Combined LDL and VLDL Electronegativity Correlates with Coronary Heart Disease Risk in Asymptomatic Individuals


 and
and
Abstract
:1. Introduction
2. Research Design and Methods
2.1. Study Participants
2.2. Sample Collection
2.3. VLDL and LDL Isolation and Separation
2.4. Cell Culture and Cellular Senescence Assay
2.5. Mice
2.6. Electrophoresis Assay
2.7. SA-β-Gal Staining and Oil Red O Staining in Aortas
2.8. Statistical Analysis
3. Results
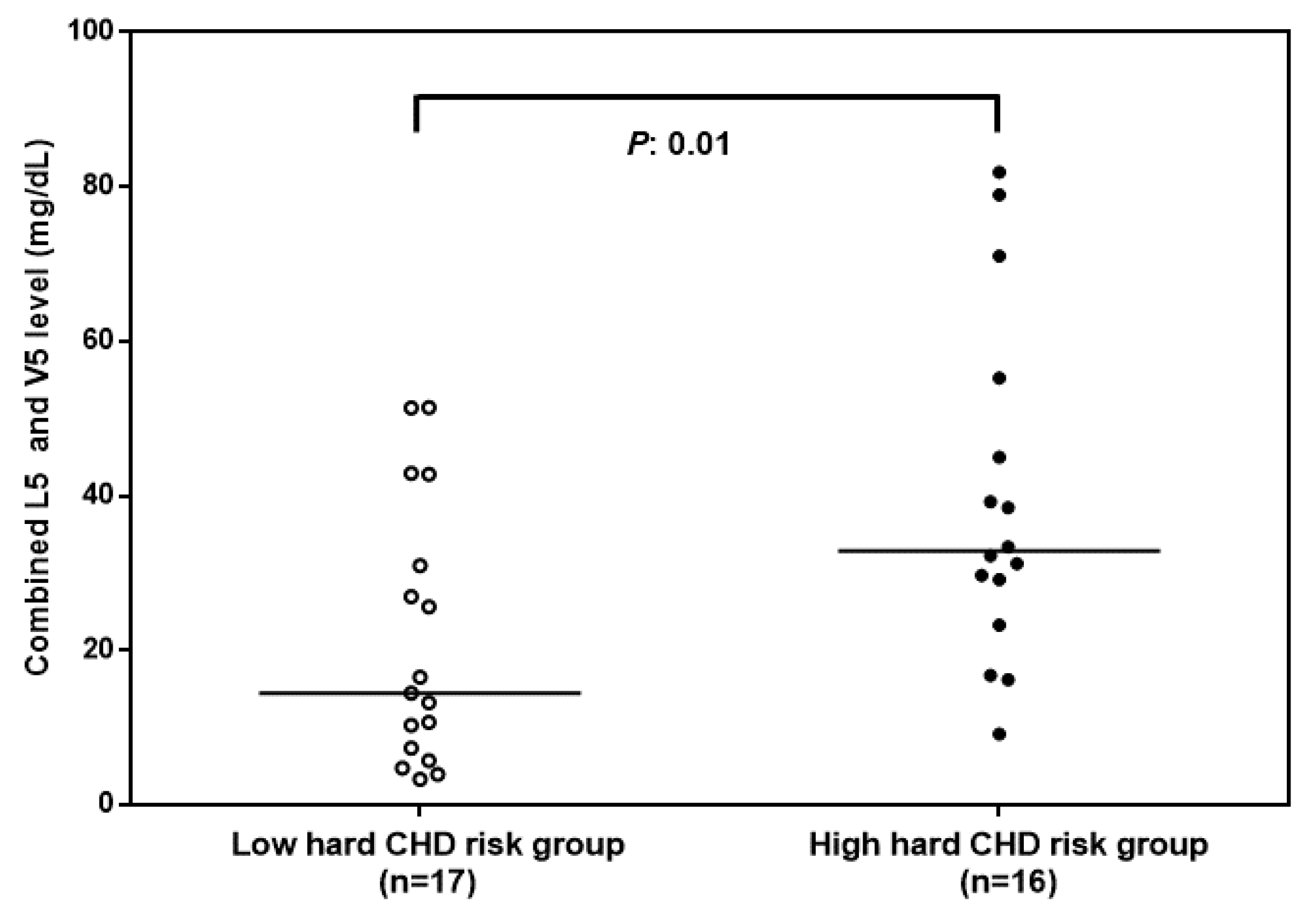
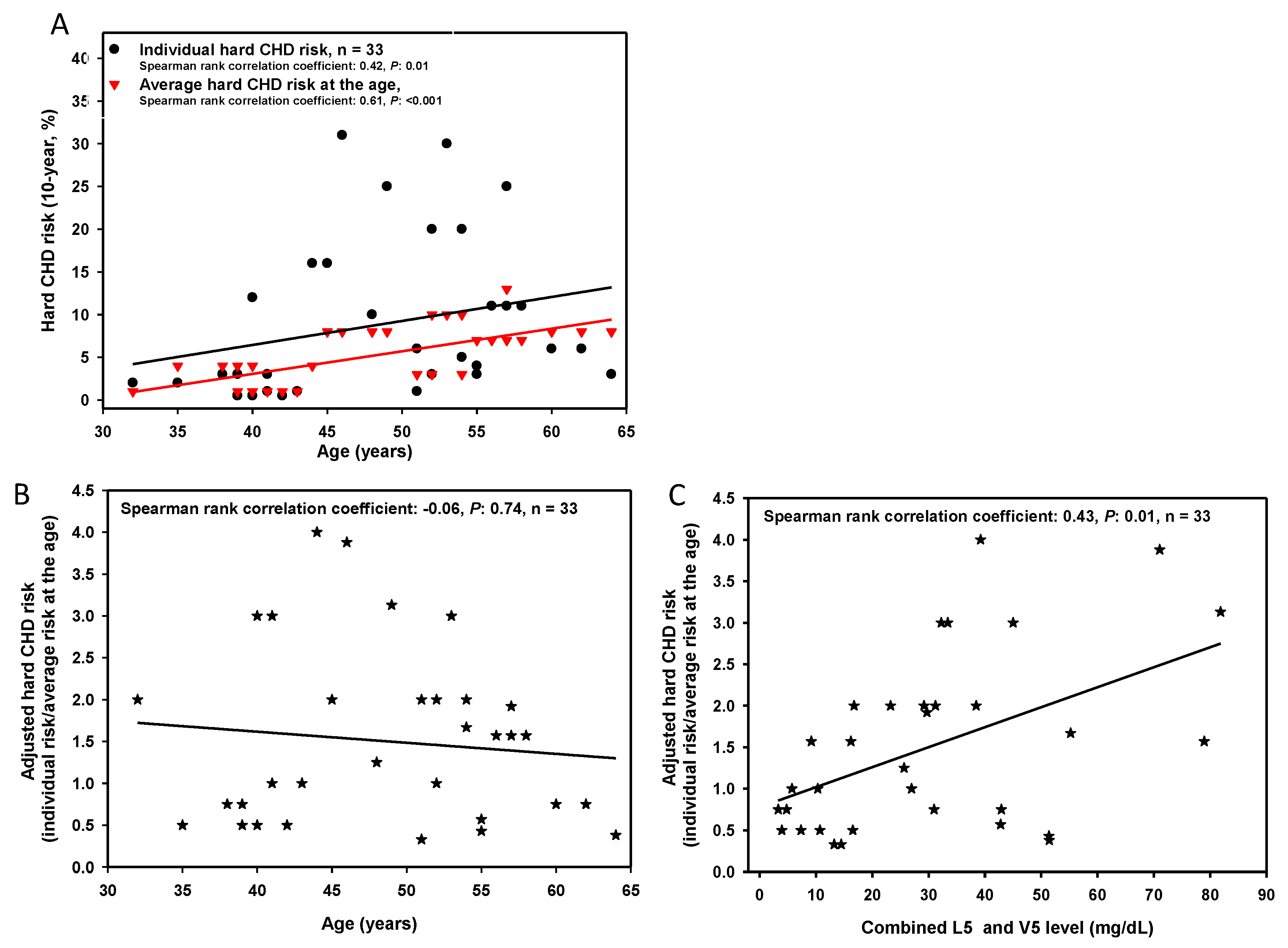
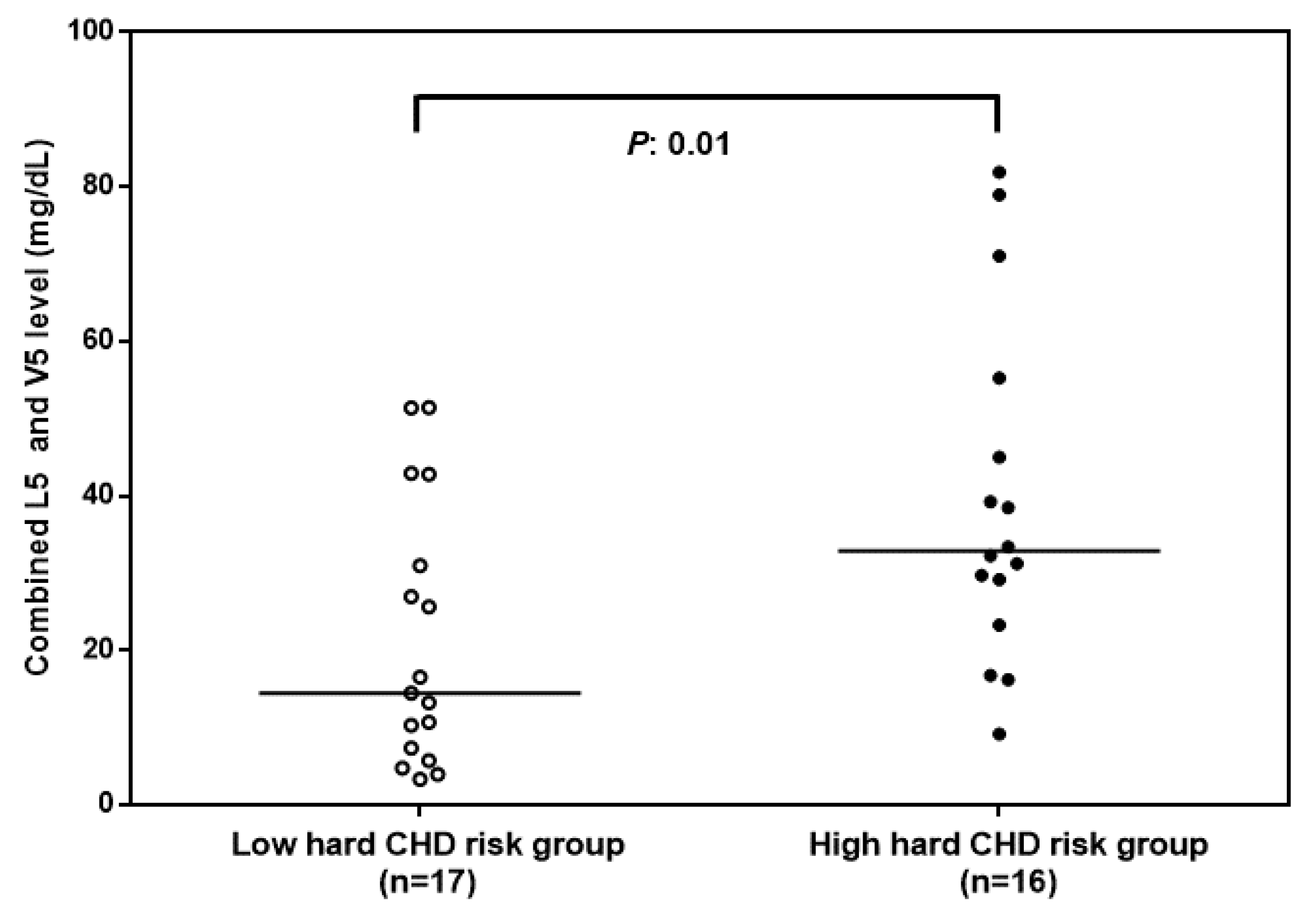
3.1. Combined L5 + V5 Plasma Levels Are Associated with Hard CHD Risk
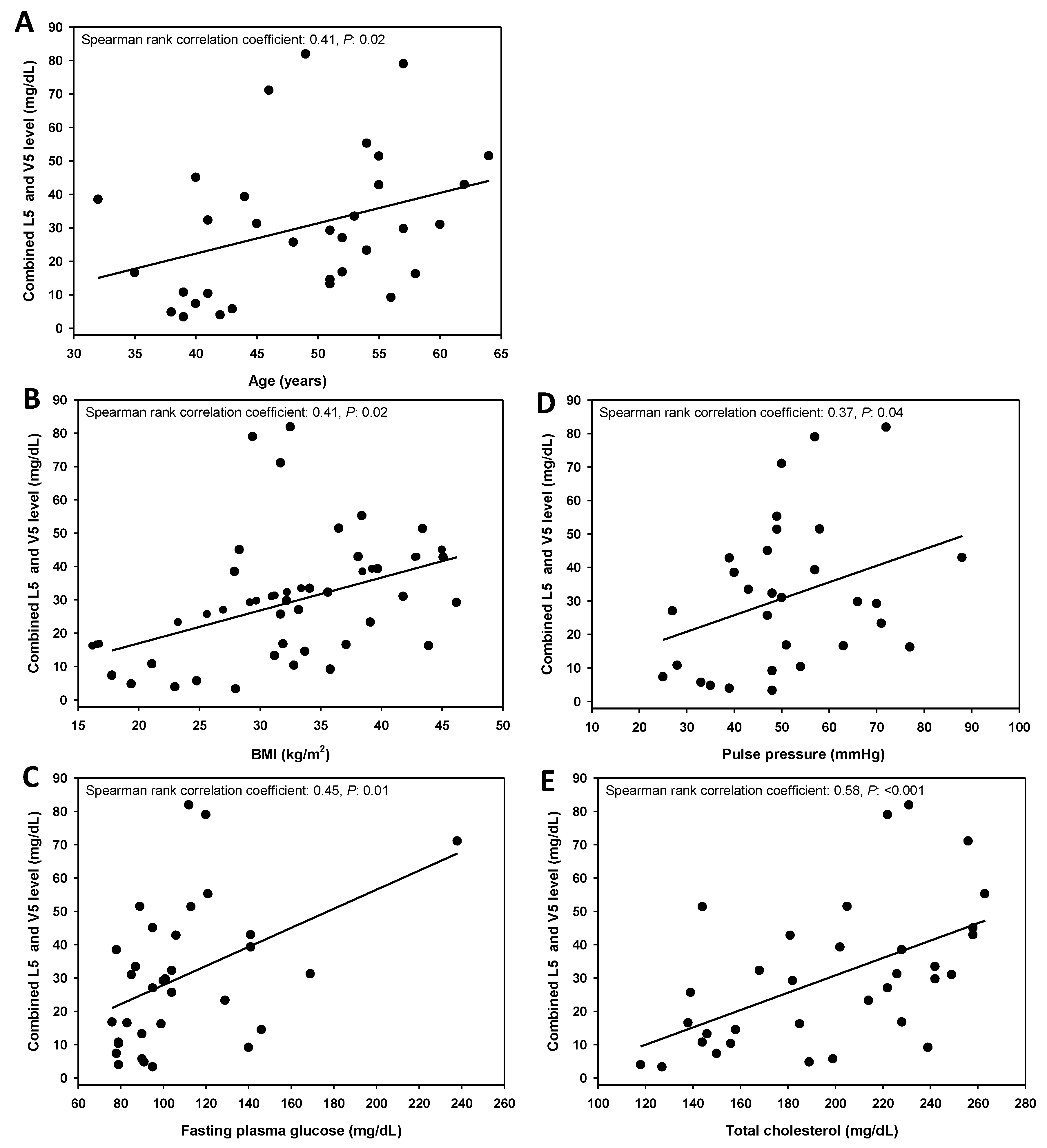
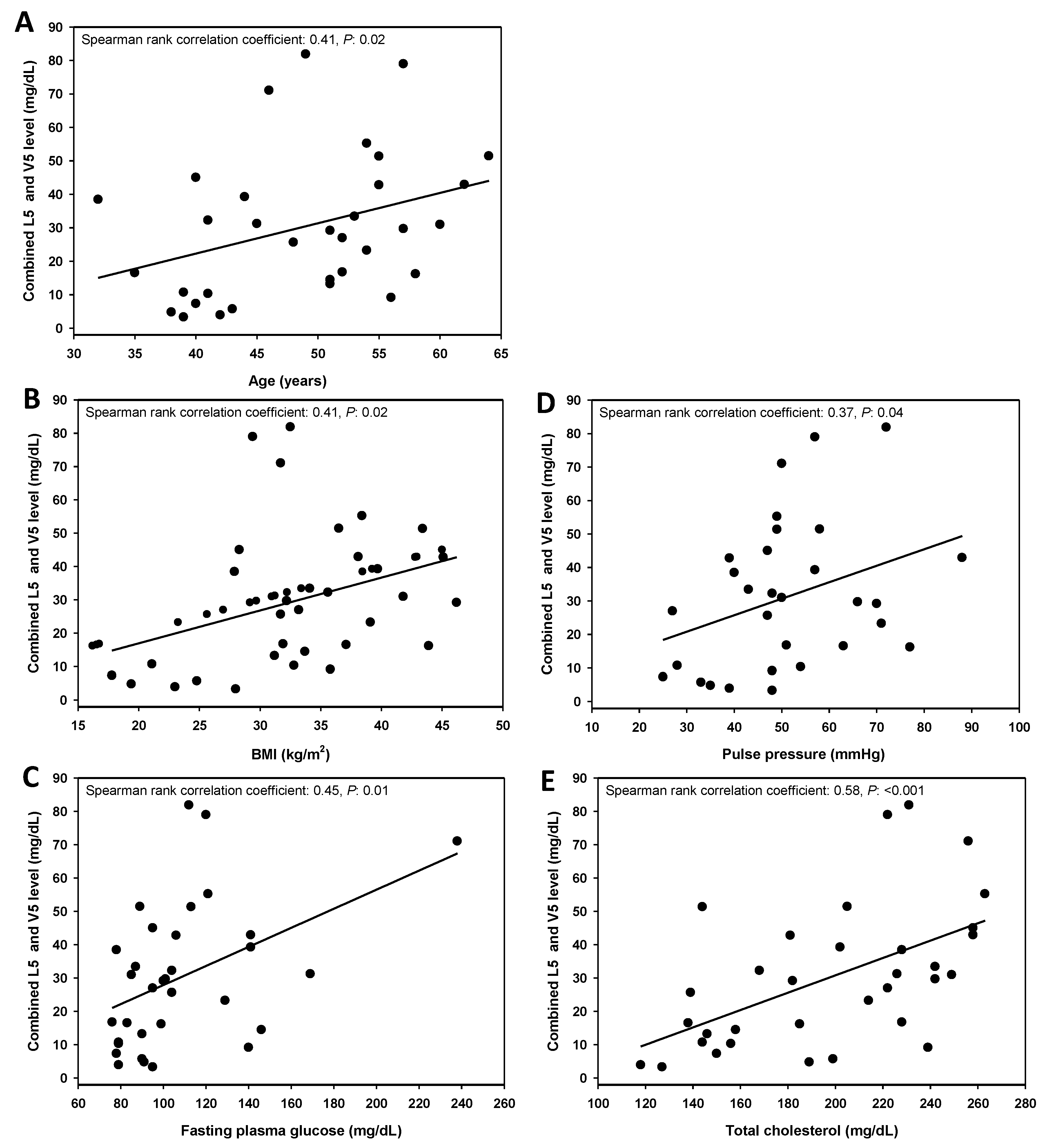
3.2. Combined L5 + V5 Plasma Levels Are Associated with Age and Select Hard CHD Risk Factors
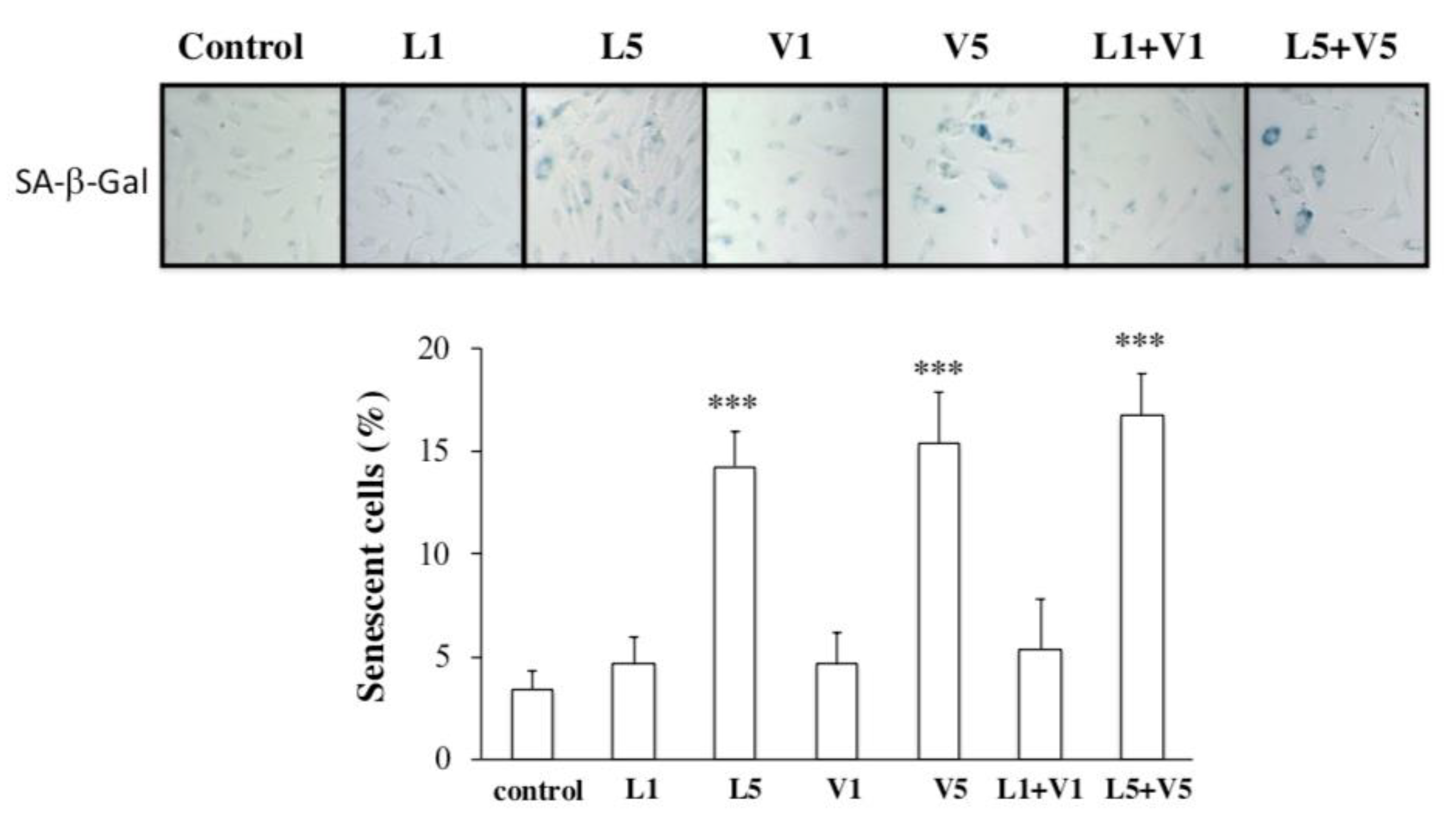
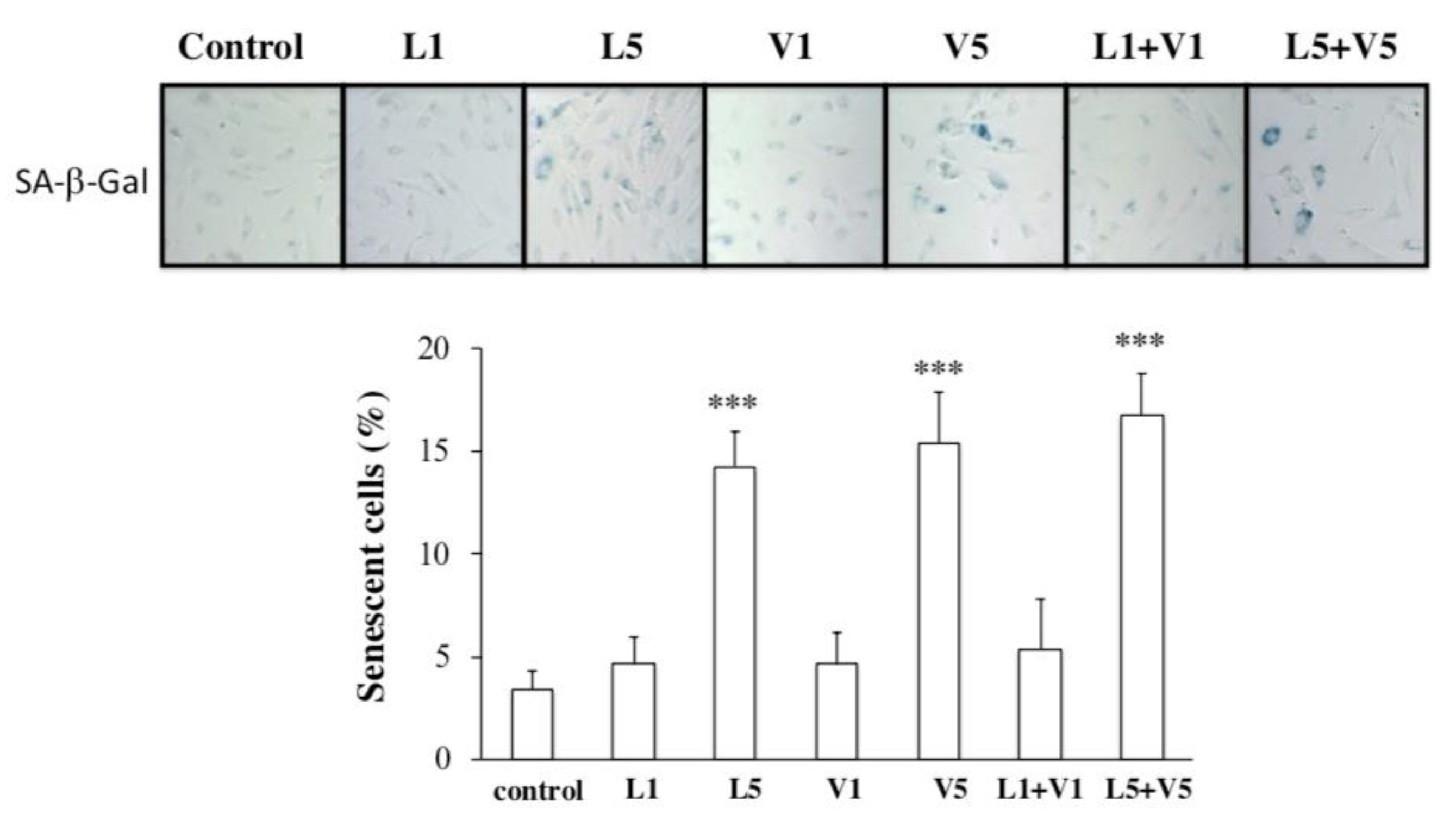
3.3. L5 + V5 Promote Cellular Senescence in HAECs
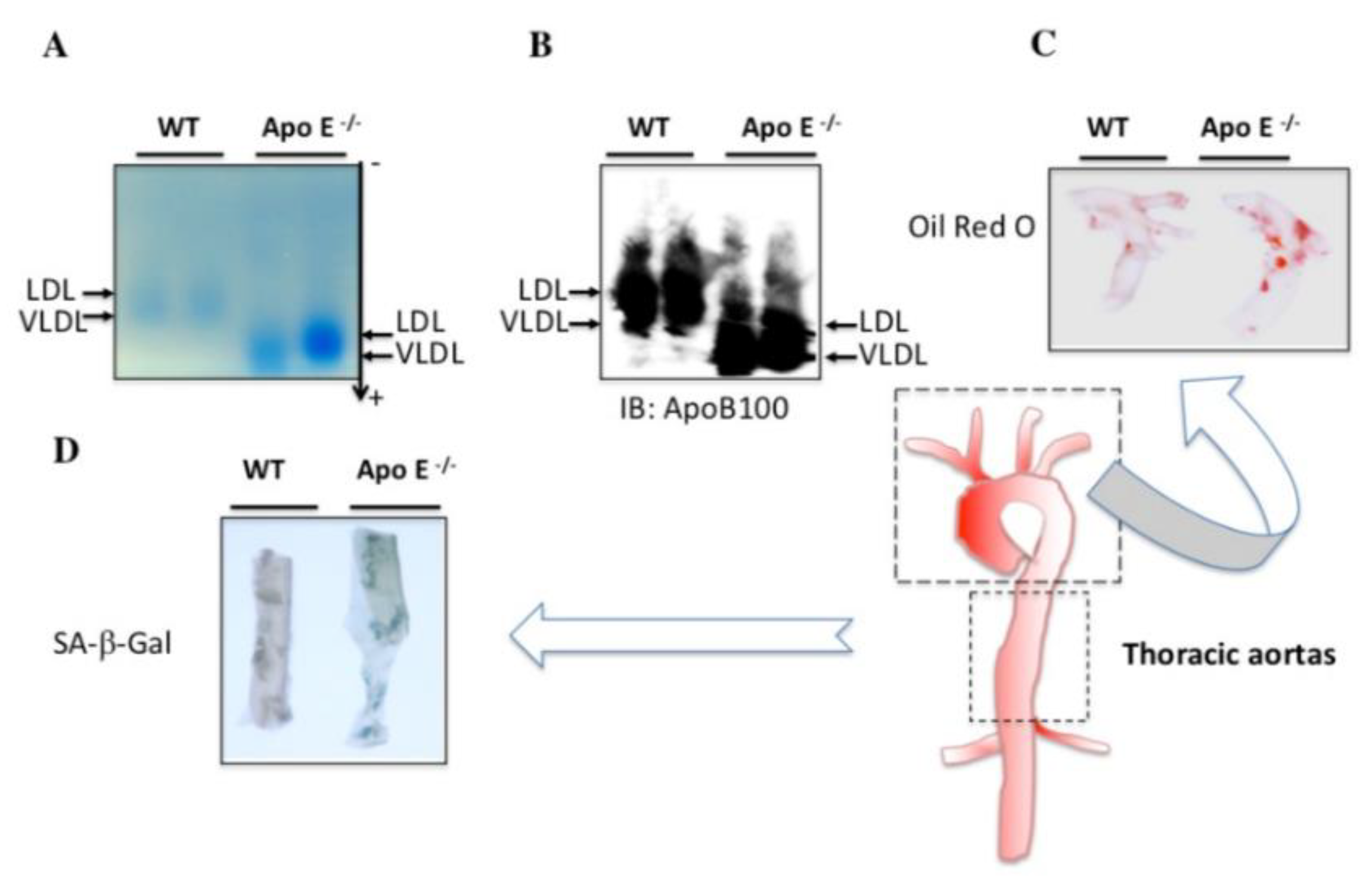
3.4. Endogenous Levels of Electronegative VLDL and LDL Are Increased in the Serum of ApoE−/− Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Huffman, M.D. Executive summary: Heart disease and stroke statistics-2015 update: A report from the American Heart Association. Circulation 2015, 131, 434–441. [Google Scholar] [CrossRef]
- Nichols, M.; Townsend, N.; Scarborough, P.; Rayner, M. Cardiovascular disease in Europe 2014: Epidemiological update. Eur. Heart. J. 2014, 35, 2950–2959. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Muntner, P.; Bittencourt, M.S. Heart Disease and Stroke Statistics-2019 Update: A Report from the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Alla, F. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef]
- Lloyd-Jones, D.M.; Larson, M.G.; Beiser, A.; Levy, D. Lifetime risk of developing coronary heart disease. Lancet 1999, 353, 89–92. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Levy, D. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises: Part I: Aging Arteries: A ‘Set Up’ for Vascular Disease. Circulation 2003, 107, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell. Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Hayflick, L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell. Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Minamino, T.; Komuro, I. Vascular Cell Senescence: Contribution to Atherosclerosis. Circ. Res. 2007, 100, 15–26. [Google Scholar] [CrossRef]
- Chen, C.H.; Jiang, T.; Yang, J.H.; Jiang, W.; Lu, J.; Marathe, G.K.; Yang, C.Y. Low-density lipoprotein in hypercholesterolemic human plasma induces vascular endothelial cell apoptosis by inhibiting fibroblast growth factor 2 transcription. Circulation 2003, 107, 2102–2108. [Google Scholar] [CrossRef]
- Chen, W.Y.; Chen, F.Y.; Lee, A.S.; Ting, K.H.; Chang, C.M.; Hsu, J.F.; Shen, M.Y. Sesamol reduces the atherogenicity of electronegative L5 LDL in vivo and in vitro. J. Nat. Prod. 2015, 78, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Hsu, H.C.; Lee, A.S.; Tang, D.; Chow, L.P.; Yang, C.Y.; Chen, C.H. The most negatively charged low-density lipoprotein L5 induces stress pathways in vascular endothelial cells. J. Vasc. Res. 2012, 49, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Chen, W.Y.; Chan, H.C.; Hsu, J.F.; Shen, M.Y.; Chang, C.M.; Chen, C.H. Gender disparity in LDL-induced cardiovascular damage and the protective role of estrogens against electronegative LDL. Cardiovasc. Diabetol. 2014, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Lee, A.S.; Lu, L.S.; Ke, L.Y.; Chen, W.Y.; Dong, J.W.; Kuzan, T.Y. Human electronegative LDL induces mitochondrial dysfunction and premature senescence of vascular cells in vivo. Aging Cell. 2018, 17, e12792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, C.S.; Chan, H.C.; Tsai, M.H.; Stancel, N.; Lee, H.C.; Cheng, K.H.; Lai, W.T. Range of L5 LDL levels in healthy adults and L5′s predictive power in patients with hyperlipidemia or coronary artery disease. Sci. Rep. 2018, 8, 11866. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.C.; Ke, L.Y.; Chu, C.S.; Lee, A.S.; Shen, M.Y.; Cruz, M.A.; Lai, W.T. Highly electronegative LDL from patients with ST-elevation myocardial infarction triggers platelet activation and aggregation. Blood 2013, 122, 3632–3641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.Y.; Chen, F.Y.; Hsu, J.F.; Fu, R.H.; Chang, C.M.; Chang, C.T.; Sheu, J.R. Plasma L5 levels are elevated in ischemic stroke patients and enhance platelet aggregation. Blood 2016, 127, 1336–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.F.; Chou, T.C.; Lu, J.; Chen, S.H.; Chen, F.Y.; Chen, C.C.; Chen, C.H. Low-density lipoprotein electronegativity is a novel cardiometabolic risk factor. PLoS ONE 2014, 9, e107340. [Google Scholar] [CrossRef]
- Sanchez-Quesada, J.L.; Benitez, S.; Ordonez-Llanos, J. Electronegative low-density lipoprotein. Curr. Opin. Lipidol. 2004, 15, 329–335. [Google Scholar] [CrossRef]
- Mello, A.P.; da Silva, I.T.; Abdalla, D.S.; Damasceno, N.R. Electronegative low-density lipoprotein: Origin and impact on health and disease. Atherosclerosis 2011, 215, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Damasceno, N.R.; Sevanian, A.; Apolinario, E.; Oliveira, J.M.; Fernandes, I.; Abdalla, D.S. Detection of electronegative low density lipoprotein (LDL-) in plasma and atherosclerotic lesions by monoclonal antibody-based immunoassays. Clin. Biochem. 2006, 39, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.C.; Wang, Y.C.; Chang, S.S.; Lo, P.H.; Lu, J.; Sawamura, T.; Chen, C.H. Abstract 13127: Expression of the Highly Electronegative LDL Receptor LOX-1 is Increased in Thrombi of STEMI Patients. Circulation 2012, 126, A13127. [Google Scholar]
- Jo-Watanabe, A.; Ohse, T.; Nishimatsu, H.; Takahashi, M.; Ikeda, Y.; Wada, T.; Hirata, Y. Glyoxalase I reduces glycative and oxidative stress and prevents age-related endothelial dysfunction through modulation of endothelial nitric oxide synthase phosphorylation. Aging Cell. 2014, 13, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Burnley, P.; Rahman, M.; Wang, H.; Zhang, Z.; Sun, X.; Zhuge, Q.; Su, D.M. Role of the p63-FoxN1 regulatory axis in thymic epithelial cell homeostasis during aging. Cell Death Dis. 2013, 4, e932. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, R.B.; Vasan, R.S.; Pencina, M.J.; Wolf, P.A.; Cobain, M.; Massaro, J.M.; Kannel, W.B. General cardiovascular risk profile for use in primary care: The Framingham Heart Study. Circulation 2008, 117, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Pencina, M.J.; D’Agostino, R.B.; Sr Larson, M.G.; Massaro, J.M.; Vasan, R.S. Predicting the 30-year risk of cardiovascular disease: The framingham heart study. Circulation 2009, 119, 3078–3084. [Google Scholar] [CrossRef] [PubMed]
- Kolovou, G.; Anagnostopoulou, K.; Mikhailidis, D.P.; Cokkinos, D.V. Apolipoprotein E knockout models. Curr. Pharm. Des. 2008, 14, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Kumazaki, T.; Kobayashi, M.; Mitsui, Y. Enhanced expression of fibronectin during in vivo cellular aging of human vascular endothelial cells and skin fibroblasts. Exp. Cell. Res. 1993, 205, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Burrig, K.F. The endothelium of advanced arteriosclerotic plaques in humans. Arter. Thromb 1991, 11, 1678–1689. [Google Scholar] [CrossRef]
- Fenton, M.; Barker, S.; Kurz, D.J.; Erusalimsky, J.D. Cellular senescence after single and repeated balloon catheter denudations of rabbit carotid arteries. Arter. Thromb. Vasc. Biol. 2001, 21, 220–226. [Google Scholar] [CrossRef]
- Nakashima, Y.; Plump, A.S.; Raines, E.W.; Breslow, J.L.; Ross, R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arter. Thromb. 1994, 14, 133–140. [Google Scholar] [CrossRef]
- Chen, C.H.; Lu, J.; Chen, S.H.; Huang, R.Y.; Yilmaz, H.R.; Dong, J.; Yang, C.Y. Effects of Electronegative VLDL on endothelium damage in metabolic syndrome. Diabetes Care 2012, 35, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, R.; Sharma, H.W.; Ramakrishnan, S.; Keith, E.; Narayanan, R. Telomerase activity in normal human endothelial cells. Anticancer Res. 1997, 17, 827–832. [Google Scholar] [PubMed]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Bennett, M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: Effects of telomerase and oxidative stress. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Low Hard CHD Risk (n = 17) c | High Hard CHD Risk (n = 16) c | p-Value d | |
|---|---|---|---|
| Age (years) | 44.9 ± 6.8 | 49.3 ± 7.4 | 0.46 |
| Gender (men: women) | 4:13 | 9:7 | 0.06 |
| Diabetes mellitus drug treatment | 2/17 | 2/16 | 0.94 |
| Hypertension drug treatment | 6/17 | 10/16 | 0.12 |
| Metabolic syndrome | 7/17 | 13/16 | 0.02 |
| Waist circumference (cm) | 97.1 ± 21.9 | 110.8 ± 11.8 | 0.17 |
| Body mass index (kg/m2) | 30.2 ± 8.4 | 35.1 ± 5.5 | 0.27 |
| Systolic blood pressure (mmHg) | 115.1 ± 21.9 | 138 ± 11.6 | 0.01 |
| Diastolic blood pressure (mmHg) | 74.5 ± 13.4 | 81.6 ± 11.5 | 0.03 |
| Pulse pressure (mmHg) a | 40.6 ± 11.8 | 56.4 ± 11.8 | 0.03 |
| Mean arterial pressure (mmHg) | 88 ± 15.8 | 100.4 ± 10.1 | 0.01 |
| Fasting plasma glucose (mg/dL) | 94.9 ± 18.4 | 119.4 ± 40.1 | 0.05 |
| Total cholesterol (mg/dL) | 157.9 ± 29.3 | 224.1 ± 27.9 | <0.001 |
| Triglyceride (mg/dL) | 96.4 ± 52 | 167.3 ± 64.4 | 0.02 |
| HDL (mg/dL) | 48.1 ± 12.2 | 47.5 ± 10.5 | 0.75 |
| LDL (mg/dL) | 90.6 ± 25.7 | 143.1 ± 27.6 | <0.001 |
| VLDL (mg/dL) | 19.3 ± 10.4 | 33.5 ± 12.9 | 0.02 |
| LDL/HDL ratio | 2 ± 0.6 | 3.1 ± 0.9 | 0.003 |
| L5 (mg/dL) | 9.2 ± 10.5 | 27.9 ± 21.8 | 0.01 |
| V5 (mg/dL) | 7.7 ± 6.2 | 11.5 ± 8.3 | 0.46 |
| L5 and V5 (mg/dL) | 16.9 ± 14.8 | 39.4 ± 22 | 0.01 |
| Hard CHD risk (%) b | 2.4 ± 2.5 | 15.3 ± 9.2 | <0.001 |
| Average hard CHD risk at the age (%) b | 3.4 ± 2.4 | 6.5 ± 3.5 | 0.07 |
| Adjusted hard CHD risk b | 0.7 ± 0.3 | 2.4 ± 0.8 | <0.001 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, M.-Y.; Hsu, J.-F.; Chen, F.-Y.; Lu, J.; Chang, C.-M.; Madjid, M.; Dean, J.; Dixon, R.A.F.; Shayani, S.; Chou, T.-C.; et al. Combined LDL and VLDL Electronegativity Correlates with Coronary Heart Disease Risk in Asymptomatic Individuals. J. Clin. Med. 2019, 8, 1193. https://doi.org/10.3390/jcm8081193
Shen M-Y, Hsu J-F, Chen F-Y, Lu J, Chang C-M, Madjid M, Dean J, Dixon RAF, Shayani S, Chou T-C, et al. Combined LDL and VLDL Electronegativity Correlates with Coronary Heart Disease Risk in Asymptomatic Individuals. Journal of Clinical Medicine. 2019; 8(8):1193. https://doi.org/10.3390/jcm8081193
Chicago/Turabian StyleShen, Ming-Yi, Jing-Fang Hsu, Fang-Yu Chen, Jonathan Lu, Chia-Ming Chang, Mohammad Madjid, Juliette Dean, Richard A. F. Dixon, Steven Shayani, Tzu-Chieh Chou, and et al. 2019. "Combined LDL and VLDL Electronegativity Correlates with Coronary Heart Disease Risk in Asymptomatic Individuals" Journal of Clinical Medicine 8, no. 8: 1193. https://doi.org/10.3390/jcm8081193