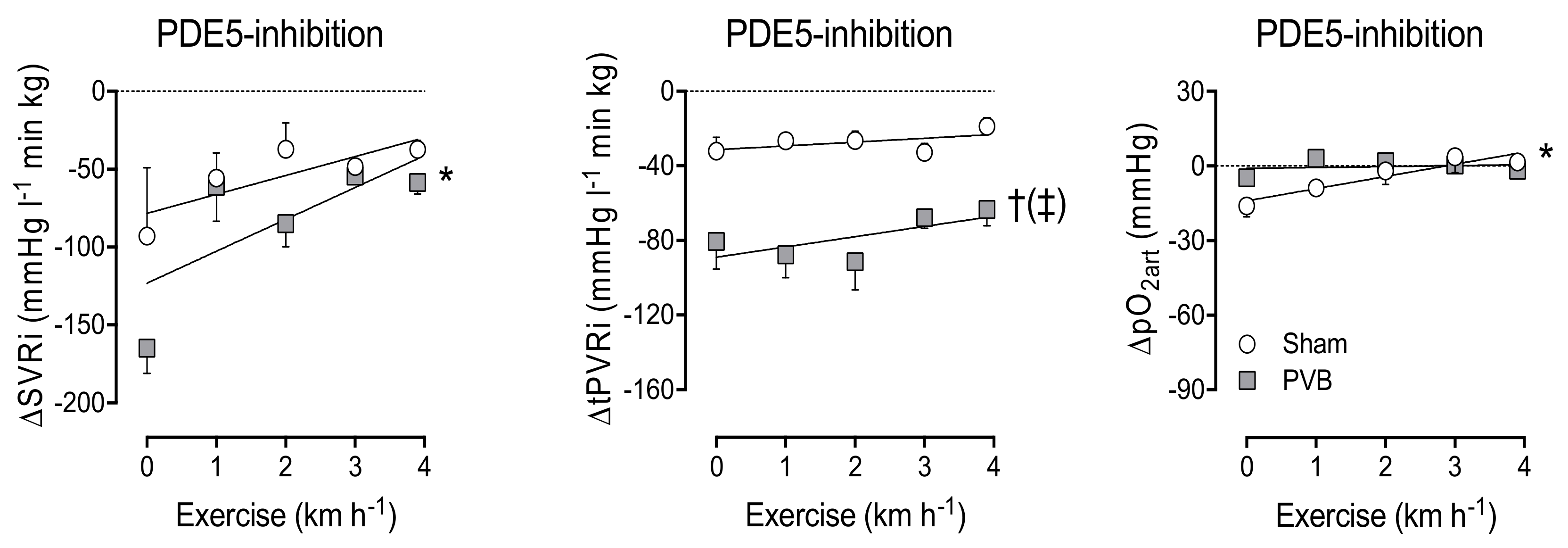
3.4. Contribution of PDE5
3.5. Regional Differences in NO Pathway
4. Discussion
4.1. Pulmonary Hypertension Associated with PVS
4.2. Clinical Relevance
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension. Rev. Esp. Cardiol. 2016, 69, 177. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, S.; Gibbs, J.S.; Wachter, R.; De Marco, T.; Vonk-Noordegraaf, A.; Vachiery, J.L. Left ventricular heart failure and pulmonary hypertension. Eur. Heart J. 2016, 37, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Cappato, R. Pulmonary Vein Stenosis Following Radiofrequency Ablation of Atrial Fibrillation: Has It Become a Clinically Negligible Complication? JACC Clin. Electrophysiol. 2017, 3, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Backes, C.H.; Nealon, E.; Armstrong, A.K.; Cua, C.L.; Mitchell, C.; Krishnan, U.; Vanderlaan, R.D.; Song, M.K.; Viola, N.; Smith, C.V.; et al. Pulmonary Vein Stenosis in Infants: A Systematic Review, Meta-Analysis, and Meta-Regression. J. Pediatr. 2018, 198, 36–45.e3. [Google Scholar] [CrossRef] [PubMed]
- Seale, A.N.; Webber, S.A.; Uemura, H.; Partridge, J.; Roughton, M.; Ho, S.Y.; McCarthy, K.P.; Jones, S.; Shaughnessy, L.; Sunnegardh, J.; et al. Pulmonary vein stenosis: The UK, Ireland and Sweden collaborative study. Heart 2009, 95, 1944–1949. [Google Scholar] [CrossRef] [PubMed]
- Gowda, S.; Bhat, D.; Feng, Z.; Chang, C.H.; Ross, R.D. Pulmonary vein stenosis with Down syndrome: A rare and frequently fatal cause of pulmonary hypertension in infants and children. Congenit. Heart Dis. 2014, 9, E90–E97. [Google Scholar] [CrossRef] [PubMed]
- Mahgoub, L.; Kaddoura, T.; Kameny, A.R.; Lopez Ortego, P.; Vanderlaan, R.D.; Kakadekar, A.; Dicke, F.; Rebeyka, I.; Calderone, C.A.; Redington, A.; et al. Pulmonary vein stenosis of ex-premature infants with pulmonary hypertension and bronchopulmonary dysplasia, epidemiology, and survival from a multicenter cohort. Pediatr. Pulmonol. 2017, 52, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Laux, D.; Rocchisani, M.A.; Boudjemline, Y.; Gouton, M.; Bonnet, D.; Ovaert, C. Pulmonary Hypertension in the Preterm Infant with Chronic Lung Disease can be Caused by Pulmonary Vein Stenosis: A Must-Know Entity. Pediatr. Cardiol. 2016, 37, 313–321. [Google Scholar] [CrossRef]
- Gerges, M.; Gerges, C.; Pistritto, A.M.; Lang, M.B.; Trip, P.; Jakowitsch, J.; Binder, T.; Lang, I.M. Pulmonary Hypertension in Heart Failure. Epidemiology, Right Ventricular Function, and Survival. Am. J. Respir. Crit. Care Med. 2015, 192, 1234–1246. [Google Scholar] [CrossRef]
- Miller, W.L.; Grill, D.E.; Borlaug, B.A. Clinical features, hemodynamics, and outcomes of pulmonary hypertension due to chronic heart failure with reduced ejection fraction: Pulmonary hypertension and heart failure. JACC Heart Fail. 2013, 1, 290–299. [Google Scholar] [CrossRef]
- Vanderpool, R.R.; Naeije, R. Progress in Pulmonary Hypertension with Left Heart Failure. Beyond New Definitions and Acronyms. Am. J. Respir. Crit. Care Med. 2015, 192, 1152–1154. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, J.; Radegran, G. Pathophysiology and potential treatments of pulmonary hypertension due to systolic left heart failure. Acta Physiol. 2014, 211, 314–333. [Google Scholar] [CrossRef] [PubMed]
- Roman, K.S.; Kellenberger, C.J.; Macgowan, C.K.; Coles, J.; Redington, A.N.; Benson, L.N.; Yoo, S.J. How is pulmonary arterial blood flow affected by pulmonary venous obstruction in children? A phase-contrast magnetic resonance study. Pediatr. Radiol. 2005, 35, 580–586. [Google Scholar] [PubMed]
- Aguero, J.; Ishikawa, K.; Hadri, L.; Santos-Gallego, C.; Fish, K.; Hammoudi, N.; Chaanine, A.; Torquato, S.; Naim, C.; Ibanez, B.; et al. Characterization of right ventricular remodeling and failure in a chronic pulmonary hypertension model. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1204–H1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Alvarez, A.; Fernandez-Friera, L.; Garcia-Ruiz, J.M.; Nuno-Ayala, M.; Pereda, D.; Fernandez-Jimenez, R.; Guzman, G.; Sanchez-Quintana, D.; Alberich-Bayarri, A.; Pastor-Escuredo, D.; et al. Noninvasive monitoring of serial changes in pulmonary vascular resistance and acute vasodilator testing using cardiac magnetic resonance. J. Am. Coll. Cardiol. 2013, 62, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Pereda, D.; Garcia-Alvarez, A.; Sanchez-Quintana, D.; Nuno, M.; Fernandez-Friera, L.; Fernandez-Jimenez, R.; Garcia-Ruiz, J.M.; Sandoval, E.; Aguero, J.; Castella, M.; et al. Swine model of chronic postcapillary pulmonary hypertension with right ventricular remodeling: Long-term characterization by cardiac catheterization, magnetic resonance, and pathology. J. Cardiovasc. Transl. Res. 2014, 7, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Van Duin, R.W.B.; Stam, K.; Cai, Z.; Uitterdijk, A.; Garcia-Alvarez, A.; Ibanez, B.; Danser, A.H.J.; Reiss, I.K.M.; Duncker, D.J.; Merkus, D. Transition from post-capillary pulmonary hypertension to combined pre- and post-capillary pulmonary hypertension in swine: A key role for endothelin. J. Physiol. 2019, 597, 1157–1173. [Google Scholar] [CrossRef]
- Keusch, S.; Bucher, A.; Muller-Mottet, S.; Hasler, E.; Maggiorini, M.; Speich, R.; Ulrich, S. Experience with exercise right heart catheterization in the diagnosis of pulmonary hypertension: A retrospective study. Multidiscip. Respir. Med. 2014, 9, 51. [Google Scholar] [CrossRef]
- De Wijs-Meijler, D.P.; Stam, K.; van Duin, R.W.; Verzijl, A.; Reiss, I.K.; Duncker, D.J.; Merkus, D. Surgical Placement of Catheters for Long-term Cardiovascular Exercise Testing in Swine. J. Vis. Exp. 2016, e53772. [Google Scholar] [CrossRef]
- Haitsma, D.B.; Merkus, D.; Vermeulen, J.; Verdouw, P.D.; Duncker, D.J. Nitric oxide production is maintained in exercising swine with chronic left ventricular dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H2198–H2209. [Google Scholar] [CrossRef] [Green Version]
- Mulvany, M.J.; Halpern, W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ. Res. 1977, 41, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; de Beer, V.J.; de Wijs-Meijler, D.; Bender, S.B.; Hoekstra, M.; Laughlin, M.H.; Duncker, D.J.; Merkus, D. Pulmonary vasoconstrictor influence of endothelin in exercising swine depends critically on phosphodiesterase 5 activity. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L442–L452. [Google Scholar] [CrossRef] [PubMed]
- Moens, A.L.; Champion, H.C.; Claeys, M.J.; Tavazzi, B.; Kaminski, P.M.; Wolin, M.S.; Borgonjon, D.J.; Van Nassauw, L.; Haile, A.; Zviman, M.; et al. High-dose folic acid pretreatment blunts cardiac dysfunction during ischemia coupled to maintenance of high-energy phosphates and reduces postreperfusion injury. Circulation 2008, 117, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Sykes, M.C.; Ireland, C.; McSweeney, J.E.; Rosenholm, E.; Andren, K.G.; Kulik, T.J. The impact of right ventricular pressure and function on survival in patients with pulmonary vein stenosis. Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingo, L.; Magdo, H.S.; Day, R.W. Acute Pulmonary Vasodilator Testing and Long-Term Clinical Course in Segmental Pulmonary Vascular Disease. Pediatr. Cardiol. 2018, 39, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Hoeper, M.M.; Humbert, M.; Torbicki, A.; Vachiery, J.L.; Barbera, J.A.; Beghetti, M.; Corris, P.; Gaine, S.; Gibbs, J.S.; et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2009, 30, 2493–2537. [Google Scholar] [PubMed]
- Rol, N.; Timmer, E.M.; Faes, T.J.; Vonk Noordegraaf, A.; Grunberg, K.; Bogaard, H.J.; Westerhof, N. Vascular narrowing in pulmonary arterial hypertension is heterogeneous: Rethinking resistance. Physiol. Rep. 2017, 5, e13159. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Week | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Group | 6 | 8 | 10 | 12 | ||||||||||||||
| MAP | 5 | Sham | Rest | 90 | ± | 1 | 81 | ± | 4 | 92 | ± | 5 | 95 | ± | 4 | ||||
| (mmHg) | 5 | Exercise | 93 | ± | 4 | 92 | ± | 5 | 98 | ± | 4 | 102 | ± | 3 | |||||
| 6 | PVB | Rest | 86 | ± | 5 | 85 | ± | 4 | 80 | ± | 4 | 86 | ± | 3 | (†) | ||||
| 6 | Exercise | 90 | ± | 2 | 88 | ± | 3 | 83 | ± | 4 | † | 86 | ± | 7 | (†) | ||||
| LAP | 5 | Sham | Rest | 6.0 | ± | 2.1 | 3.8 | ± | 1.2 | 4.0 | ± | 1.2 | 3.1 | ± | 1.0 | ||||
| (mmHg) | 5 | Exercise | 9.5 | ± | 1.2 | 9.0 | ± | 0.8 | * | 7.9 | ± | 0.9 | (*) | 10.1 | ± | 1.1 | * | ||
| 6 | PVB | Rest | 4.1 | ± | 0.9 | 3.5 | ± | 1.3 | 6.4 | ± | 1.3 | 7.1 | ± | 1.6 | |||||
| 6 | Exercise | 7.2 | ± | 0.9 | * | 5.9 | ± | 0.8 | † | 10.1 | ± | 1.7 | 12.8 | ± | 1.9 | * (‡) | |||
| mPAP | 5 | Sham | Rest | 20 | ± | 1 | 17 | ± | 2 | 22 | ± | 1 | 20 | ± | 1 | ||||
| (mmHg) | 5 | Exercise | 34 | ± | 1 | * | 31 | ± | 1 | * | 32 | ± | 2 | * | 35 | ± | 3 | * | |
| 6 | PVB | Rest | 30 | ± | 2 | † | 31 | ± | 2 | † | 36 | ± | 3 | † | 39 | ± | 2 | † ‡ | |
| 6 | Exercise | 51 | ± | 2 | * † | 56 | ± | 3 | * † | 56 | ± | 2 | * † | 62 | ± | 3 | * † ‡ | ||
| HR | 5 | Sham | Rest | 152 | ± | 12 | 136 | ± | 10 | 138 | ± | 12 | 132 | ± | 9 | ||||
| (bpm) | 5 | Exercise | 259 | ± | 10 | * | 228 | ± | 15 | * | 243 | ± | 19 | * | 251 | ± | 10 | * | |
| 5 | PVB | Rest | 147 | ± | 8 | 139 | ± | 4 | 136 | ± | 10 | 135 | ± | 5 | |||||
| 5 | Exercise | 246 | ± | 11 | * | 238 | ± | 11 | * | 206 | ± | 8 | * (†) ‡ | 207 | ± | 6 | * † ‡ | ||
| CI | 5 | Sham | Rest | 0.19 | ± | 0.01 | 0.19 | ± | 0.01 | 0.19 | ± | 0.01 | 0.17 | ± | 0.01 | ||||
| (L min−1 kg−1) | 5 | Exercise | 0.32 | ± | 0.01 | * | 0.30 | ± | 0.01 | * | 0.29 | ± | 0.02 | * | 0.28 | ± | 0.02 | * | |
| 5 | PVB | Rest | 0.21 | ± | 0.01 | 0.19 | ± | 0.01 | 0.18 | ± | 0.02 | 0.16 | ± | 0.01 | (‡) | ||||
| 5 | Exercise | 0.32 | ± | 0.01 | * | 0.28 | ± | 0.02 | * | 0.25 | ± | 0.01 | * ‡ | 0.23 | ± | 0.01 | * † ‡ | ||
| SVi | 5 | Sham | Rest | 1.30 | ± | 0.09 | 1.39 | ± | 0.09 | 1.41 | ± | 0.13 | 1.31 | ± | 0.11 | ||||
| (mL kg−1) | 5 | Exercise | 1.26 | ± | 0.06 | 1.36 | ± | 0.11 | 1.23 | ± | 0.14 | 1.11 | ± | 0.10 | |||||
| 5 | PVB | Rest | 1.41 | ± | 0.09 | 1.35 | ± | 0.09 | 1.38 | ± | 0.05 | 1.21 | ± | 0.06 | |||||
| 5 | Exercise | 1.26 | ± | 0.06 | 1.19 | ± | 0.05 | 1.25 | ± | 0.06 | 1.10 | ± | 0.06 | ||||||
| SVRi | 5 | Sham | Rest | 472 | ± | 18 | 439 | ± | 28 | 488 | ± | 24 | 562 | ± | 26 | (‡) | |||
| (mmHg L−1 min kg) | 5 | Exercise | 288 | ± | 13 | * | 309 | ± | 22 | * | 340 | ± | 22 | * | 378 | ± | 27 | * (‡) | |
| 5 | PVB | Rest | 390 | ± | 32 | † | 471 | ± | 52 | 455 | ± | 59 | 534 | ± | 38 | (‡) | |||
| 5 | Exercise | 270 | ± | 13 | * | 319 | ± | 27 | * | 325 | ± | 24 | (*) | 391 | ± | 45 | * | ||
| tPVRi | 5 | Sham | Rest | 107 | ± | 9 | 94 | ± | 11 | 118 | ± | 7 | 116 | ± | 10 | ||||
| (mmHg L−1 min kg) | 5 | Exercise | 106 | ± | 6 | 105 | ± | 7 | 111 | ± | 10 | 128 | ± | 11 | |||||
| 5 | PVB | Rest | 132 | ± | 4 | † | 169 | ± | 14 | † | 192 | ± | 12 | † ‡ | 244 | ± | 26 | † ‡ | |
| 5 | Exercise | 155 | ± | 7 | * † | 201 | ± | 16 | † (‡) | 218 | ± | 11 | † ‡ | 274 | ± | 14 | † ‡ | ||
| pO2art | 5 | Sham | Rest | 98 | ± | 4 | 102 | ± | 4 | 103 | ± | 4 | 102 | ± | 4 | ||||
| (mmHg) | 5 | Exercise | 84 | ± | 4 | * | 86 | ± | 3 | * | 87 | ± | 3 | * | 92 | ± | 5 | ||
| 6 | PVB | Rest | 92 | ± | 5 | 87 | ± | 5 | † | 88 | ± | 3 | † | 88 | ± | 3 | † | ||
| 6 | Exercise | 68 | ± | 3 | * † | 63 | ± | 2 | * † | 66 | ± | 3 | * † | 65 | ± | 3 | * † | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Duin, R.W.B.; Stam, K.; Uitterdijk, A.; Bartelds, B.; Danser, A.H.J.; Reiss, I.K.M.; Duncker, D.J.; Merkus, D. Intervening with the Nitric Oxide Pathway to Alleviate Pulmonary Hypertension in Pulmonary Vein Stenosis. J. Clin. Med. 2019, 8, 1204. https://doi.org/10.3390/jcm8081204
van Duin RWB, Stam K, Uitterdijk A, Bartelds B, Danser AHJ, Reiss IKM, Duncker DJ, Merkus D. Intervening with the Nitric Oxide Pathway to Alleviate Pulmonary Hypertension in Pulmonary Vein Stenosis. Journal of Clinical Medicine. 2019; 8(8):1204. https://doi.org/10.3390/jcm8081204
Chicago/Turabian Stylevan Duin, Richard W. B., Kelly Stam, André Uitterdijk, Beatrijs Bartelds, A. H. Jan Danser, Irwin K. M. Reiss, Dirk J. Duncker, and Daphne Merkus. 2019. "Intervening with the Nitric Oxide Pathway to Alleviate Pulmonary Hypertension in Pulmonary Vein Stenosis" Journal of Clinical Medicine 8, no. 8: 1204. https://doi.org/10.3390/jcm8081204