The Regulatory Role of Rac1, a Small Molecular Weight GTPase, in the Development of Diabetic Retinopathy


1
Department of Ophthalmology, Visual and Anatomical Sciences, Detroit, MI, 48201, USA
2
Pharmaceutical Sciences, Wayne State University, Detroit, MI, 48201, USA
3
Wayne State University, and John D Dingell VA Medical Center, Detroit, MI, 48201, USA
*
Author to whom correspondence should be addressed.
J. Clin. Med. 2019, 8(7), 965; https://doi.org/10.3390/jcm8070965
Submission received: 22 May 2019
/
Revised: 24 June 2019
/
Accepted: 27 June 2019
/
Published: 3 July 2019
(This article belongs to the Special Issue Diabetic Retinopathy: Biomolecules and Pathophysiology)
{kind=link}
{kind=link}
Abstract
:Diabetic retinopathy, a microvascular complication of diabetes, remains the leading cause of vision loss in working age adults. Hyperglycemia is considered as the main instigator for its development, around which other molecular pathways orchestrate. Of these multiple pathways, oxidative stress induces many metabolic, functional and structural changes in the retinal cells, leading to the development of pathological features characteristic of this blinding disease. An increase in cytosolic reactive oxygen species (ROS), produced by cytosolic NADPH oxidase 2 (Nox2), is an early event in the pathogenesis of diabetic retinopathy, which leads to mitochondrial damage and retinal capillary cell apoptosis. Activation of Nox2 is mediated through an obligatory small molecular weight GTPase, Ras-related C3 botulinum toxin substrate 1 (Rac1), and subcellular localization of Rac1 and its activation are regulated by several regulators, rendering it a complex biological process. In diabetes, Rac1 is functionally activated in the retina and its vasculature, and, via Nox2-ROS, contributes to mitochondrial damage and the development of retinopathy. In addition, Rac1 is also transcriptionally activated, and epigenetic modifications play a major role in this transcriptional activation. This review focusses on the role of Rac1 and its regulation in the development and progression of diabetic retinopathy, and discusses some possible avenues for therapeutic interventions.
1. Introduction
Diabetes has become a major public health burden, and is now considered to be an epidemic in the 21st century. In 2015, diabetes affected 415 million people globally, and these numbers are expected to reach 642 million by 2040 (International Diabetes Federation. IDF Diabetes Atlas, 7th Edition). Diabetes leads to structural and functional changes in both the macro- and micro-vasculature of tissue; the macrovascular alterations are observed mainly in cardiovascular, cerebrovascular and peripheral artery diseases, and the microvascular alterations result in retinopathy, nephropathy and neuropathy [1]. Though hyperglycemia has been implicated as a causal link between diabetes and its complications [2,3,4], a clear understanding of the mechanism(s) of the development of these complications remain complex as the genetic and environmental factors also orchestrate these mechanism(s) to varied degrees.
Chronic hyperglycemia damages the retina, leading to progressive vision loss and ultimately blindness. It is the leading cause of blindness in working age adults around the world, and its incidence is expected to increase owing to the increasing global diabetes burden each year [5]. The global prevalence of diabetic retinopathy was about 126.6 million in 2010, and this number is expected to escalate to 191 million by 2030 [6]. In about one third of diabetic patients, retinopathy progresses to vision threatening stage, and 2% of the patients become blind. The etiology of diabetic retinopathy follows a distinctive trend, where one feature succumbs to another as the disease progresses; while the early clinical signs are microaneurysms, small hemorrhages and leakage of lipids into the retina, but with time this progresses to capillary closure and retinal hypoxia [1,7]. Inevitably, excessive capillary closure leads to abnormal proliferation of retinal vessels, accompanied by vitreous hemorrhage and fibrosis resulting in proliferative diabetic retinopathy [8].
Several modifiable risk factors such as hyperglycemia, hypertension, dyslipidemia and obesity, have been associated with diabetic retinopathy, but the significance of hyperglycemia transcends any other factor [9]. The Diabetes Control and Complications Trial (DCCT) has clearly documented a close association between tight glycemic control (HbA1c < 7%) and reduced risk of the development/progression of diabetic retinopathy in type 1 diabetic patients. The incidence of retinopathy is reduced by 76%, and that of progression to advanced retinopathy, by 54%, if the intensive glycemic control is maintained [2].The United Kingdom Prospective Diabetes Study also documented similar association between the severity of hyperglycemia and the incidence of diabetic retinopathy in type 2 diabetic patients [4]. However, maintenance of intensive control is difficult for this life-long disease, which requires modification of life style, dedication by the patient and the loved ones, and carries an increased risk of hypoglycemic seizure and possible weight gain [10]. Thus, understanding the molecular mechanism of its development is essential for identifying potential future therapies for retinopathy in diabetes.
2. Molecular Mechanisms of Diabetic Retinopathy
The extensive research conducted over the years has shed enormous light on the various molecular players and pathways that orchestrate the development and progression of diabetic retinopathy. The persistent hyperglycemia is considered as the primary causative factor that leads to its development. Activation of polyol pathway, protein kinase C, increased oxidative stress and advanced glycation product formation are some of the pathways that are considered to play a role in the development diabetic retinopathy. Of these multiple pathways, oxidative stress is considered to be one of the major pathways in the complex pathophysiological of diabetic retinopathy [11,12,13].
Hyperglycemia can generate reactive oxygen species (ROS) via autoxidizing glucose or increasing cytosolic NADPH oxidase (Nox) activity, or by affecting mitochondrial respiratory chain, and ROS damage biomolecules including DNA, lipids, proteins and carbohydrates [11,14,15]. Experimental models have shown that an increase in cytosolic ROS, produced by Nox2, is an early event in the pathogenesis of diabetic retinopathy, which precedes mitochondrial damage, further increasing free radicals [15]. Continuous ROS production damages mitochondrial DNA (mtDNA), damaged mtDNA further compromises the electron transport chain by reducing the transcription of mtDNA-encoded genes critical for its functioning, and the vicious cycle of free radicals continues to self-propagate. Damage to mitochondrial membranes by ROS renders them permeable, releasing cytochrome C into the cytoplasm. This serves as a signal of cell apoptosis, and capillary cells begin to undergo accelerated apoptosis [16,17]. Persistent hyperglycemia, excessive free radicals and dysfunctional mitochondria form a conundrum that further propagates these events, leading to cell apoptosis and the development and progression of diabetic retinopathy.
3. NADPH Oxidases
The cytosolic Nox is the primary enzyme responsible for the generation of cellular ROS [18]. The Nox family comprises of seven different isoforms, each with multiple large transmembrane catalytic subunits, catalyzing the reduction of molecular oxygen to superoxide anion by oxidizing cytosolic NADPH to NADP. The seven Nox genes have been categorized as Nox1, Nox2, Nox3, Nox4, Nox5, Duox1 and Duox 2 [19]. Of these isoforms, Nox2 is present in numerous cell types, including retina. Though, initially regarded as a phagocytic enzyme, its altered activity in non-phagocytic cells has been associated with the development of several pathologies including beta cell loss [20]. In addition to Nox2, other members of the Nox family, specifically Nox1 and Nox4, are also involved in cellular dysfunction leading to pathological conditions, and altered activities of Nox1, Nox2 and Nox4 are seen in specific retinal cell types including endothelial cells and pericytes, in diabetes, implicating them in the development and progression of diabetic retinopathy [15,21,22]. Moreover, a recent genome wide association study has demonstrated the association of the Nox4 gene with severe diabetic retinopathy in type 2 diabetes patients [23]. The present review focuses on Rac1-Nox2, and the role of other isoforms of Nox in diabetic retinopathy is beyond the scope of this review.
Nox2 is constitutively associated with p22phox on the membrane but requires a complex process of protein-protein interactions for its activation. The p47phos subunit localizes to the membrane, recruiting the p67phox (activator subunit) and p40phox to the complex. Further, the activation of Nox2 is mediated through an obligatory small molecular weight GTPase, Ras-related C3 botulinum toxin substrate 1 (Rac1), which interacts initially with Nox2 and subsequently with p67phox [20,24]. Rac1 belongs to the Ras superfamily of GTP binding proteins, and its activity is manipulated at various levels due to the inherent and external factors orchestrating its regulation [18,25]. The inherent stipulations are that these are small molecular switches alternating between active and inactive states.
4. Rac1
Rac1 promotes ROS generation that participates in the regulation of complex cell biological processes such as transcription factor activation, proliferation, transformation, immune response and apoptosis [26,27,28]. The small GTPases are molecular switches regulating almost every facet of cell biology, and one of the mechanisms involves modulating the gene expression of several transcription factors [29]. Rac1 also activates transcription factors, including nuclear transcription factor B (NF-kB) and activator protein 1 (Ap1) [28,30]. Typically, Rac1 is associated with the modulation of actin cytoskeleton, inducing actin polymerization at the membrane to internalize the microorganism and elicit phagocytosis [31,32,33]. However, it is also an integral part of the Nox2 holoenzyme, and thus is also important in regulating ROS levels, and the associated intracellular oxidative stress.
The regulation and action of Rac1 are not limited to its active and inactive state, but rather extends to its distribution at various sub-cellular spaces where it interacts with different effectors eliciting diverse biological responses. Rac1 cycles between the cytoplasm and the plasma membrane to regulate cellular processes. The cytoplasmic Rac1 is hydrophilic, lacking a transmembrane domain and is rendered hydrophobic by its post-translational modification (e.g., lipidation, prenylation) that drives it to the plasma membrane [18,31].
The mitochondrial localization of Rac1 is one of the most recent and novel finding that is gaining scientific interest; Rac1 in the mitochondria elicits varied response in different cell types; the same process could be cytoprotective in one cell type, and toxic in the others. Rac1 interacts with anti-apoptotic mitochondrial outer membrane protein Bcl2, and these interactions play a significant role in determining the fate of a cell [34]. In lymphoma cells, overexpression of Bcl2 inhibits Rac1-mediated apoptosis [34], but in pheochromocytoma-12 cells, these interactions are considered to be protective [35]. A critical regulatory role for Rac1 in the onset of pulmonary fibrosis is considered due to increased H2O2 production in alveolar macrophages, and Cys-189 of Rac1 is necessary for its mitochondrial import [36]. In diabetic retinopathy, inhibition of Rac1 activity protects retinal endothelial cells from hyperglycemia-induced mitochondrial damage and accelerated apoptosis [15].
Nuclear localization of Rac1 is considered to be cell cycle-dependent; it regulates the centrosome separation and mitotic entry [37]. Nuclear Rac1 also interacts with transcription factors such as STAT3 and NF-kB [30]. Several mechanisms have been suggested for nuclear translocation (or import) of Rac1, including post-translational modifications; for example phosphorylation of Rac1 at Thr-108 by extracellular-regulated kinase is considered to be necessary for Rac1 nuclear translocation and accumulation [38].
5. The Role of Rac1 in Diabetic Retinopathy
Rac1 plays a critical role in the pathophysiology of diabetic retinopathy by activating Nox2, which leads to excessive free radical generation. As mentioned above, in the pathogenesis of diabetic retinopathy, increased cytosolic ROS produced by Nox2 is an early event, which initiates a cascade of events that elicit mitochondrial damage and capillary cell apoptosis [15,21,39,40]. Activation of Rac1-Nox2-ROS also results in multiple downstream effects, and ROS-mediated alterations of many cellular processes lead to the pathological development and disease [15,41,42]. Experimental models have shown that ROS generated via Nox2 activation precedes mitochondrial damage. Once the mitochondria are damaged, the caspase signaling pathway is activated, resulting in cell apoptosis and the diabetic retinopathy pathology [15,43].
Rac1 also activates stress kinases such as p38MAPK, and in addition to their role in mitochondrial damage, stress kinases alter several facets of the retinal physiology that contribute to the pathological development of diabetic retinopathy including alteration in the tight junctions, breakdown of blood retinal barrier and activation of matrix metalloproteinases (MMPs) [44,45,46,47].
As detailed above, Rac1 is pro-apoptotic in the initial stages of diabetic retinopathy, however, Rac1 activation is also associated with aberrant retinal neovascularization; animals models of retinal vein occlusion have demonstrated beneficial effects of silencing Rac1 on retinal neovascularization including choroidal neovascularization [48,49], suggesting it also has a role in the advanced stages of diabetic retinopathy.
6. Regulation of Rac1 in Diabetic Retinopathy
The gene expression and activity of a protein is a remarkably complex and regulated process. The complexity lies in the organization of the genetic material and the diversity of players/processes that regulate it. Primarily, the protein levels and its activity are regulated at transcriptional and post-translational level [50,51]. However, in addition, small GTPases exist in alternating GTP-bound active and GDP-bound inactive states, and GTP- and GDP- bound states are regulated by two distinct classes of proteins. While guanine nucleotide exchange factors (GEFs) facilitate GTP binding by releasing the bound GDP, GTPase activating proteins (GAPs) inactivate Rac1 by hydrolyzing the bound GTP. In addition, guanine nucleotide dissociation inhibitors (GDIs) help keep Rac1 in an inactive state by sequestering it away from GEFs [29,39,52]. In mammals, around 70 GAPs and 80 GEFs have been known to regulate the small GTPases, of which over 30 GEFs and several GAPs have been known to regulate Rac1 alone [29].
The following sections discuss how Rac1 regulation is altered in diabetes, and its role in the development of diabetic retinopathy.
6.1. Functional Regulation of Rac1
6.1.1. Guanine Nucleotide Exchange Factors (GEFs)
Rac1, like most other small G-proteins, is activated by GEFs that facilitate GTP binding by releasing bound GDP [52]. In diabetes, GEF, T cell lymphoma invasion and metastasis (Tiam1), is shown to be critical in orchestrating Rac1 activation; activation of Tiam1-Rac1-Nox2 signaling axis, an early event in the pathogenesis of diabetes, leads to mitochondrial damage and accelerated capillary cell apoptosis. Inhibition of Tiam1 by its specific inhibitor NSC23766, markedly attenuates Rac1 activation, and protects mitochondrial damage and acceleration of capillary cell apoptosis [15].
The same G-protein can also be modulated by multiple GEFs, and Vav2 is considered as another important GEF for Rac1 activation [53,54]; our recent work has implicated Vav2 in the activation of Rac1-Nox2 signaling in diabetic retinopathy. We have shown that the inhibition of Vav2 by its pharmacological inhibitor EHop, in addition to regulating diabetes-induced activation of Rac1-Nox2, also prevents mitochondrial damage, vascular leakage and capillary cell apoptosis. Furthermore, administration of this inhibitor, soon after establishment of diabetes in mice, also inhibits the development of retinopathy [55].
Another GEF, Son of sevenless homolog 1 (Sos1), is also implicated in the regulation of Rac1 activation in diabetic milieu. The activity of Sos1 is regulated by 66kDa proto-oncogene Src homologous-collagen homologue (p66Shc) adaptor protein [26,56]. The expression of p66Shc in the human retinal endothelial cells is upregulated in hyperglycemic milieu, and its overexpression displaces Sos1 protein from Grb2, leading to Rac1 activation. P66Shc-mediated activation of Rac1 is facilitated by decreased binding of Sos1 with the growth factor receptor-bound protein 2 (Grb2) [57]. Thus, these pathways seem to be critical in regulating significant downstream signals implicated in the development of diabetic retinopathy.
6.1.2. Guanine Nucleotide Dissociation Inhibitors
Though, GEFs are important regulators of Rac1, additional regulatory control is also rendered by their association with GDIs. In addition to keeping Rac1 away from GEFs, GDIs inhibit their dissociation from the bound GDP, and maintain the small GTPases in the cytosolic compartment [52]. The dissociation of the small GTPases from the GDIs, followed by their post-translational modification, are essential for these GTPases to target the plasma membrane [58]. Experimental models of diabetic retinopathy have demonstrated that in hyperglycemic milieu, while the levels of the prenylating enzyme, farnesyltransferase (FNTA) and GEF Vav2 are increased in the retinal vasculature, that of GDI are decreased, suggesting the importance of GDIs in diabetic retinopathy (Figure 1) [55].
6.1.3. Post-Translational Modifications
As detailed above, Rac1 shuttles between the cytoplasm and plasma membrane, and a critical balance is maintained under normal physiological condition. Posttranslational modification of Rac1 is important to drive Rac1 to the plasma membrane; geranylgeranylation and palmitoylation are some of the major post-translational modifications [31,59]. The lipid raft localization of the Rac1 is its major signaling site, and palmitoylation of Rac1 drives it to the lipid raft region of the plasma membrane. However, Rac1 palmitoylation can only be accomplished if it is prenylated and has an intact PBR (C-terminal polybasic region) region [31]. Further, the PBR region helps clustering of the Rac1 at the plasma membrane, increasing protein-protein interaction [60,61]. In the pathogenesis of diabetic retinopathy, the Rac1-Nox2 signaling pathway is activated in the retina leading to oxidative stress, suggesting that Rac1 must be post-translationally modified to be more hydrophobic. In this context, palmitoylation of Rac1 is observed as a prerequisite for its plasma membrane targeting in high glucose condition, and 2-bromopalmitate (2-BP), and inhibitor of palmitoylaton, attenuates the Rac1-Nox2 signaling pathway in retinal endothelial cells [15,59,62]. Furthermore, the prenylation enzyme is increased in hyperglycemic condition, and inhibition of FNTA prevents the activation of Rac1-Nox2-ROS signaling, suggesting prenylation is also an important posttranslational modification for Rac1 activation and the development and progression of diabetic retinopathy [15,63]. Although not examined in the context of the onset of diabetic retinopathy, Rac1 functions have also been shown to be regulated by RNA splicing and other post-translational modifications such as ubiquitination, adenylation, phosphorylation and SUMOylation [64,65], and mutations of Rho GTPases (e.g., Cdc42, Rho and Rac1) have also been reported in pathological states, including immonodeficiency and cancer [64].
6.2. Transcriptional Regulation and Epigenetic Modifications
Diabetic environment alters the expression of several genes associated with the development and progression of retinopathy. Gene expression can also be influenced by external factors and disease state, without altering the DNA sequence, and these epigenetics changes define the dynamic and intricate regulatory and functional interactions between DNA, RNA, and protein, ultimately resulting in phenotypes [66,67,68,69]. These modifications can be transmitted to the daughter cells- thus epigenetic modifications can be considered as ‘inheritance, which is not mediated via DNA sequence of genes [70]. However, depending on the regulation of external factors and life style, these epigenetic changes can also be erased/reversed, which make them good therapeutic targets for chronic diseases [71]. Modification of histones, e.g., lysine or arginine residues, methylation of cytosine and noncoding RNAs are some of the major epigenetic modifications. The type of histone modification, e.g., methylation and acetylation, and also the site of methylation, determine whether histone modifications close or open the chromatin structure to regulate transcription factor binding [72]. Interestingly, DNA methylation and histone modifications can also influence each other; histone methylation can help to direct DNA methylation patterns, and DNA methylation may serve as a template for rebuilding histone modification patterns [73].
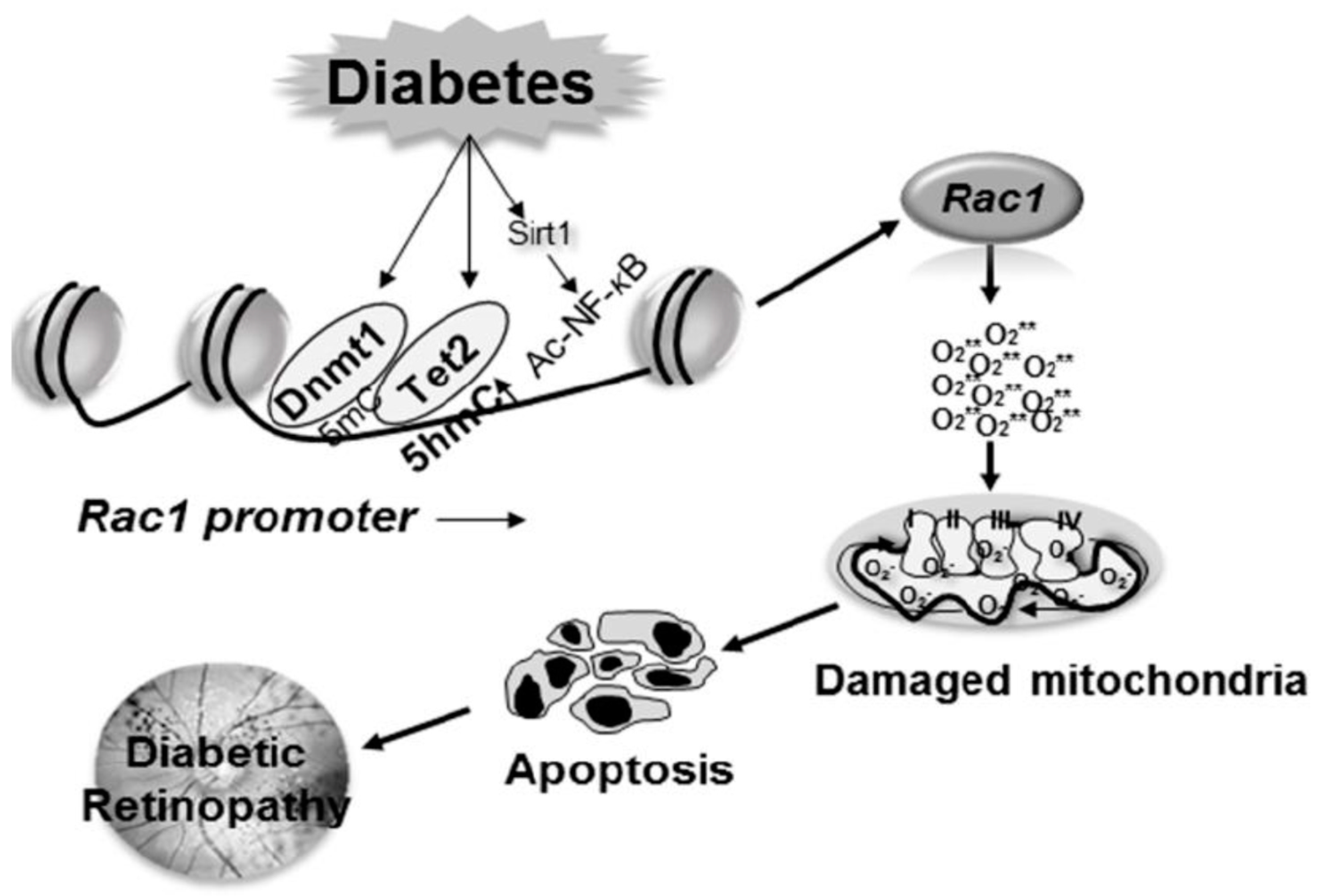
One of the most widely studied epigenetic modifications is the methylation of cytosine bases. In mammals, methylation is restricted to cytosine residues and mainly encountered in cytosine-guanine dinucleotides (CpG) [74]. Although CpG is usually underrepresented throughout the genome, stretches of 0.3–3 kb (CpG islands) are generally present in the promoter region of a gene [75,76], and their methylation is associated with gene silencing. The addition of methyl group to cytosine is mediated by a family of enzymes, the DNA methyltransferases (Dnmts). Among this family, while Dnmt3a and Dnmt3b are associated with the establishment of de novo DNA methylation patterns, Dnmt1 is responsible for the maintenance of established DNA methylation patterns [77]. DNA methylation is a dynamic process, and to maintain proper DNA methylation status, DNA demethylation can be reversed by the conversion of methylcytosine to hydroxymethylcytosine (5hmC) [78]. The demethylation process can either be passive or active, or a combination of both. Passive DNA demethylation is usually on newly synthesized DNA strands, and occurs during replication rounds, and active DNA demethylation is mediated via the sequential modification by the ten-eleven translocation (TET) family of enzymes; 5mC can be converted to 5hmC, 5hmC to 5-formylcytosine (5fC), and 5fC to 5-carboxylcytosine [79]. Among these, the conversion of 5mC to 5hmC is the most common hydroxymethylation. In diabetes, enzyme activities of both Dnmts and Tets are increased in the retina and its vasculature, and among these two families of enzymes, Dnmt1 and Tet2 are the only respective isoforms that are upregulated [80,81,82].
Rac1 transcription is mediated by many transcriptional factors including NF-kB and Sp1 [43]. In the pathogenesis of diabetic retinopathy, gene transcripts of Rac1 are increased in the retina and its vasculature. In diabetes, the binding of both Dnmt1 and Tet2 is increased at its promoter, suggesting an active methylation-hydroxymethylation process of the Rac1 promoter. 5mC formed by increased Dnmt1 binding is immediately hydroxymethylated by Tet2, and this interplay between two opposing enzymes leaves the promoter hypomethylated. The hypomethylated promoter region allows the transcription factor to bind, ultimately resulting in increased Rac1 transcription [80]. In addition to regulating the DNA methylation status of Rac1 promoter, inhibition of Dnmts/Tets prevents NF-kB binding. NF-kB is also activated in the retina in diabetes, and acetylation of its p65 subunit plays an important role in regulating NF-kB-dependent transcription [83,84]. In diabetes, due to hypermethylation of promoter DNA, Sirtuin 1 (Sirt1), a NAD-dependent deacetylase, is also inhibited [43,85]. Inhibition of Sirt1 results in impaired deacetylation mechanism, and increases the binding of acetylated NF-kB at the promoter, further facilitating Rac1 transcription (Figure 2).
As mentioned above, many epigenetic modifications are interrelated; DNA methylation can alter H3K9 methylation, and histone modifications can regulate Dnmt1 recruitment to promoter CpG sites [86,87]. Recent studies from our laboratory have shown that due to increased binding of methyltransferase Suv39H1 at the Rac1 promoter in diabetes, H3K9me3 levels are significantly elevated, and this assists in the recruitment of the DNA methylation machinery, altering the DNA methylation status of Rac1 and increasing its expression [88].
7. Therapeutic Targets
The above discussion clearly shows Rac1 as an important participant in the development of diabetic retinopathy; the diabetic environment functionally activates Rac1 in the retina and its vasculature, and alterations in the epigenetic machinery influence its transcriptional activation, making Rac1 as an excellent target for therapeutical intervention. As Rac1 is regulated via several processes, it provides an opportunity to identify the critical processes and mediators essential for its activation and, thus, their regulation to inhibit Rac1 activation. Several pharmacological approaches have been used to target these and inhibit Rac1 activation, and one of the most obvious among these is targeting the GEFs. Administration of inhibitors of Tiam1 and Vav2, inhibit Rac1-Nox2 signaling in diabetic animal models, and prevent the development of diabetic retinopathy [15,55]. Pharmacological and molecular inhibitors of posttranslational modifications, and the enzymes associated with such modification provide another opportunity to regulate Rac1 activation [42,89].
Rac1-Nox2 is also regulated by the inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA); simvastatin has been shown to decrease Rac1 activation and increase in nitric oxide in spontaneously hypertensive rats [90]. Although human trials investigating the effect of statins on Rac1 are very limited, they have shown positive effects on blood vessels [91,92,93]. A recent population-based cohort study conducted in Taiwan has shown a decreased risk of diabetic retinopathy, and its progression to vision-threatening diabetic retinopathy in Taiwanese patients receiving statin therapy [94]; additional studies are needed to ascertain the link between statins and diabetic retinopathy. Furthermore, since nitric oxide can also activate Rac1 [95], several nitric oxide releasing molecules have been analyzed for their protective effect against oxidative stress, and VP10/39 (caffeic acid phenethyl ester derivative) has been shown to protect retinal pigment epithelial cells against oxidative stress [96]; however, the effect of such molecules on diabetic retinopathy remains to be investigated.
As stated above, pharmacological investigations involving Rac1 inhibitors have suggested Tiam1 (NSC23766), Vav2 (Ehop-016) and SoS1 as GEFs involved in sustained activation of Rac1 in the retina in diabetes (Figure 1). It is important to note that other pathological conditions including lipotoxicity (ceramide-mediated) or chronic inflammation (proinflammatory cytokines, such as IL-1β) could also mediate their cytotoxic effects via activation of Tiam1/Vav2-Rac1-Nox2 pathway, thus suggesting that this signaling axis might be “druggable” to prevent/halt metabolic defects. However, some aspects remain speculative and needs to be confirmed experimentally including potential cross-talk and/or inter-dependence, if any, between Tiam1, Vav2 and SoS1 in mediating their effects on Rac1 activation, and potential regulatory mechanisms upstream to the activation of these GEFs. Furthermore, it is critical to understand the subcellular compartmentalization of these GEFs and Rac1 in diabetes and/or glucolipotoxic conditions that could potentially lead to mistargeting/mislocalization of Rac1 (e.g., translocation to nuclear compartment), culminating in cellular dysfunction and loss. We recognize that Rap1, which represents one of the components of the membranous core of Nox2, could also contribute significantly in the subcellular-localization (e.g., plasma membrane) of GEFs in the steps leading to Rac1 activation and optimal assembly of the Nox2 complex, possibly providing additional therapeutic targets.
Since Rac1 is also transcriptionally activated in diabetes, and epigenetic modifications modulate its transcriptional activation, targeting the enzymes responsible for such modifications provides another opportunity to regulate Rac1 activation. As mentioned above, increased Dnmt binding, through forms more 5mC, but simultaneous activation of Tet2, hydroxymethylates Rac1 promoter. This makes Dnmts as a good target to inhibit Dnmt binding- hydroxymethylation. Over the past decade, many epigenetic-modifying agents have been developed, and are being employed in the clinical management of patients with malignancies [97,98]. Demethylating agents, azacytidine and decitabine, are being used for the treatment of myelodysplastic syndromes, but these agents carry a burden of high toxicity [99,100]. Some of the synthetic DNA methylation inhibitors such as hydralazine and procainamide are now being used in clinical trials for tumors, and these inhibitors have less adverse side effects than azacytidine and decitabine [101]. In addition, since histone methylation also plays an important role, inhibitors of methyl transferases also carry a therapeutic advantage [102,103]. Downregulation of deacetylase Sirt1 allows NF-kB to remain acetylated, suggesting the possibility of increasing Sirt1 activity could refrain NF-kB for being in acetylated state to bind at the promoter. This raises the possibility of use of dietary flavonoids and polyphenols such as resveratrol and catechins, which may upregulate Sirt1, and also directly inhibit Dnmts [104,105]. Although dietary flavonoids and polyphenols seem to be attractive therapeutic models, due to their varying pharmacokinetic profiles, their physiological relevance with supporting clinical findings needs to be investigated. Furthermore, modulation of epigenetic enzymes has many limitations including silencing/activation of other genes, and less than desirable drug specificity [106]. Thus, the selection and rational prioritization of epigenetic agents are important for identifying the most appropriate agents for patients in clinical practice. Further, the blood-retinal barrier poses another challenge for these drugs to reach to the retina, which should always be considered.
Computational systems biology is gaining the interest of the scientific community for identifying drug targets, and has potential considering the complex pathology of diabetic retinopathy. The analysis of transcriptomics data (retrieved from Gene Expression Omnibus Dataset repository datasets) has identified some genes and biological pathways related with inflammation, fibrosis and G protein-coupled receptors involved in the development of this blinding complication of diabetes [107]. However, additional analysis using different data sources are warranted to gain interesting insights/leads into the molecular pathways implicated in the pathogenesis of diabetic retinopathy.
Retina has an organized blood-retina barrier [108], which makes topical delivery of drugs to the posterior segment of the eye a challenging task [109]. However, the use of nanocarriers to formulate sustained delivery of the compounds [110], to prevent functional and/or transcriptional activation of Rac1 remains an attractive avenue.
In conclusion, as detailed above, in the pathogenesis of diabetic retinopathy once mtDNA is damaged, due to dysfunctional electron transport chain, the vicious cycle of ROS continues to self-propagate, but activation of Rac1-Nox2 precedes mitochondrial damage. Thus, inhibition of Rac1 activation, an early event in the pathogenesis of diabetic retinopathy, will halt the vicious cycle of ROS accumulation, and ameliorate further progression of the disease. Furthermore, Rac1 is also associated with aberrant retinal neovascularization, and its inhibition during the advanced stages of retinopathy would also slow down neovascularization, a hallmark of proliferative diabetic retinopathy [48]. Optimistically, efforts are being put into developing strategies to regulate Rac1 activation for the treatment of other chronic diseases, and Dnmt inhibitors are also being used in clinical trials for tumors. The delivery of these drugs, along with maintenance of sensible glycemic control, holds promise to retard the development/progression of retinopathy, and prevent vision loss in diabetic patients.
Author Contributions
N.S., A.K. and R.A.K. prepared and edited the review article, and N.S. also prepared the figures.
Funding
This work was supported in parts by grants from the National Institutes of Health (RAK: EY014370 and EY017313; RAK & AK: EY022230), Thomas Foundation (RAK), Department of Veterans Affairs (AK: 1BX000469), and an unrestricted grant to the Ophthalmology Department from Research to Prevent Blindness. AK is the recipient of a Senior Research Career Scientist Award from the Department of Veterans Affairs (13S-RCS-006).
Acknowledgments
R.A.K. and A.K. thank their laboratory associates (past and present) for their contributions to the work cited in this review article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Frank, R.N. Diabetic retinopathy. N. Engl. J. Med. 2004, 350, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Orasanu, G.; Plutzky, J. The pathologic continuum of diabetic vascular disease. J. Am. Coll. Cardiol. 2009, 53, S35–S42. [Google Scholar] [CrossRef] [PubMed]
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes. Lancet 1998, 352, 837–853. [Google Scholar] [CrossRef]
- Klein, B.E. Overview of epidemiologic studies of diabetic retinopathy. Ophthalmic Epidemiol. 2007, 14, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; He, M.; Congdon, N. The worldwide epidemic of diabetic retinopathy. Indian J. Ophthalmol. 2012, 60, 428–431. [Google Scholar] [PubMed]
- Cunha-Vaz, J.; Faria de Abreu, J.R.; Campos, A.J. Early breakdown of the blood-retinal barrier in diabetes. Br. J. Ophthalmol. 1975, 59, 649–656. [Google Scholar] [CrossRef] [PubMed]
- De La Cruz, J.P.; Gonzalez-Correa, J.A.; Guerrero, A.; De La Cuesta, F.S. Pharmacological approach to diabetic retinopathy. Diabetes Metab. Res. Rev. 2004, 20, 91–113. [Google Scholar] [CrossRef]
- Ting, D.S.; Cheung, G.C.; Wong, T.Y. Diabetic retinopathy: Global prevalence, major risk factors, screening practices and public health challenges: A review. Clin. Exp. Ophthalmol. 2016, 44, 260–277. [Google Scholar] [CrossRef]
- Kalra, S.; Mukherjee, J.J.; Venkataraman, S.; Bantwal, G.; Shaikh, S.; Saboo, B.; Das, A.K.; Ramachandran, A. Hypoglycemia: The neglected complication. Indian J. Endocrinol. Metab. 2013, 17, 819–834. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A. Diabetic retinopathy: Mitochondrial dysfunction and retinal capillary cell death. Antioxid. Redox Signal. 2005, 7, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.G. Oxidative stress profiling: Part I. Its potential importance in the optimization of human health. Ann. N. Y. Acad. Sci. 2005, 1055, 93–135. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Kowluru, A.; Veluthakal, R.; Mohammad, G.; Syed, I.; Santos, J.M.; Mishra, M. Tiam1-rac1 signalling axis-mediated activation of nadph oxidase-2 initiates mitochondrial damage in the development of diabetic retinopathy. Diabetologia 2014, 57, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Abbas, S.N. Diabetes-induced mitochondrial dysfunction in the retina. Invesig. Ophtahlmol. Vis. Sci. 2003, 44, 5327–5334. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Mishra, M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim. Biophys. Acta 2015, 1852, 2474–2483. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, A. Small g proteins in islet beta-cell function. Endocr. Rev. 2010, 31, 52–78. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The nox family of ros-generating nadph oxidases: Physiology and pathophysiology. Phys. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Kowluru, A. Friendly, and not so friendly, roles of rac1 in islet beta-cell function: Lessons learnt from pharmacological and molecular biological approaches. Biochem. Pharmacol. 2011, 81, 965–975. [Google Scholar] [CrossRef]
- Ibrahim, A.S.; Elshafey, S.; Sellak, H.; Hussein, K.A.; El-Sherbiny, M.; Abdelsaid, M.; Rizk, N.; Beasley, S.; Tawfik, A.M.; Smith, S.B.; et al. A lipidomic screen of hyperglycemia-treated hrecs links 12/15-lipoxygenase to microvascular dysfunction during diabetic retinopathy via nadph oxidase. J. Lipid Res. 2015, 56, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, K.M.; Kim, C.S.; Sohn, E.; Lee, Y.M.; Jo, K.; Kim, J.S. Puerarin inhibits the retinal pericyte apoptosis induced by advanced glycation end products in vitro and in vivo by inhibiting nadph oxidase-related oxidative stress. Free Radic. Biol. Med. 2012, 53, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Shah, K.P.; Pollack, S.; Toppila, I.; Hebert, H.L.; McCarthy, M.I.; Groop, L.; Ahlqvist, E.; Lyssenko, V.; Agardh, E.; et al. A genome-wide association study suggests new evidence for an association of the nadph oxidase 4 (nox4) gene with severe diabetic retinopathy in type 2 diabetes. Acta Ophthalmol. 2018, 96, e811–e819. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Banfi, B.; Jesaitis, A.J.; Dinauer, M.C.; Allen, L.A.; Nauseef, W.M. Critical roles for p22phox in the structural maturation and subcellular targeting of nox3. Biochem. J. 2007, 403, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Matos, P.; Skaug, J.; Marques, B.; Beck, S.; Verissimo, F.; Gespach, C.; Boavida, M.G.; Scherer, S.W.; Jordan, P. Small gtpase rac1: Structure, localization, and expression of the human gene. Biochem. Biophys. Res. Commun. 2000, 277, 741–751. [Google Scholar] [CrossRef]
- Bhat, H.F.; Baba, R.A.; Adams, M.E.; Khanday, F.A. Role of snta1 in rac1 activation, modulation of ros generation, and migratory potential of human breast cancer cells. Br. J. Cancer 2014, 110, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Biro, M.; Munoz, M.A.; Weninger, W. Targeting rho-gtpases in immune cell migration and inflammation. Br. J. Pharmacol. 2014, 171, 5491–5506. [Google Scholar] [CrossRef]
- Cuadrado, A.; Martin-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors nrf2 and nf-kappab are coordinated effectors of the rho family, gtp-binding protein rac1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef]
- Jaffe, A.B.; Hall, A. Rho gtpases: Biochemistry and biology. Ann. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef]
- Simon, A.R.; Vikis, H.G.; Stewart, S.; Fanburg, B.L.; Cochran, B.H.; Guan, K.L. Regulation of stat3 by direct binding to the rac1 gtpase. Science 2000, 290, 144–147. [Google Scholar] [CrossRef]
- Bustelo, X.R.; Ojeda, V.; Barreira, M.; Sauzeau, V.; Castro-Castro, A. Rac-ing to the plasma membrane: The long and complex work commute of rac1 during cell signaling. Small GTPases 2012, 3, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.; Chang, P.; Zhang, Q.; Reddy, P.G.; Bokoch, G.M.; Greenberg, S. Requirements for both rac1 and cdc42 in membrane ruffling and phagocytosis in leukocytes. J. Exp. Med. 1997, 186, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, T.; Bokoch, G.M.; Waterman-Storer, C.M. Regulation of leading edge microtubule and actin dynamics downstream of rac1. J. Cell Biol. 2003, 161, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Velaithan, R.; Kang, J.; Hirpara, J.L.; Loh, T.; Goh, B.C.; Le Bras, M.; Brenner, C.; Clement, M.V.; Pervaiz, S. The small gtpase rac1 is a novel binding partner of bcl-2 and stabilizes its antiapoptotic activity. Blood 2011, 117, 6214–6226. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, N.; Xia, P.; Wang, E.; Guo, Q.; Ye, Z. Inhibition of rac1 ameliorates neuronal oxidative stress damage via reducing bcl-2/rac1 complex formation in mitochondria through pi3k/akt/mtor pathway. Exp. Neurobiol. 2018, 300, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Chong, S.J.; Ooi, V.Z.; Vali, S.; Kumar, A.; Kapoor, S.; Abbasi, T.; Hirpara, J.L.; Loh, T.; Goh, B.C.; et al. Overexpression of bcl-2 induces stat-3 activation via an increase in mitochondrial superoxide. Oncotarget 2015, 6, 34191–34205. [Google Scholar] [CrossRef] [PubMed]
- May, M.; Schelle, I.; Brakebusch, C.; Rottner, K.; Genth, H. Rac1-dependent recruitment of pak2 to g2 phase centrosomes and their roles in the regulation of mitotic entry. Cell Cycle 2014, 13, 2211–2221. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Li, L.; Ballermann, B.; Wang, Z. Phosphorylation of rac1 t108 by extracellular signal-regulated kinase in response to epidermal growth factor: A novel mechanism to regulate rac1 function. Mol. Cell. Biol. 2013, 33, 4538–4551. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, A.; Kowluru, R.A. Phagocyte-like nadph oxidase [nox2] in cellular dysfunction in models of glucolipotoxicity and diabetes. Biochem. Pharmacol. 2014, 88, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.J.; Yu, Q.; Chen, K.; Mahadev, K.; Zhang, S.X. Inhibition of reactive oxygen species by lovastatin downregulates vascular endothelial growth factor expression and ameliorates blood-retinal barrier breakdown in db/db mice: Role of nadph oxidase 4. Diabetes 2010, 59, 1528–1538. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Chan, P.S. Oxidative stress and diabetic retinopathy. Exp. Diabetes Res. 2007, 2007. [Google Scholar] [CrossRef] [PubMed]
- Veluthakal, R.; Kumar, B.; Mohammad, G.; Kowluru, A.; Kowluru, R.A. Tiam1-rac1 axis promotes activation of p38 map kinase in the development of diabetic retinopathy: Evidence for a requisite role for protein palmitoylation. Cell. Phys. Biochem. 2015, 36, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mishra, M.; Kumar, B. Diabetic retinopathy and transcriptional regulation of a small molecular weight g-protein, rac1. Exp. Eye Res. 2016, 147, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Leal, E.C.; Martins, J.; Voabil, P.; Liberal, J.; Chiavaroli, C.; Bauer, J.; Cunha-Vaz, J.; Ambrosio, A.F. Calcium dobesilate inhibits the alterations in tight junction proteins and leukocyte adhesion to retinal endothelial cells induced by diabetes. Diabetes 2010, 59, 2637–2645. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Flaga, J.; Kowluru, R.A. Molecular mechanism of transcriptional regulation of matrix metalloproteinase-9 in diabetic retinopathy. J. Cell. Physiol. 2016, 231, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.; Kowluru, R.A. Matrix metalloproteinase-2 in the development of diabetic retinopathy and mitochondrial dysfunction. Lab. Investig. 2010, 90, 1365–1372. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lai, D.; Bao, S.; Hambly, B.D.; Gillies, M.C. Triamcinolone acetonide inhibits p38mapk activation and neuronal apoptosis in early diabetic retinopathy. Curr. Mol. Med. 2013, 13, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Y.; Zhang, M.; Hu, Z. Silencing of rac1 expression via rna interference inhibits retinal neovascularization in rats. Mol. Vis. 2012, 18, 1354–1360. [Google Scholar]
- Wang, H.; Fotheringham, L.; Wittchen, E.S.; Hartnett, M.E. Rap1 gtpase inhibits tumor necrosis factor-alpha-induced choroidal endothelial migration via nadph oxidase- and nf-kappab-dependent activation of rac1. Am. J. Pathol. 2015, 185, 3316–3325. [Google Scholar] [CrossRef]
- Kowluru, A. Regulatory roles for small g proteins in the pancreatic beta-cell: Lessons from models of impaired insulin secretion. Am. J. Phys. Endocr. Metab. 2003, 285, E669–E684. [Google Scholar] [CrossRef]
- Kowluru, A. Protein prenylation in glucose-induced insulin secretion from the pancreatic islet beta cell: A perspective. J. Cell Mol. Med. 2008, 12, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Cherfils, J.; Zeghouf, M. Regulation of small gtpases by gefs, gaps, and gdis. Physiol Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, A. Tiam1/vav2-rac1 axis: A tug-of-war between islet function and dysfunction. Biochem. Pharmacol. 2017, 132, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, N.; Waschke, J. Camp with other signaling cues converges on rac1 to stabilize the endothelial barrier- a signaling pathway compromised in inflammation. Cell Tissue Res. 2014, 355, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.; Duraisamy, A.J.; Kowluru, A.; Kowluru, R.A. Functional regulation of an oxidative stress mediator, rac1, in diabetic retinopathy. Mol. Neurobiol 2019, in press. [Google Scholar]
- Ingersoll, M.A.; Chou, Y.W.; Lin, J.S.; Yuan, T.C.; Miller, D.R.; Xie, Y.; Tu, Y.; Oberley-Deegan, R.E.; Batra, S.K.; Lin, M.F. P66shc regulates migration of castration-resistant prostate cancer cells. Cell. Signal. 2018, 46, 1–14. [Google Scholar] [CrossRef]
- Mishra, M.; Duraisamy, A.J.; Bhattacharjee, S.; Kowluru, R.A. Adaptor protein p66shc: A link between cytosolic and mitochondrial dysfunction in the development of diabetic retinopathy. Antioxid. Redox Signal. 2019, 30, 1621–1634. [Google Scholar] [CrossRef]
- DerMardirossian, C.; Bokoch, G.M. Gdis: Central regulatory molecules in rho gtpase activation. Trends Cell Biol. 2005, 15, 356–363. [Google Scholar] [CrossRef]
- Navarro-Lerida, I.; Sanchez-Perales, S.; Calvo, M.; Rentero, C.; Zheng, Y.; Enrich, C.; Del Pozo, M.A. A palmitoylation switch mechanism regulates rac1 function and membrane organization. EMBO J. 2012, 31, 534–551. [Google Scholar] [CrossRef]
- Li, L.; Shi, X.; Guo, X.; Li, H.; Xu, C. Ionic protein-lipid interaction at the plasma membrane: What can the charge do? Trends Biochem. Sci. 2014, 39, 130–140. [Google Scholar] [CrossRef]
- Remorino, A.; De Beco, S.; Cayrac, F.; Di Federico, F.; Cornilleau, G.; Gautreau, A.; Parrini, M.C.; Masson, J.B.; Dahan, M.; Coppey, M. Gradients of rac1 nanoclusters support spatial patterns of rac1 signaling. Cell Rep. 2017, 21, 1922–1935. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.M.; Syeda, K.; Hadden, T.; Kowluru, A. Upregulation of phagocyte-like nadph oxidase by cytokines in pancreatic beta-cells: Attenuation of oxidative and nitrosative stress by 2-bromopalmitate. Biochem. Pharmacol. 2013, 85, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Di-Poi, N.; Faure, J.; Grizot, S.; Molnar, G.; Pick, E.; Dagher, M.C. Mechanism of nadph oxidase activation by the rac/rho-gdi complex. Biochemistry 2001, 40, 10014–10022. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.F. Rho gtpases, their post-translational modifications, disease-associated mutations and pharmacological inhibitors. Small GTPases 2018, 9, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Lluva, S.; Tatham, M.H.; Jones, R.C.; Jaffray, E.G.; Edmondson, R.D.; Hay, R.T.; Malliri, A. Sumoylation of the gtpase rac1 is required for optimal cell migration. Nat. Cell Biol. 2010, 12, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Fraga, M.F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Holliday, R. Epigenetics: A historical overview. Epigenetics 2006, 1, 76–80. [Google Scholar] [CrossRef]
- Zhang, T.Y.; Meaney, M.J. Epigenetics and the environmental regulation of the genome and its function. Annu. Rev. Psychol. 2010, 61, 439–466. [Google Scholar] [CrossRef]
- Turner, B.M. Epigenetic responses to environmental change and their evolutionary implications. Philos. Trans. R. Soc. 2009, 364, 3403–3418. [Google Scholar] [CrossRef] [Green Version]
- Weinhold, B. Epigenetics: The science of change. Environ. Health Perspect. 2006, 114, A160–A167. [Google Scholar] [CrossRef] [PubMed]
- Javaid, N.; Choi, S. Acetylation- and methylation-related epigenetic proteins in the context of their targets. Genes 2017, 8, 196. [Google Scholar] [CrossRef] [PubMed]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Willbanks, A.; Leary, M.; Greenshields, M.; Tyminski, C.; Heerboth, S.; Lapinska, K.; Haskins, K.; Sarkar, S. The evolution of epigenetics: From prokaryotes to humans and its biological consequences. Genet. Epigent. 2016, 8, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P.; Taggart, M.H.; Nicholls, R.D.; Higgs, D.R. Non-methylated cpg-rich islands at the human alpha-globin locus: Implications for evolution of the alpha-globin pseudogene. EMBO J. 1987, 6, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Hisano, M.; Ohta, H.; Nishimune, Y.; Nozaki, M. Methylation of cpg dinucleotides in the open reading frame of a testicular germ cell-specific intronless gene, tact1/actl7b, represses its expression in somatic cells. Nucleic Acids Res. 2003, 31, 4797–4804. [Google Scholar] [CrossRef] [PubMed]
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Zhang, Y. Reversing DNA methylation: Mechanisms, genomics, and biological functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef]
- Duraisamy, A.J.; Mishra, M.; Kowluru, A.; Kowluru, R.A. Epigenetics and regulation of oxidative stress in diabetic retinopathy. Invesig. Ophtahlmol. Vis. Sci. 2018, 59, 4831–4840. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Shan, Y.; Mishra, M. Dynamic DNA methylation of matrix metalloproteinase-9 in the development of diabetic retinopathy. Lab. Investig. 2016, 96, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Kowluru, R.A. The role of DNA methylation in the metabolic memory phenomenon associated with the continued progression of diabetic retinopathy. Investig. Ophtahlmol. Vis. Sci. 2016, 57, 5748–5757. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Cooper, N.G. Glutamate-induced nfkappab activation in the retina. Investig. Ophtahlmol. Vis. Sci. 2009, 50, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Santos, J.M.; Zhong, Q. Sirt1, a negative regulator of matrix metalloproteinase-9 in diabetic retinopathy. Investig. Ophtahlmol. Vis. Sci. 2014, 55, 5653–5660. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Duraisamy, A.J.; Kowluru, R.A. Sirt1-a guardian of the development of diabetic retinopathy. Diabetes 2018, 67, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, B.; Li, Y.; Robertson, K.D. DNA methylation: Superior or subordinate in the epigenetic hierarchy? Genes Cancer 2011, 2, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Duraisamy, A.D.; Kowluru, A.; Kowluru, R.A. Epigenetic regulation of an oxidative stress mediator Rac1, in the development of diabetic retinopathy. Free Rad. Biol. Med. 2018, S137. [Google Scholar] [CrossRef]
- Payapilly, A.; Malliri, A. Compartmentalisation of rac1 signalling. Curr. Opin. Cell Biol. 2018, 54, 50–56. [Google Scholar] [CrossRef]
- Cheng, W.H.; Ho, W.Y.; Chang, C.F.; Lu, P.J.; Cheng, P.W.; Yeh, T.C.; Hong, L.Z.; Sun, G.C.; Hsiao, M.; Tseng, C.J. Simvastatin induces a central hypotensive effect via ras-mediated signalling to cause enos up-regulation. Br. J. Pharmacol. 2013, 170, 847–858. [Google Scholar] [CrossRef]
- Liang, S.L.; Liu, H.; Zhou, A. Lovastatin-induced apoptosis in macrophages through the rac1/cdc42/jnk pathway. J. Immunol. 2006, 177, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic effects of statins on the cardiovascular system. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Oesterle, A.; Liao, J.K. The pleiotropic effects of statins-from coronary artery disease and stroke to atrial fibrillation and ventricular tachyarrhythmia. Curr. Vasc. Pharmacol. 2019, 17, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.Y.; Chen, T.H.; Garg, S.J.; Sun, C.C.; Kang, J.H.; Wu, W.C.; Hung, M.J.; Lai, C.C.; Cherng, W.J.; Hwang, Y.S. Association of statin therapy with prevention of vision-threatening diabetic retinopathy. JAMA Ophthalmol. 2019, 137, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Eller-Borges, R.; Batista, W.L.; da Costa, P.E.; Tokikawa, R.; Curcio, M.F.; Strumillo, S.T.; Sartori, A.; Moraes, M.S.; de Oliveira, G.A.; Taha, M.O.; et al. Ras, rac1, and phosphatidylinositol-3-kinase (pi3k) signaling in nitric oxide induced endothelial cell migration. Nitric Oxide 2015, 47, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Pittala, V.; Fidilio, A.; Lazzara, F.; Platania, C.B.M.; Salerno, L.; Foresti, R.; Drago, F.; Bucolo, C. Effects of novel nitric oxide-releasing molecules against oxidative stress on retinal pigmented epithelial cells. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef]
- Fardi, M.; Solali, S.; Farshdousti Hagh, M. Epigenetic mechanisms as a new approach in cancer treatment: An updated review. Genes Dis. 2018, 5, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Lapinska, K.; Snyder, N.; Leary, M.; Rollinson, S.; Sarkar, S. Use of epigenetic drugs in disease: An overview. Genet. Epigenet. 2014, 6, 9–19. [Google Scholar] [CrossRef]
- Robak, T. New nucleoside analogs for patients with hematological malignancies. Expert Opin. Investig. Drugs 2011, 20, 343–359. [Google Scholar] [CrossRef]
- Silverman, L.R.; Demakos, E.P.; Peterson, B.L.; Kornblith, A.B.; Holland, J.C.; Odchimar-Reissig, R.; Stone, R.M.; Nelson, D.; Powell, B.L.; DeCastro, C.M.; et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: A study of the cancer and leukemia group b. J. Clin. Oncol. 2002, 20, 2429–2440. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Treatment of cardiovascular pathology with epigenetically active agents: Focus on natural and synthetic inhibitors of DNA methylation and histone deacetylation. Int. J. Cardiol. 2017, 227, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Foulks, J.M.; Parnell, K.M.; Nix, R.N.; Chau, S.; Swierczek, K.; Saunders, M.; Wright, K.; Hendrickson, T.F.; Ho, K.K.; McCullar, M.V.; et al. Epigenetic drug discovery: Targeting DNA methyltransferases. J. Biolmol. Screen. 2012, 17, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Pechalrieu, D.; Etievant, C.; Arimondo, P.B. DNA methyltransferase inhibitors in cancer: From pharmacology to translational studies. Biochem. Pharmacol. 2017, 129, 1–13. [Google Scholar] [CrossRef] [PubMed]
- De Boer, V.C.; de Goffau, M.C.; Arts, I.C.; Hollman, P.C.; Keijer, J. Sirt1 stimulation by polyphenols is affected by their stability and metabolism. Mech. Ageing Dev. 2006, 127, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Sarubbo, F.; Esteban, S.; Miralles, A.; Moranta, D. Effects of resveratrol and other polyphenols on sirt1: Relevance to brain function during aging. Curr. Neuropharmacol. 2018, 16, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Purrucker, J.C.; Mahlknecht, U. Targeting the epigenome: Effects of epigenetic treatment strategies on genomic stability in healthy human cells. Clin. Epigenet. 2010, 1, 45–54. [Google Scholar] [CrossRef]
- Platania, C.B.M.; Leggio, G.M.; Drago, F.; Salomone, S.; Bucolo, C. Computational systems biology approach to identify novel pharmacological targets for diabetic retinopathy. Biochem. Pharmacol. 2018, 158, 13–26. [Google Scholar] [CrossRef]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef]
- Bucolo, C.; Drago, F.; Salomone, S. Ocular drug delivery: A clue from nanotechnology. Front. Pharmacol. 2012, 3, 188. [Google Scholar] [CrossRef]
- Joseph, R.R.; Venkatraman, S.S. Drug delivery to the eye: What benefits do nanocarriers offer? Nanomedicine 2017, 12, 683–702. [Google Scholar] [CrossRef]
Figure 1.
Rac1 activation is regulated by GEFs and GAP. In diabetes Tiam1 and Vav2 are upregulated, and these GEFs activate Rac1 leading to increased Rac1-Nox2-ROS signaling pathway. Furthermore, increase in p66Shc displaces Sos1 from Grb2, leading to Rac1 activation. Hyperglycemia also increases prenylation enzyme FNTA and decreases GDI, that further helps Rac1 translocation to the membrane for Nox2 holoenzyme assembly. Increased ROS production by Nox2 augments mitochondrial damage, leading to capillary cell apoptosis and the development of diabetic retinopathy
Figure 1.
Rac1 activation is regulated by GEFs and GAP. In diabetes Tiam1 and Vav2 are upregulated, and these GEFs activate Rac1 leading to increased Rac1-Nox2-ROS signaling pathway. Furthermore, increase in p66Shc displaces Sos1 from Grb2, leading to Rac1 activation. Hyperglycemia also increases prenylation enzyme FNTA and decreases GDI, that further helps Rac1 translocation to the membrane for Nox2 holoenzyme assembly. Increased ROS production by Nox2 augments mitochondrial damage, leading to capillary cell apoptosis and the development of diabetic retinopathy

Figure 2.
Epigenetic modifications of Rac1 promoter increase its gene transcription. Activation of Dnmts in diabetes increases methylation of Rac1 promoter, but concomitant increased binding of the hydroxymethylating enzyme Tet2, 5mC is hydroxymethylated, opening up the chromatin for the binding of the transcription factors. In addition, due to upregulation of SUV39H1, increase in H3K9me3 levels at the promoter further helps the recruitment of Dnmts and the methylation-hydroxymethylation process. Diabetes also inhibits Sirt1, which allows NF-kB to be acetylated, and facilitating the binding of the acetylated NF-kB at the Rac1 promoter, further helping in Rac1 transcription.
Figure 2.
Epigenetic modifications of Rac1 promoter increase its gene transcription. Activation of Dnmts in diabetes increases methylation of Rac1 promoter, but concomitant increased binding of the hydroxymethylating enzyme Tet2, 5mC is hydroxymethylated, opening up the chromatin for the binding of the transcription factors. In addition, due to upregulation of SUV39H1, increase in H3K9me3 levels at the promoter further helps the recruitment of Dnmts and the methylation-hydroxymethylation process. Diabetes also inhibits Sirt1, which allows NF-kB to be acetylated, and facilitating the binding of the acetylated NF-kB at the Rac1 promoter, further helping in Rac1 transcription.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sahajpal, N.; Kowluru, A.; Kowluru, R.A. The Regulatory Role of Rac1, a Small Molecular Weight GTPase, in the Development of Diabetic Retinopathy. J. Clin. Med. 2019, 8, 965. https://doi.org/10.3390/jcm8070965
AMA Style
Sahajpal N, Kowluru A, Kowluru RA. The Regulatory Role of Rac1, a Small Molecular Weight GTPase, in the Development of Diabetic Retinopathy. Journal of Clinical Medicine. 2019; 8(7):965. https://doi.org/10.3390/jcm8070965
Chicago/Turabian StyleSahajpal, Nikhil, Anjan Kowluru, and Renu A. Kowluru. 2019. "The Regulatory Role of Rac1, a Small Molecular Weight GTPase, in the Development of Diabetic Retinopathy" Journal of Clinical Medicine 8, no. 7: 965. https://doi.org/10.3390/jcm8070965
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.