A Mutation in ZNF143 as a Novel Candidate Gene for Endothelial Corneal Dystrophy

 , , , , add
Show full author list
, , , , add
Show full author list
Abstract
:1. Introduction
2. Experimental Section
2.1. Ethics Statement
2.2. Subject and Ophthalmologic Examination
2.3. Sanger Sequencing of OVOL2, COL8A2, ZEB1, SLC4A11 and GRHL2
2.4. Array-Comparative Genomic Hybridization (CGH)
2.5. Haplotype Analysis
2.6. Whole Exome Sequencing
2.7. Modeling of ZNF143 p.Asp313His
2.8. In Vitro Functional Studies of ZNF143 p.Asp313His
2.8.1. Isolation and Culture of Human Corneal Endothelial Cells (hCECs)
2.8.2. siRNA-Mediated Silencing of Human ZNF143
2.8.3. Transfection of Human ZNF143 (Wild Type and Mutant)
2.8.4. Gene Expression Analysis
2.8.5. Immunoblotting
2.9. Immunohistochemical Staining
3. Results
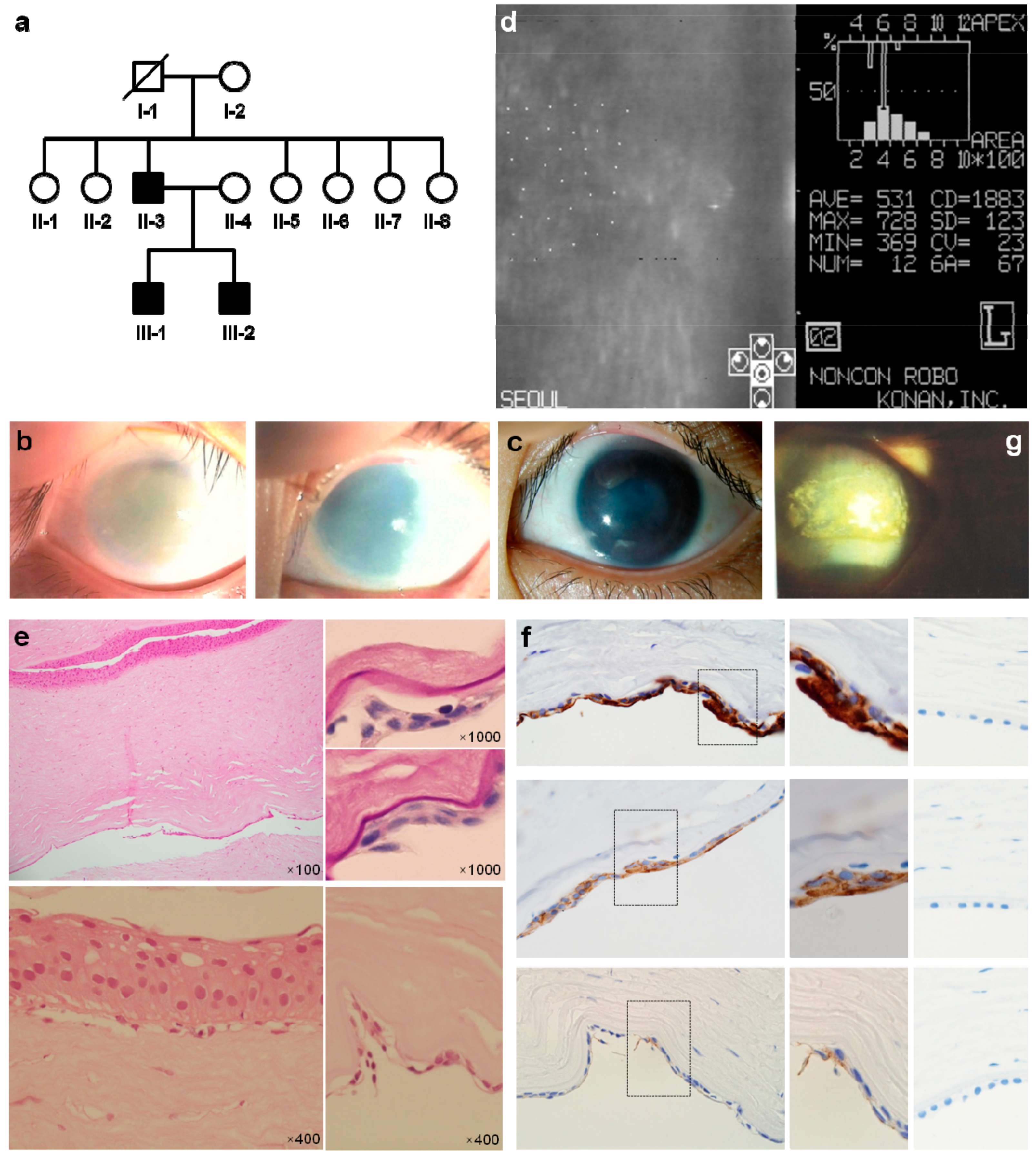
3.1. Patients Characteristics
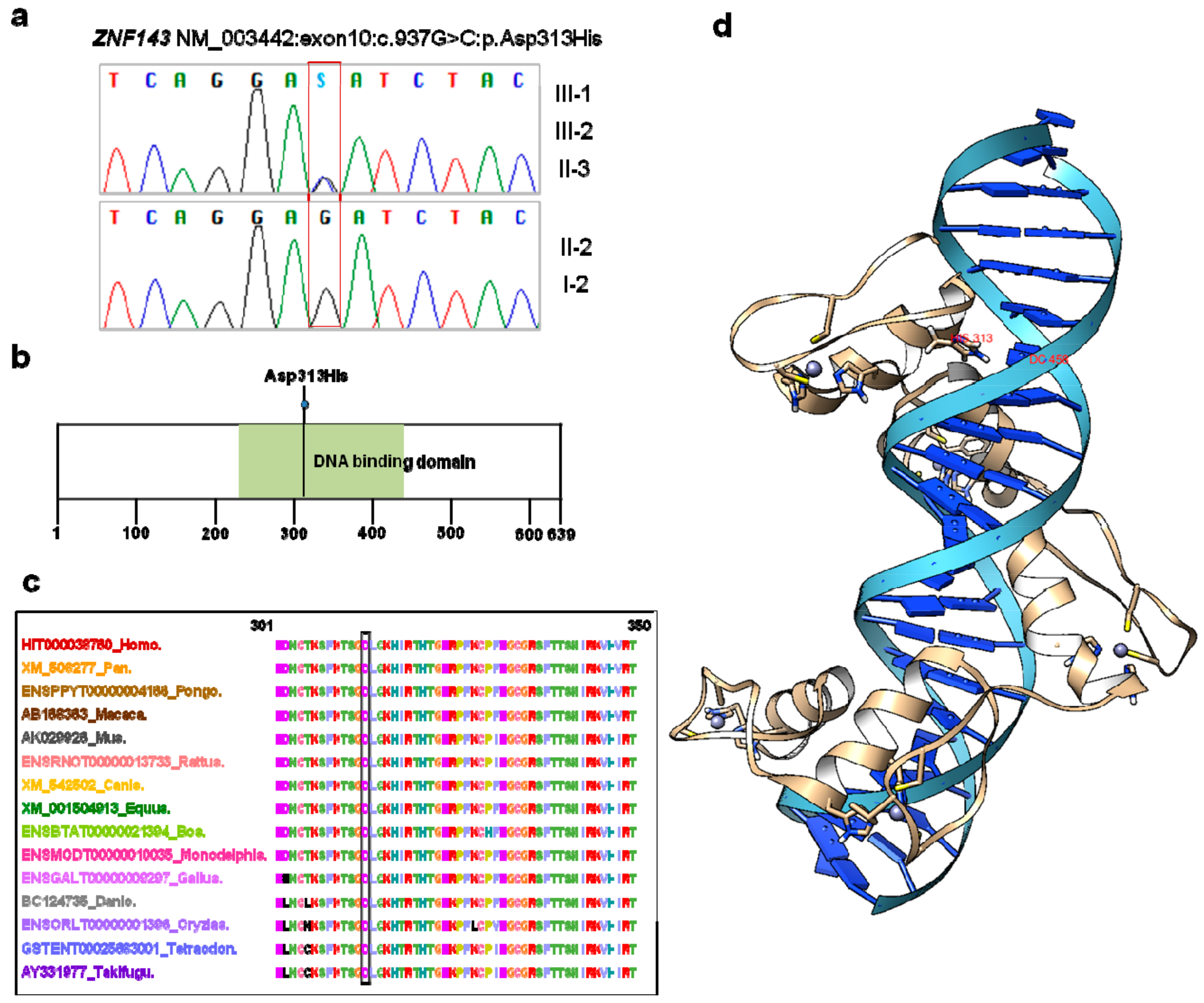
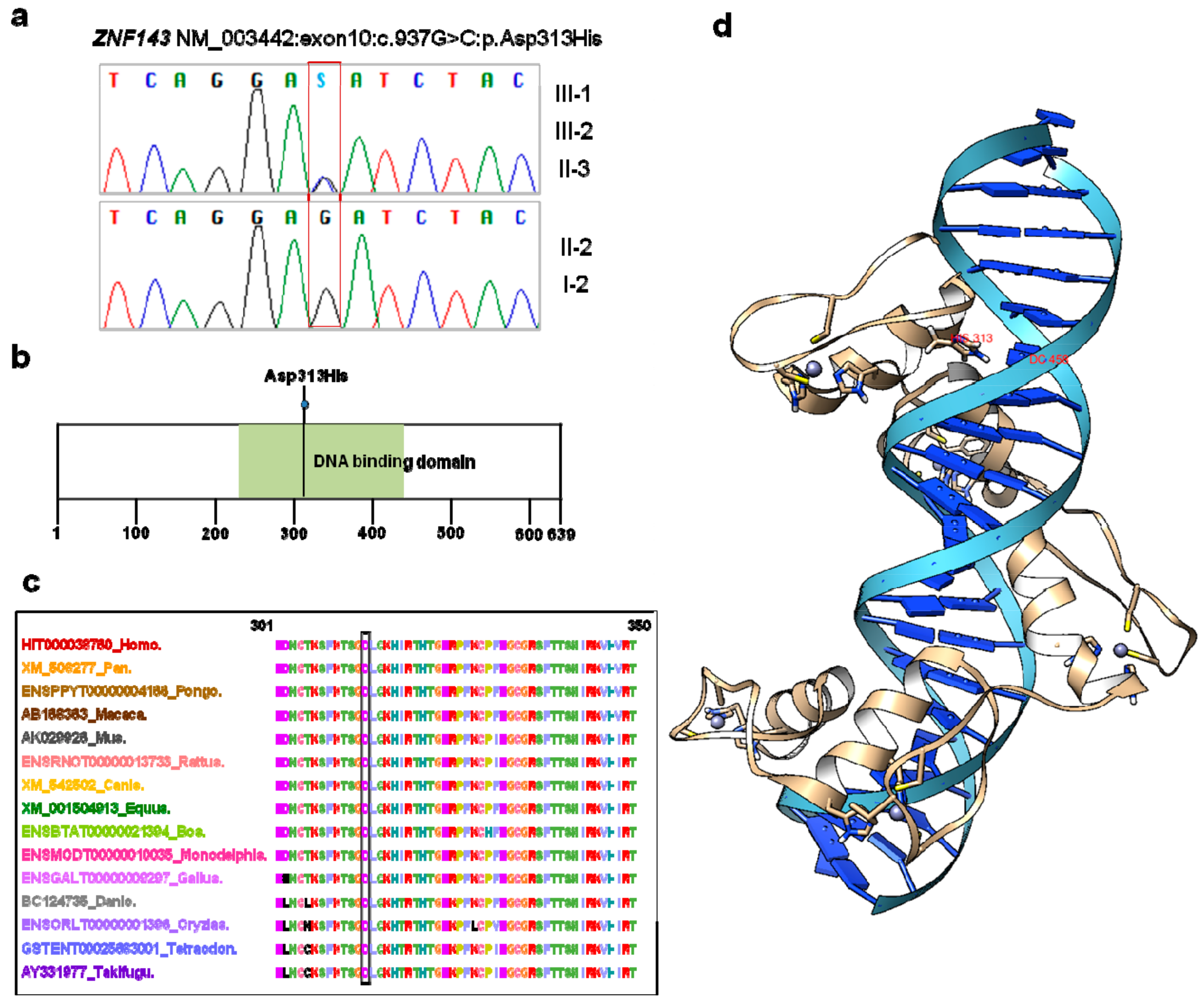
3.2. ZNF143 c.937G>C p.(Asp313His) Mutation Was Identified as a New Candidate for Endothelial CD
3.3. Three-Dimensional Modeling of ZNF143 p.Asp313His
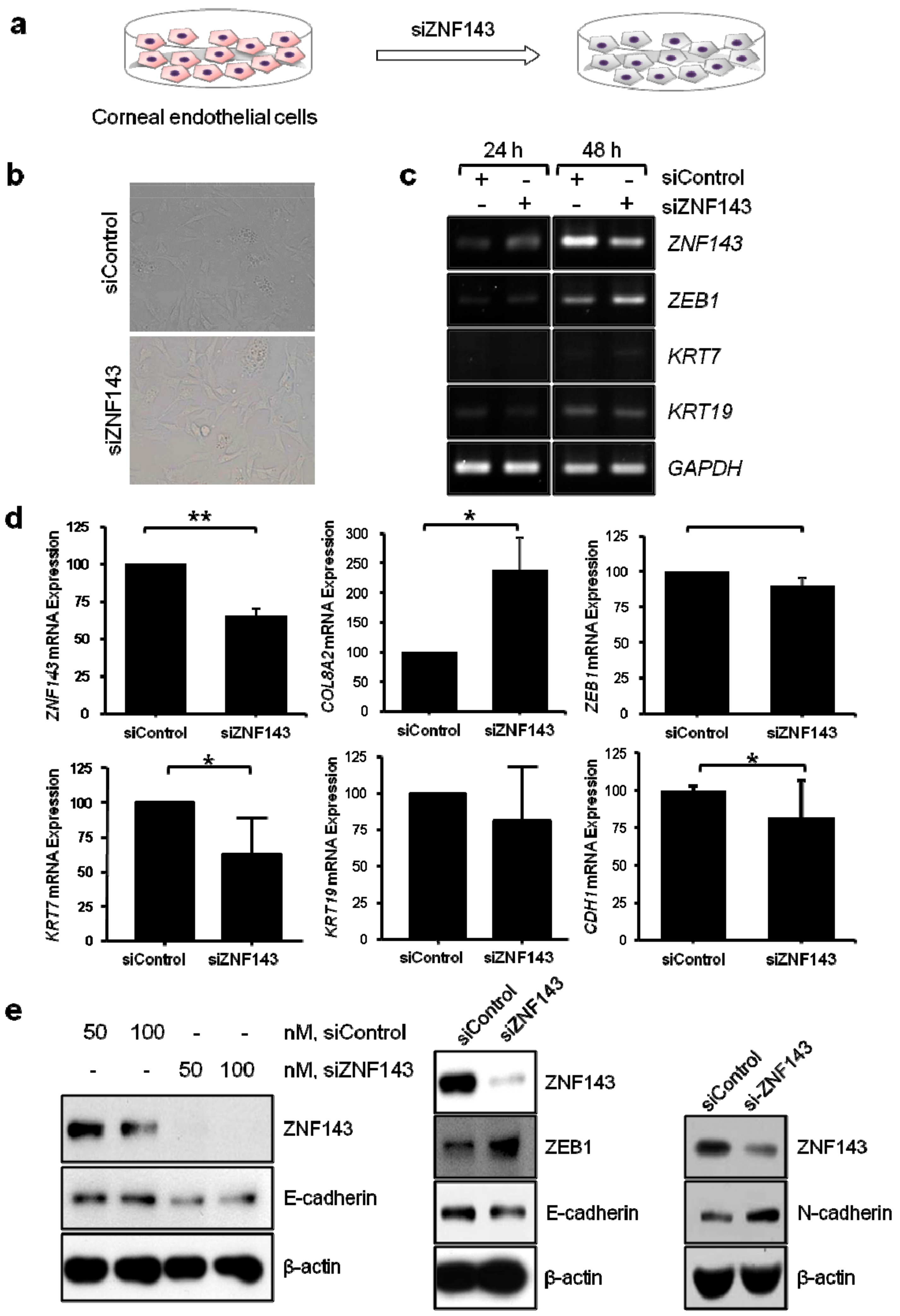
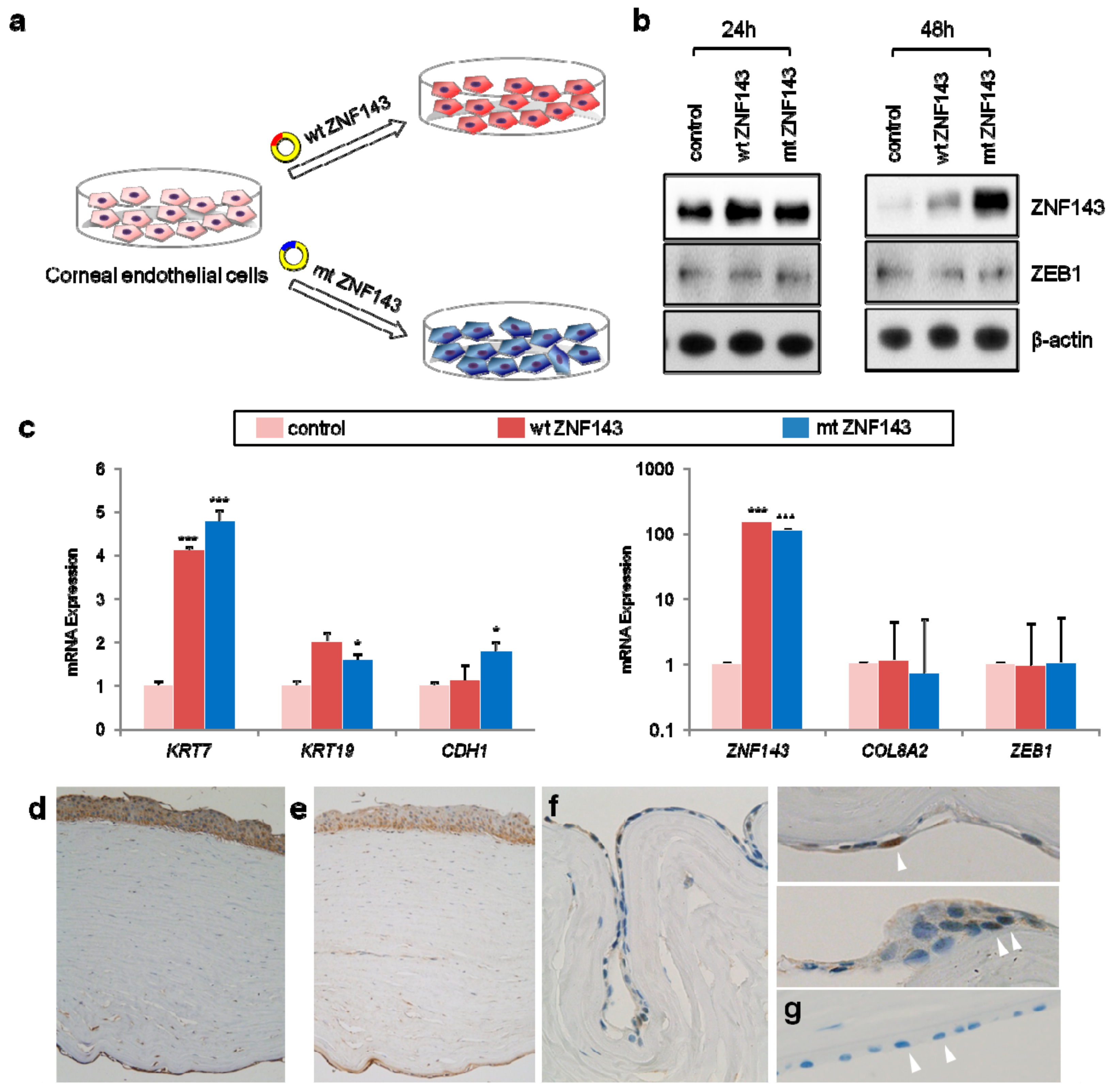
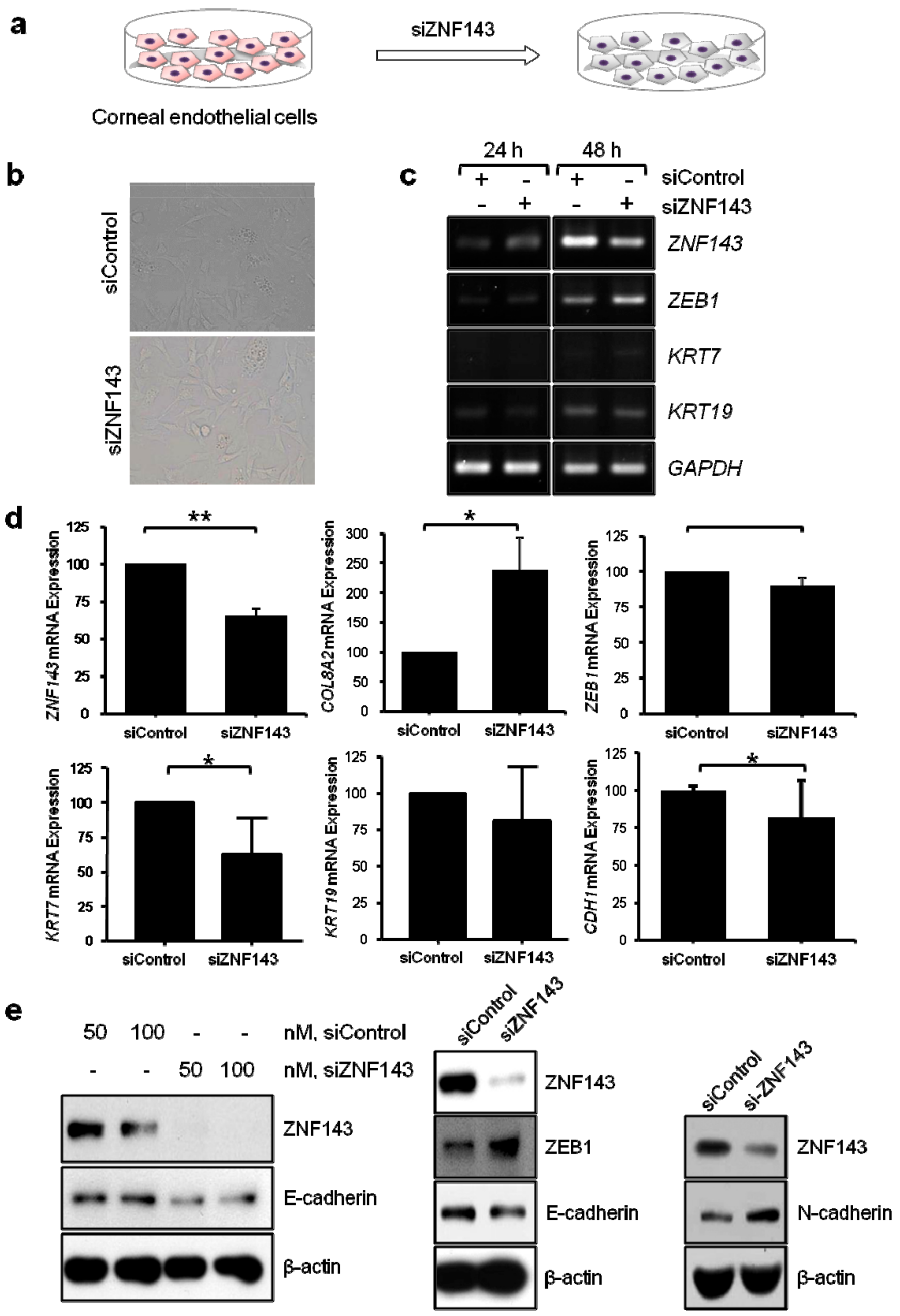
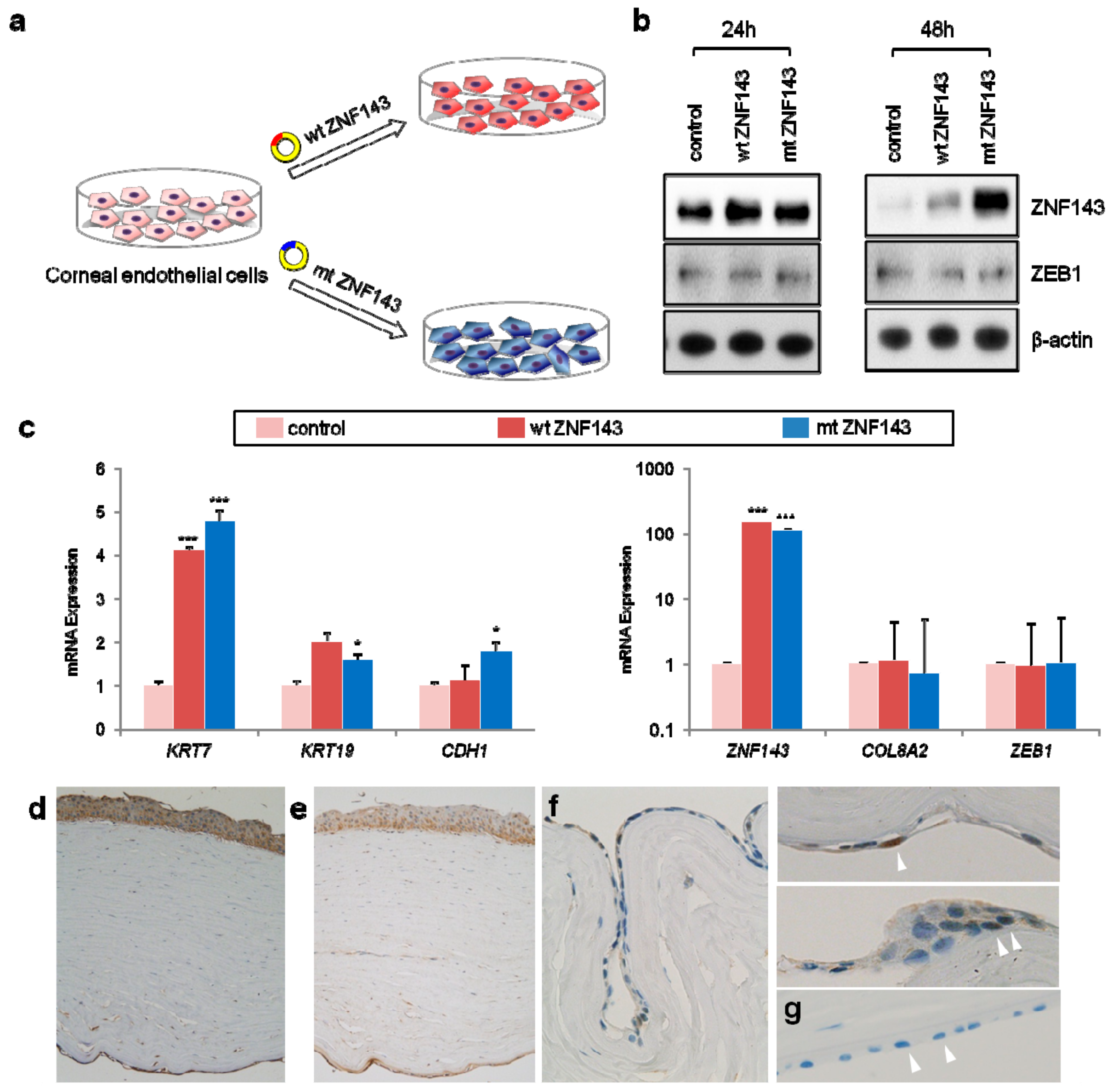
3.4. In Vitro Functional Studies of ZNF143 c.937G>C p.Asp313His
3.5. Network Analysis Reveals Activation of Epithelialization and Proinflammatory Signals
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weiss, J.S.; Moller, H.U.; Aldave, A.J.; Seitz, B.; Bredrup, C.; Kivela, T.; Munier, F.L.; Rapuano, C.J.; Nischal, K.K.; Kim, E.K.; et al. IC3D classification of corneal dystrophies—Edition 2. Cornea 2015, 34, 117–159. [Google Scholar] [CrossRef] [PubMed]
- Vemuganti, G.K.; Rathi, V.M.; Murthy, S.I. Histological landmarks in corneal dystrophy: Pathology of corneal dystrophies. Dev. Ophthalmol. 2011, 48, 24–50. [Google Scholar] [CrossRef] [PubMed]
- Klintworth, G.K. Corneal dystrophies. Orphanet J. Rare Dis. 2009, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.; Kim, M.; Kim, Y.; Kim, J.; Kwon, A.; Choi, H.; Park, J.; Jang, W.; Lee, Y.S.; Park, S.H.; et al. Mutational spectrum of Korean patients with corneal dystrophy. Clin. Genet. 2016, 89, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Schmedt, T.; Silva, M.M.; Ziaei, A.; Jurkunas, U. Molecular bases of corneal endothelial dystrophies. Exp. Eye Res. 2012, 95, 24–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.; Munier, F.L.; Yardley, J.; Hart-Holden, N.; Perveen, R.; Cousin, P.; Sutphin, J.E.; Noble, B.; Batterbury, M.; Kielty, C.; et al. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum. Mol. Genet. 2001, 10, 2415–2423. [Google Scholar] [CrossRef] [Green Version]
- Krafchak, C.M.; Pawar, H.; Moroi, S.E.; Sugar, A.; Lichter, P.R.; Mackey, D.A.; Mian, S.; Nairus, T.; Elner, V.; Schteingart, M.T.; et al. Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells. Am. J. Hum. Genet. 2005, 77, 694–708. [Google Scholar] [CrossRef]
- Aldave, A.J.; Han, J.; Frausto, R.F. Genetics of the corneal endothelial dystrophies: An evidence-based review. Clin. Genet. 2013, 84, 109–119. [Google Scholar] [CrossRef]
- Liskova, P.; Dudakova, L.; Evans, C.J.; Rojas Lopez, K.E.; Pontikos, N.; Athanasiou, D.; Jama, H.; Sach, J.; Skalicka, P.; Stranecky, V.; et al. Ectopic GRHL2 Expression Due to Non-coding Mutations Promotes Cell State Transition and Causes Posterior Polymorphous Corneal Dystrophy 4. Am. J. Hum. Genet. 2018, 102, 447–459. [Google Scholar] [CrossRef] [Green Version]
- Kirkness, C.M.; McCartney, A.; Rice, N.S.; Garner, A.; Steele, A.D. Congenital hereditary corneal oedema of Maumenee: Its clinical features, management, and pathology. Br. J. Ophthalmol. 1987, 71, 130–144. [Google Scholar] [CrossRef]
- Davidson, A.E.; Liskova, P.; Evans, C.J.; Dudakova, L.; Nosková, L.; Pontikos, N.; Hartmannová, H.; Hodaňová, K.; Stránecký, V.; Kozmík, Z.; et al. Autosomal-Dominant Corneal Endothelial Dystrophies CHED1 and PPCD1 Are Allelic Disorders Caused by Non-coding Mutations in the Promoter of OVOL2. Am. J. Hum. Genet. 2016, 98, 75–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vithana, E.N.; Morgan, P.; Sundaresan, P.; Ebenezer, N.D.; Tan, D.T.; Mohamed, M.D.; Anand, S.; Khine, K.O.; Venkataraman, D.; Yong, V.H.; et al. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2). Nat. Genet. 2006, 38, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Bhattacharjee, S.; Prakash, D.R.; Sadanand, C.S. Genetic analysis of two Indian families affected with congenital hereditary endothelial dystrophy: Two novel mutations in SLC4A11. Mol. Vis. 2007, 13, 39–46. [Google Scholar] [PubMed]
- Heon, E.; Mathers, W.D.; Alward, W.L.; Weisenthal, R.W.; Sunden, S.L.; Fishbaugh, J.A.; Taylor, C.M.; Krachmer, J.H.; Sheffield, V.C.; Stone, E.M. Linkage of posterior polymorphous corneal dystrophy to 20q11. Hum. Mol. Genet. 1995, 4, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Aldave, A.J.; Yellore, V.S.; Vo, R.C.; Kamal, K.M.; Rayner, S.A.; Plaisier, C.L.; Chen, M.C.; Damani, M.R.; Pham, M.N.; Gorin, M.B.; et al. Exclusion of positional candidate gene coding region mutations in the common posterior polymorphous corneal dystrophy 1 candidate gene interval. Cornea 2009, 28, 801–807. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Joo, K.; Joung, I.; Lee, S.Y.; Kim, J.Y.; Cheng, Q.; Manavalan, B.; Joung, J.Y.; Heo, S.; Lee, J.; Nam, M.; et al. Template based protein structure modeling by global optimization in CASP11. Proteins 2016, 84 (Suppl. 1), 221–232. [Google Scholar] [CrossRef]
- Modi, V.; Xu, Q.; Adhikari, S.; Dunbrack, R.L., Jr. Assessment of template-based modeling of protein structure in CASP11. Proteins 2016, 84 (Suppl. 1), 200–220. [Google Scholar] [CrossRef] [Green Version]
- Joo, K.; Lee, J.; Kim, I.; Lee, S.J.; Lee, J. Multiple sequence alignment by conformational space annealing. Biophys. J. 2008, 95, 4813–4819. [Google Scholar] [CrossRef]
- Joo, K.; Lee, J.; Seo, J.H.; Lee, K.; Kim, B.G.; Lee, J. All-atom chain-building by optimizing MODELLER energy function using conformational space annealing. Proteins 2009, 75, 1010–1023. [Google Scholar] [CrossRef]
- Lee, J.; Scheraga, H.A.; Rackovsky, S. New optimization method for conformational energy calculations on polypeptides: Conformational space annealing. J. Comput. Chem. 1997, 18, 1222–1232. [Google Scholar] [CrossRef]
- Weiner, P.K.; Kollman, P.A. AMBER: Assisted model building with energy refinement. A general program for modeling molecules and their interactions. J. Comput. Chem. 1981, 2, 287–303. [Google Scholar] [CrossRef]
- Peters, M.B.; Yang, Y.; Wang, B.; Füsti-Molnár, L.; Weaver, M.N.; Merz, K.M., Jr. Structural Survey of Zinc Containing Proteins and the Development of the Zinc AMBER Force Field (ZAFF). J. Chem. Theory Comput. 2010, 6, 2935–2947. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Paek, A.R.; Lee, C.H.; You, H.J. A role of zinc-finger protein 143 for cancer cell migration and invasion through ZEB1 and E-cadherin in colon cancer cells. Mol. Carcinog. 2014, 53 (Suppl. 1), E161–E168. [Google Scholar] [CrossRef]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goh, G.; Choi, M. Application of whole exome sequencing to identify disease-causing variants in inherited human diseases. Genom. Inform. 2012, 10, 214–219. [Google Scholar] [CrossRef]
- De Ruiter, A.; Zagrovic, B. Absolute binding-free energies between standard RNA/DNA nucleobases and amino-acid sidechain analogs in different environments. Nucleic Acids Res. 2015, 43, 708–718. [Google Scholar] [CrossRef]
- Myslinski, E.; Krol, A.; Carbon, P. ZNF76 and ZNF143 are two human homologs of the transcriptional activator Staf. J. Biol. Chem. 1998, 273, 21998–22006. [Google Scholar] [CrossRef]
- Bailey, S.D.; Zhang, X.; Desai, K.; Aid, M.; Corradin, O.; Cowper-Sal Lari, R.; Akhtar-Zaidi, B.; Scacheri, P.C.; Haibe-Kains, B.; Lupien, M. ZNF143 provides sequence specificity to secure chromatin interactions at gene promoters. Nat. Commun. 2015, 2, 6186. [Google Scholar] [CrossRef] [PubMed]
- Bitner-Glindzicz, M.; Lindley, K.J.; Rutland, P.; Blaydon, D.; Smith, V.V.; Milla, P.J.; Hussain, K.; Furth-Lavi, J.; Cosgrove, K.E.; Shepherd, R.M.; et al. A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene. Nat. Genet. 2000, 26, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Pupavac, M.; Watkins, D.; Petrella, F.; Fahiminiya, S.; Janer, A.; Cheung, W.; Gingras, A.C.; Pastinen, T.; Muenzer, J.; Majewski, J.; et al. Inborn Error of Cobalamin Metabolism Associated with the Intracellular Accumulation of Transcobalamin-Bound Cobalamin and Mutations in ZNF143, Which Codes for a Transcriptional Activator. Hum. Mutat. 2016, 37, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Halbig, K.M.; Lekven, A.C.; Kunkel, G.R. The transcriptional activator ZNF143 is essential for normal development in zebrafish. BMC Mol. Biol. 2012, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Kawatsu, Y.; Kitada, S.; Uramoto, H.; Zhi, L.; Takeda, T.; Kimura, T.; Horie, S.; Tanaka, F.; Sasaguri, Y.; Izumi, H.; et al. The combination of strong expression of ZNF143 and high MIB-1 labelling index independently predicts shorter disease-specific survival in lung adenocarcinoma. Br. J. Cancer 2014, 110, 2583–2592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.K.; Nagpal, V.; Covington, J.W.; Michaels, M.A.; Vaughan, D.E. Molecular basis of cardiac endothelial-to-mesenchymal transition (EndMT): Differential expression of microRNAs during EndMT. Cell. Signal. 2012, 24, 1031–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr | Gene | Annotation | SIFT (Score) | PolyPhen (Score) | Mutation Taster | Evola | dbSNP | GnomAD Exomes | KRG DB | ACMG Criteria | ACMG Classification |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | TXNIP | NM_006472:exon5: c.A805G:p.I269V | deleterious 0.04 | probably damaging 0.996 | disease causing | conserved (12/12) | - | 0.000003977 | - | BS2, PP3, PP4 | VUS |
| 2 | EPC2 | NM_015630:exon8: c.G1208A:p.R403Q | deleterious 0.01 | benign 0.383 | disease causing | conserved (11/15) | rs761809638 | 0.00008181 | - | BS2, PP4 | VUS |
| 9 | KCNV2 | NM_133497:exon1: c.G217C:p.E73Q | tolerated 0.08 | benign 0.005 | polymorphism | conserved (12/13) | rs752013234 | 0.00001593 | - | BS2, PP4 | VUS |
| 9 | NUTM2F | NM_017561:exon7: c.C1766G:p.A589G | deleterious 0.05 | probably damaging 0.985 | polymorphism | no data | rs201719890 | 0.04351 | 0.2363 | BA1, PP4 | Benign (I) |
| 9 | AOPEP | NM_001193329: exon10:c.A1960G:p.S654G | benign 0.018 | polymorphism | conserved (7/15) | - | - | - | PM2, PP4 | VUS | |
| 9 | TNC | NM_002160:exon3: c.C1063T:p.R355W | deleterious 0 | possibly damaging 0.677 | disease causing | conserved (8/15) | rs779621288 | 0.00001195 | - | BS2, PP3, PP4 | VUS |
| 9 | RABGAP1 | NM_012197:exon10: c.G1221A:p.M407I | tolerated 0.07 | benign 0.015 | disease causing | N/A | rs769879519 | 0.000008117 | - | BS2, PP4 | VUS |
| 11 | ZNF143 | NM_003442:exon10: c.G937C:p.D313H | deleterious 0.02 | probably damaging 0.949 | disease causing | conserved (15/15) | - | - | - | PM2, PP3, PP4 | Likely pathogenic (II) |
| 13 | ERICH6B | NM_182542:exon3: c.C302A:p.A101E | Tolerated 1 | unknown | polymorphism | no data | - | - | - | PM2, PP4 | VUS |
| 19 | MZF1 | NM_003422:exon6: c.G1952A:p.R651Q | deleterious 0.04 | probably damaging 0.994 | polymorphism | conserved (9/9) | rs201221836 | 0.0002058 | 0.0105 | BS2, PP4 | VUS |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; You, H.J.; Park, S.H.; Kim, M.S.; Chae, H.; Park, J.; Jekarl, D.W.; Kim, J.; Kwon, A.; Choi, H.; et al. A Mutation in ZNF143 as a Novel Candidate Gene for Endothelial Corneal Dystrophy. J. Clin. Med. 2019, 8, 1174. https://doi.org/10.3390/jcm8081174
Kim Y, You HJ, Park SH, Kim MS, Chae H, Park J, Jekarl DW, Kim J, Kwon A, Choi H, et al. A Mutation in ZNF143 as a Novel Candidate Gene for Endothelial Corneal Dystrophy. Journal of Clinical Medicine. 2019; 8(8):1174. https://doi.org/10.3390/jcm8081174
Chicago/Turabian StyleKim, Yonggoo, Hye Jin You, Shin Hae Park, Man Soo Kim, Hyojin Chae, Joonhong Park, Dong Wook Jekarl, Jiyeon Kim, Ahlm Kwon, Hayoung Choi, and et al. 2019. "A Mutation in ZNF143 as a Novel Candidate Gene for Endothelial Corneal Dystrophy" Journal of Clinical Medicine 8, no. 8: 1174. https://doi.org/10.3390/jcm8081174