CO-Releasing Molecule-2 Induces Nrf2/ARE-Dependent Heme Oxygenase-1 Expression Suppressing TNF-α-Induced Pulmonary Inflammation


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Reagents and Antibodies
2.2. Animal Care and Experimental Procedures
2.3. Cell Culture and Treatment
2.4. Transient Transfection with siRNAs
2.5. Real-Time RT-PCR
2.6. Preparation of Cell Extracts and Western Blot
2.7. Isolation of Subcellular Fractions
2.8. Immunofluorescence Staining
2.9. Chromatin Immunoprecipitation (ChIP) Assay
2.10. Co-Immunoprecipitation Assay
2.11. ARE Promoter Activity
2.12. Statistical Analysis of Data
3. Results
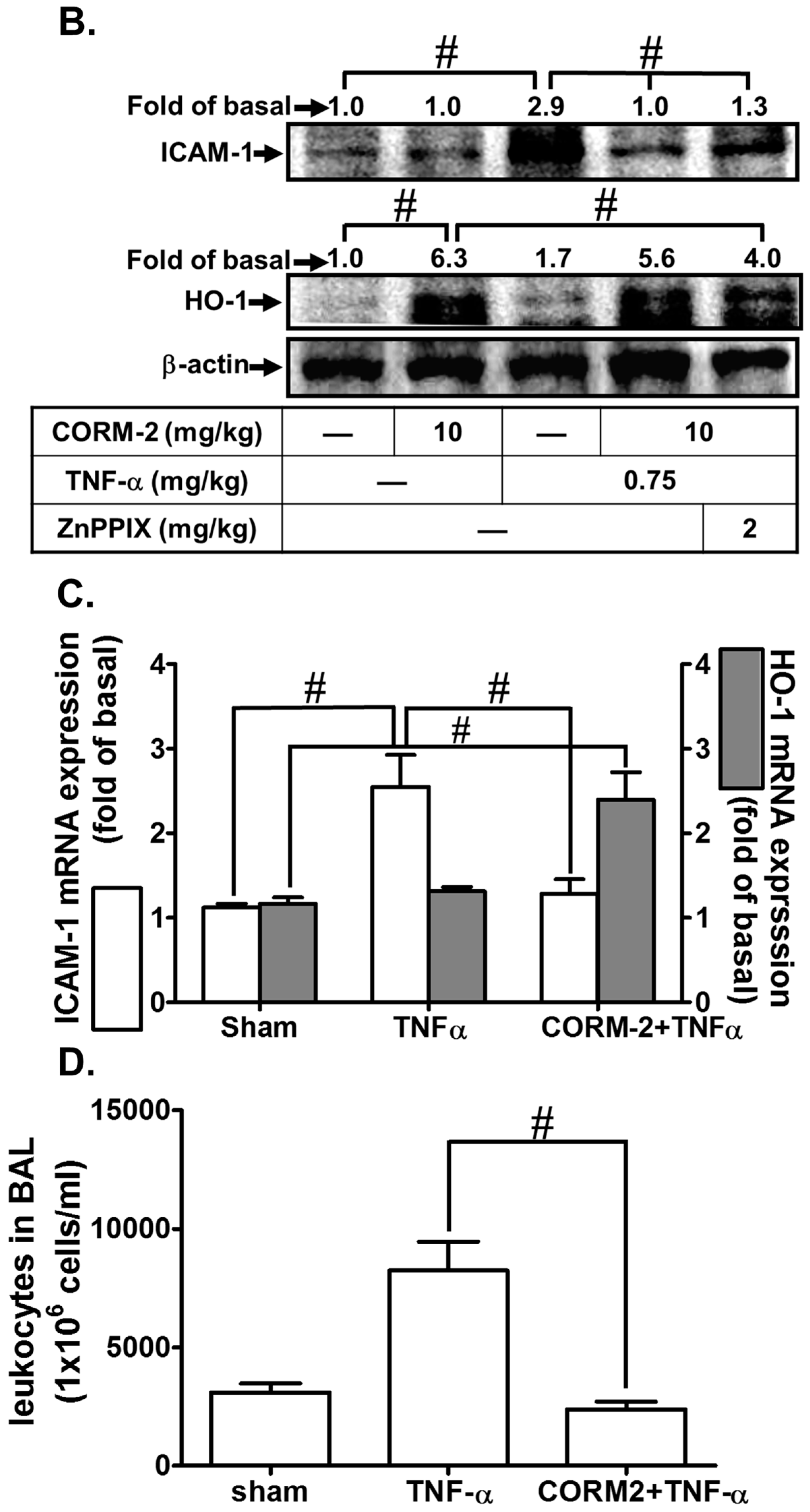
3.1. CORM-2 Inhibits TNF-α-Induced Lung Inflammation in Mice
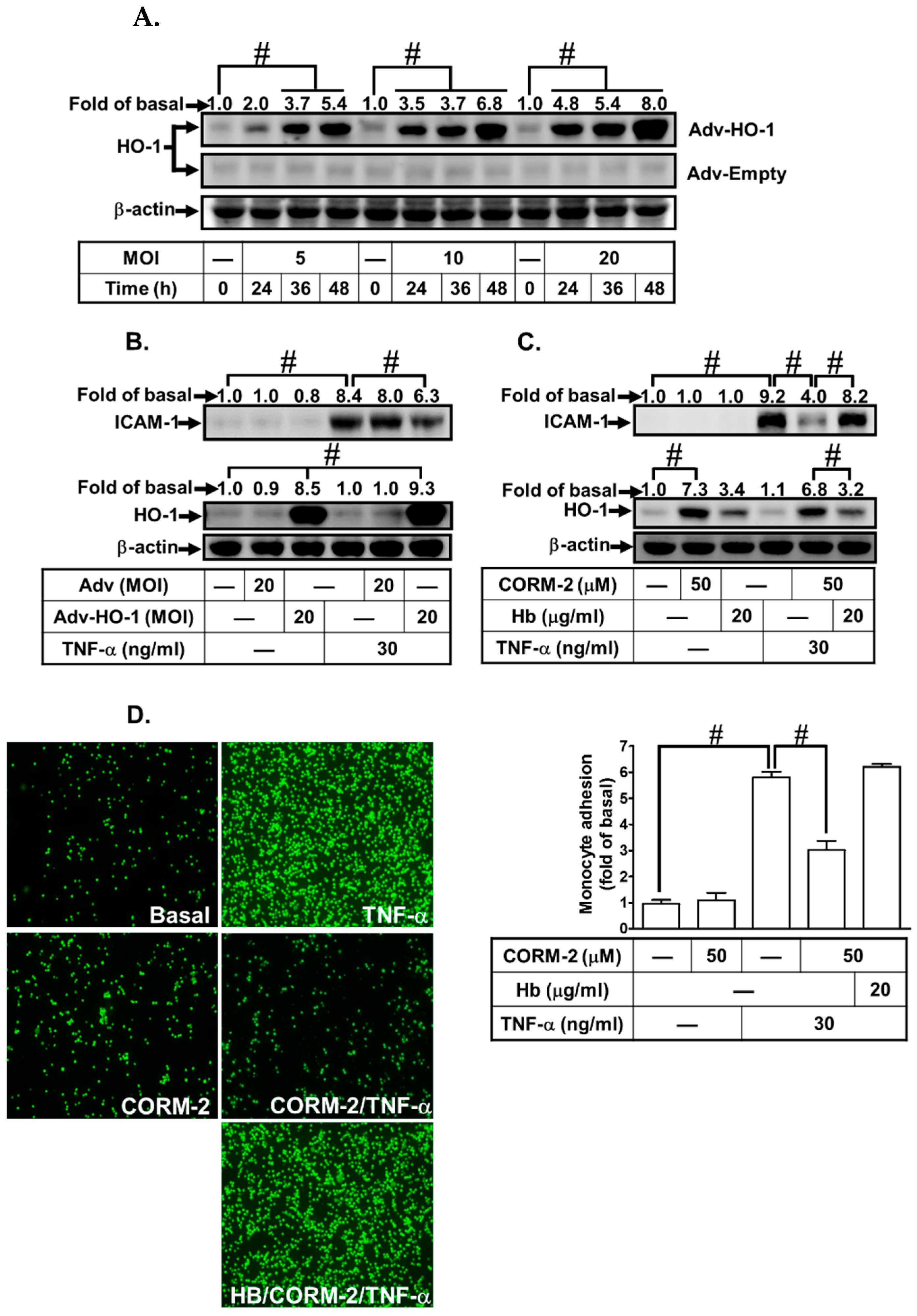
3.2. Adenovirus-Mediated HO-1 Expression Attenuates TNF-α-Induced Inflammatory Responses In Vitro
3.3. CORM-2 Stimulates Nox2/ROS-Dependent HO-1 Expression
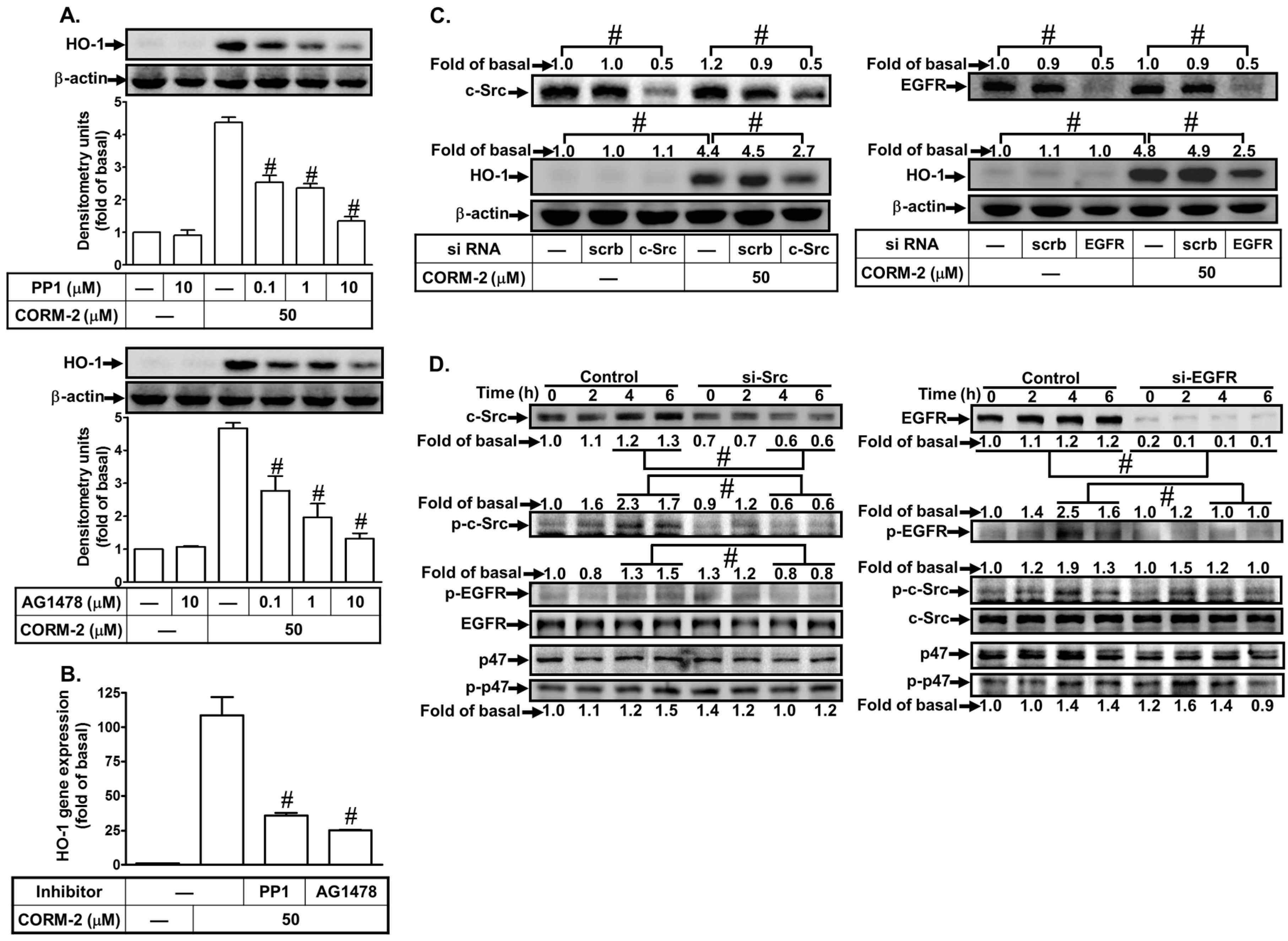
3.4. CORM-2 Induces HO-1 Expression via a c-Src/EGFR Pathway
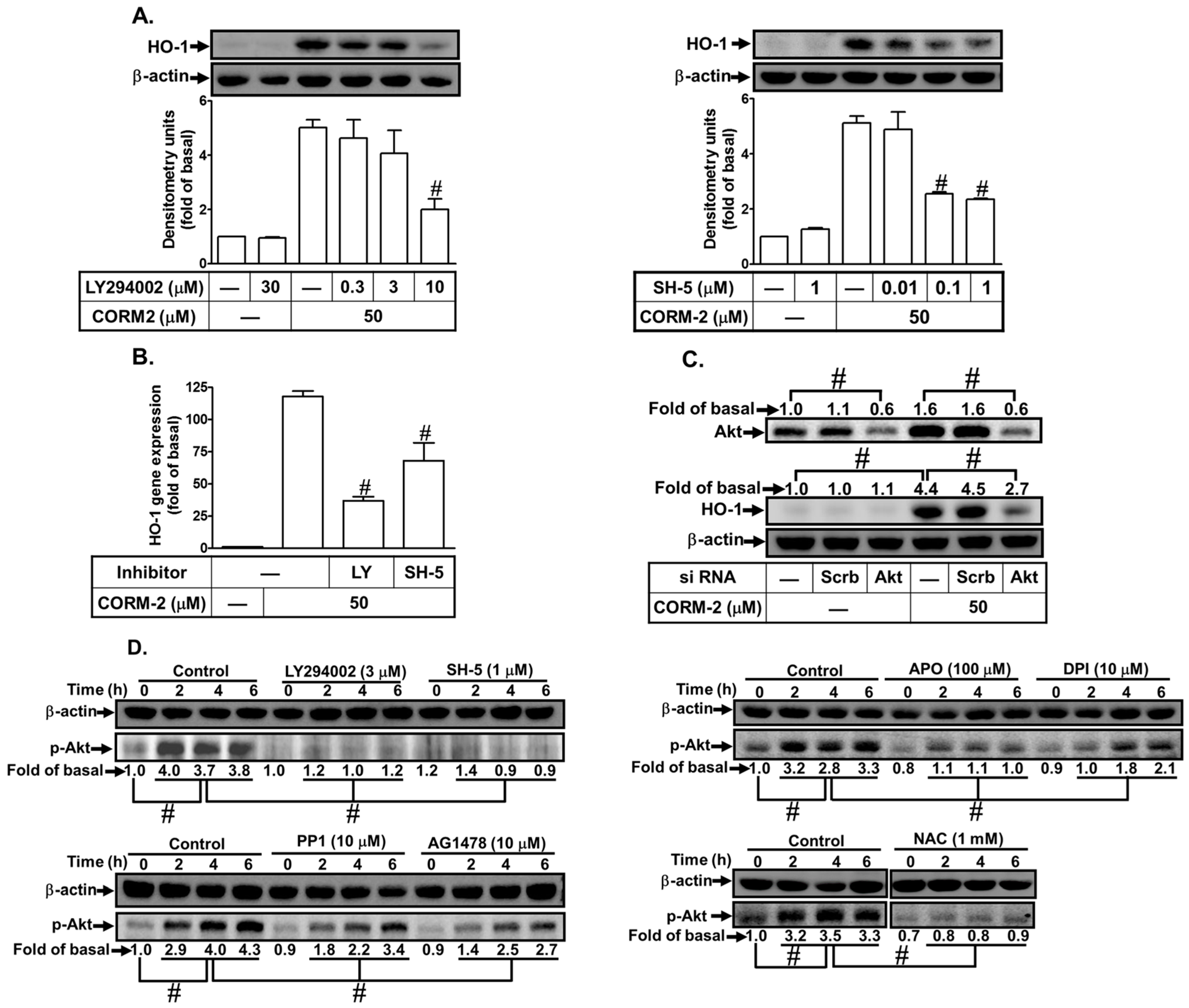
3.5. CORM-2 Stimulates PI3K/Akt-Dependent HO-1 Expression
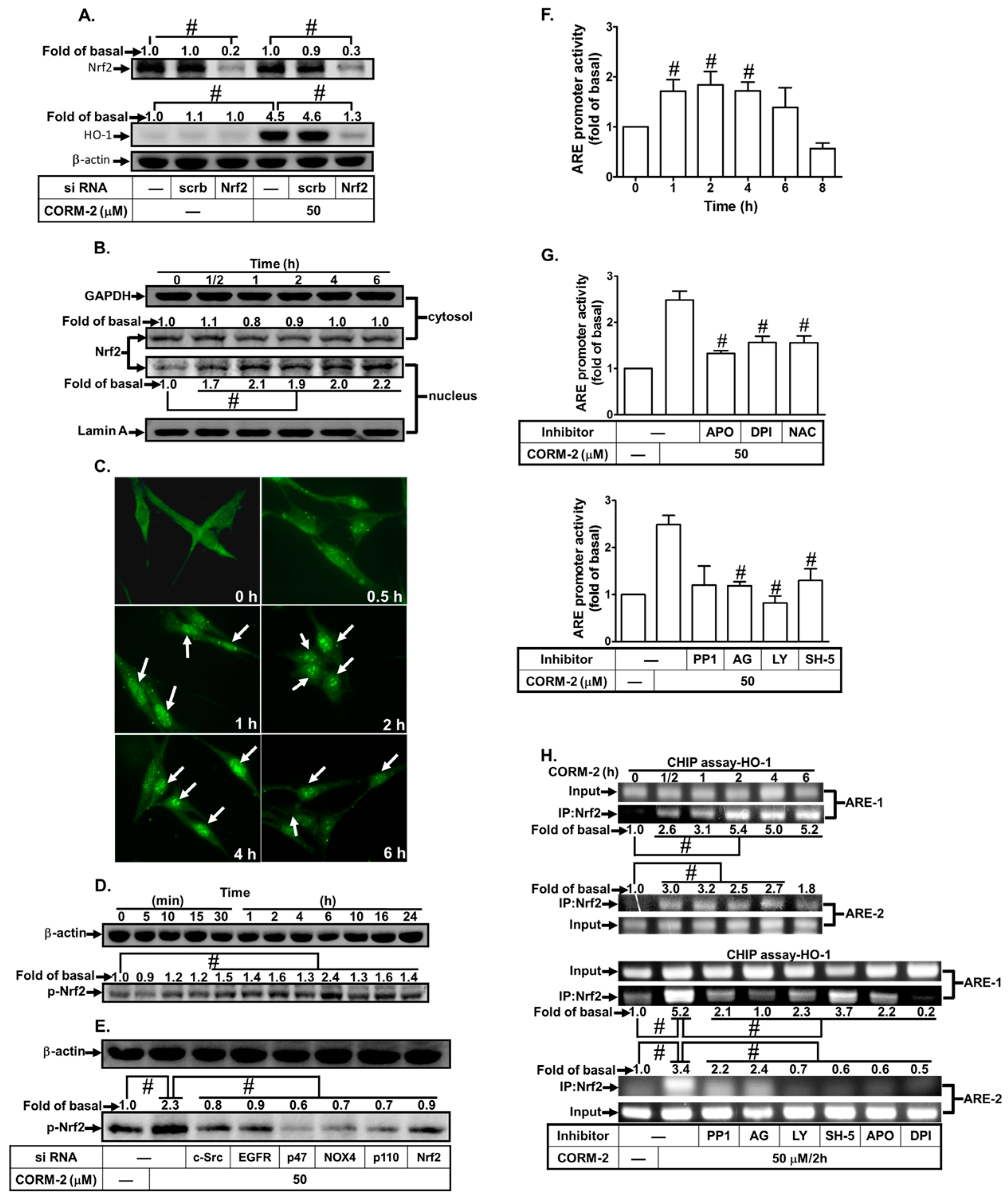
3.6. CORM-2 Stimulates Nrf2/ARE-Dependent HO-1 Expression
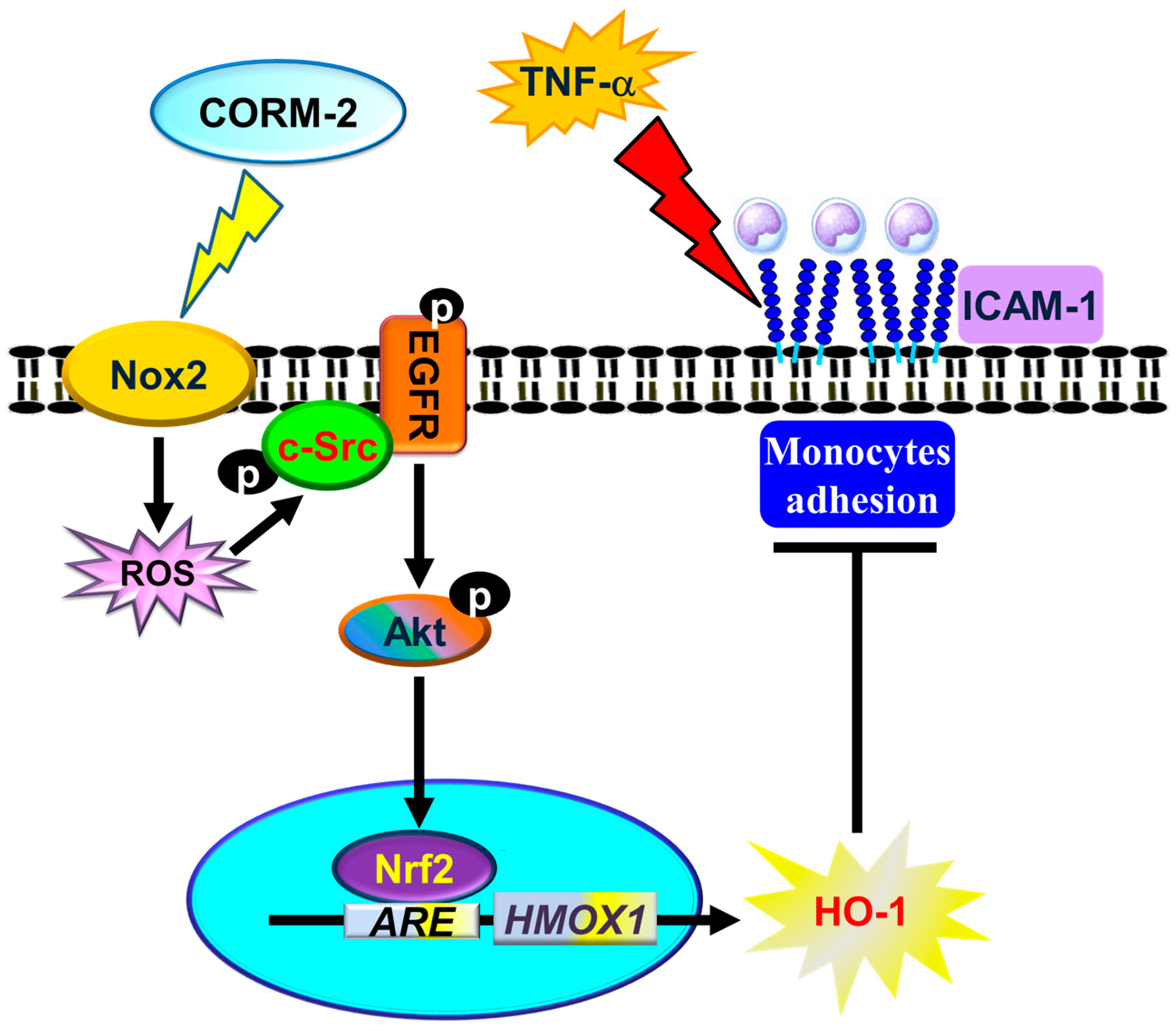
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef] [PubMed]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Otterbein, L.E.; Soares, M.P.; Yamashita, K.; Bach, F.H. Heme oxygenase-1: Unleashing the protective properties of heme. Trends Immunol. 2003, 24, 449–455. [Google Scholar] [CrossRef]
- Matsumoto, H.; Ishikawa, K.; Itabe, H.; Maruyama, Y. Carbon monoxide and bilirubin from heme oxygenase-1 suppresses reactive oxygen species generation and plasminogen activator inhibitor-1 induction. Mol. Cell. Biochem. 2006, 291, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Jamal Uddin, M.; Joe, Y.; Kim, S.K.; Oh Jeong, S.; Ryter, S.W.; Pae, H.O.; Chung, H.T. IRG1 induced by heme oxygenase-1/carbon monoxide inhibits LPS-mediated sepsis and pro-inflammatory cytokine production. Cell. Mol. Immunol. 2016, 13, 170–179. [Google Scholar] [CrossRef]
- Sawle, P.; Foresti, R.; Mann, B.E.; Johnson, T.R.; Green, C.J.; Motterlini, R. Carbon monoxide-releasing molecules (CO-RMs) attenuate the inflammatory response elicited by lipopolysaccharide in RAW264.7 murine macrophages. Br. J. Pharmacol. 2005, 145, 800–810. [Google Scholar] [CrossRef] [Green Version]
- Rushworth, S.A.; Chen, X.L.; Mackman, N.; Ogborne, R.M.; O’Connell, M.A. Lipopolysaccharide-induced heme oxygenase-1 expression in human monocytic cells is mediated via Nrf2 and protein kinase C. J. Immunol. 2005, 175, 4408–4415. [Google Scholar] [CrossRef] [PubMed]
- Aggeli, I.K.; Gaitanaki, C.; Beis, I. Involvement of JNKs and p38-MAPK/MSK1 pathways in H2O2-induced upregulation of heme oxygenase-1 mRNA in H9c2 cells. Cell Signal. 2006, 18, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Fredenburgh, L.E.; Perrella, M.A.; Mitsialis, S.A. The role of heme oxygenase-1 in pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2007, 36, 158–165. [Google Scholar] [CrossRef]
- Ferrandiz, M.L.; Devesa, I. Inducers of heme oxygenase-1. Curr. Pharm. Des. 2008, 14, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.T.; Luo, S.F.; Lee, C.W.; Wang, S.W.; Lin, C.C.; Chang, C.C.; Chen, Y.L.; Chau, L.Y.; Yang, C.M. Overexpression of HO-1 protects against TNF-α-mediated airway inflammation by down-regulation of TNFR1-dependent oxidative stress. Am. J. Pathol. 2009, 175, 519–532. [Google Scholar] [CrossRef]
- Lee, I.T.; Yang, C.M. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem. Pharmacol. 2012, 84, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Otenbaker, N.P.; Rose, B.A.; Salisbury, K.S. Molecular mechanisms of reactive oxygen species-related pulmonary inflammation and asthma. Mol. Immunol. 2013, 56, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Otterbein, L.E.; Kolls, J.K.; Mantell, L.L.; Cook, J.L.; Alam, J.; Choi, A.M. Exogenous administration of heme oxygenase-1 by gene transfer provides protection against hyperoxia-induced lung injury. J. Clin. Investig. 1999, 103, 1047–1054. [Google Scholar] [CrossRef] [Green Version]
- Li, F.J.; Duggal, R.N.; Oliva, O.M.; Karki, S.; Surolia, R.; Wang, Z.; Watson, R.D.; Thannickal, V.J.; Powell, M.; Watts, S.; et al. Heme oxygenase-1 protects corexit 9500A-induced respiratory epithelial injury across species. PLoS ONE 2015, 10, e0122275. [Google Scholar] [CrossRef] [PubMed]
- Magierowska, K.; Magierowski, M.; Hubalewska-Mazgaj, M.; Adamski, J.; Surmiak, M.; Sliwowski, Z.; Kwiecien, S.; Brzozowski, T. Carbon Monoxide (CO) Released from Tricarbonyldichlororuthenium (II) Dimer (CORM-2) in Gastroprotection against Experimental Ethanol-Induced Gastric Damage. PLoS ONE 2015, 10, e0140493. [Google Scholar] [CrossRef]
- Srisook, K.; Han, S.S.; Choi, H.S.; Li, M.H.; Ueda, H.; Kim, C.; Cha, Y.N. CO from enhanced HO activity or from CORM-2 inhibits both O2- and NO production and downregulates HO-1 expression in LPS-stimulated macrophages. Biochem. Pharmacol. 2006, 71, 307–318. [Google Scholar] [CrossRef]
- Lee, I.T.; Wang, S.W.; Lee, C.W.; Chang, C.C.; Lin, C.C.; Luo, S.F.; Yang, C.M. Lipoteichoic acid induces HO-1 expression via the TLR2/MyD88/c-Src/NADPH oxidase pathway and Nrf2 in human tracheal smooth muscle cells. J. Immunol. 2008, 181, 5098–5110. [Google Scholar] [CrossRef]
- Cheng, S.E.; Lee, I.T.; Lin, C.C.; Kou, Y.R.; Yang, C.M. Cigarette smoke particle-phase extract induces HO-1 expression in human tracheal smooth muscle cells: Role of the c-Src/NADPH oxidase/MAPK/Nrf2 signaling pathway. Free Radic. Biol. Med. 2010, 48, 1410–1422. [Google Scholar] [CrossRef]
- Choi, Y.K.; Por, E.D.; Kwon, Y.G.; Kim, Y.M. Regulation of ROS production and vascular function by carbon monoxide. Oxid. Med. Cell. Longev. 2012, 2012, 794237. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.L.; Lin, C.C.; Chen, Y.W.; Hsiao, L.D.; Yang, C.M. CO Induces Nrf2-Dependent Heme Oxygenase-1 Transcription by Cooperating with Sp1 and c-Jun in Rat Brain Astrocytes. Mol. Neurobiol. 2015, 52, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Taille, C.; El-Benna, J.; Lanone, S.; Boczkowski, J.; Motterlini, R. Mitochondrial respiratory chain and NAD(P)H oxidase are targets for the antiproliferative effect of carbon monoxide in human airway smooth muscle. J. Biol. Chem. 2005, 280, 25350–25360. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R.; Green, C.J.; Foresti, R. Regulation of heme oxygenase-1 by redox signals involving nitric oxide. Antioxid. Redox Signal. 2002, 4, 615–624. [Google Scholar] [CrossRef]
- Lee, I.T.; Yang, C.M. Inflammatory signalings involved in airway and pulmonary diseases. Mediators Inflamm. 2013, 2013, 791231. [Google Scholar] [CrossRef]
- Shih, R.H.; Lee, I.T.; Hsieh, H.L.; Kou, Y.R.; Yang, C.M. Cigarette smoke extract induces HO-1 expression in mouse cerebral vascular endothelial cells: Involvement of c-Src/NADPH oxidase/PDGFR/JAK2/STAT3 pathway. J. Cell. Physiol. 2010, 225, 741–750. [Google Scholar] [CrossRef]
- Motterlini, R.; Otterbein, L.E. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 2010, 9, 728–743. [Google Scholar] [CrossRef]
- Sun, L.; Li, W.; Li, W.; Xiong, L.; Li, G.; Ma, R. Astragaloside IV prevents damage to human mesangial cells through the inhibition of the NADPH oxidase/ROS/Akt/NFkappaB pathway under high glucose conditions. Int. J. Mol. Med. 2014, 34, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Yayeh, T.; Hong, M.; Jia, Q.; Lee, Y.C.; Kim, H.J.; Hyun, E.; Kim, T.W.; Rhee, M.H. Pistacia chinensis inhibits NO production and upregulates HO-1 induction via PI-3K/Akt pathway in LPS stimulated macrophage cells. Am. J. Chin. Med. 2012, 40, 1085–1097. [Google Scholar] [CrossRef]
- Alam, J.; Cook, J.L. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am. J. Respir. Cell Mol. Biol. 2007, 36, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Cottin, Y.; Zeller, M.; Vergely, C. Carbon monoxide: Mechanisms of action and potential clinical implications. Pharmacol. Ther. 2013, 137, 133–152. [Google Scholar] [CrossRef] [Green Version]
- Padiya, R.; Chowdhury, D.; Borkar, R.; Srinivas, R.; Pal Bhadra, M.; Banerjee, S.K. Garlic attenuates cardiac oxidative stress via activation of PI3K/AKT/Nrf2-Keap1 pathway in fructose-fed diabetic rat. PLoS ONE 2014, 9, e94228. [Google Scholar] [CrossRef]
- Wu, Z.; Uchi, H.; Morino-Koga, S.; Nakamura-Satomura, A.; Kita, K.; Shi, W.; Furue, M. Z-Ligustilide inhibits benzo(a)pyrene-induced CYP1A1 upregulation in cultured human keratinocytes via ROS-dependent Nrf2 activation. Exp. Dermatol. 2014, 23, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Constantin, M.; Choi, A.J.; Cloonan, S.M.; Ryter, S.W. Therapeutic potential of heme oxygenase-1/carbon monoxide in lung disease. Int. J. Hypertens. 2012, 2012, 859235. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Habtezion, A. Carbon monoxide-based therapy ameliorates acute pancreatitis via TLR4 inhibition. J. Clin. Investig. 2014, 124, 437–447. [Google Scholar] [CrossRef]
- Cheng, S.E.; Lee, I.T.; Lin, C.C.; Hsiao, L.D.; Yang, C.M. Thrombin induces ICAM-1 expression in human lung epithelial cells via c-Src/PDGFR/PI3K/Akt-dependent NF-κB/p300 activation. Clin. Sci. 2014, 127, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.M.; Lin, C.C.; Lee, I.T.; Hsu, C.K.; Tai, Y.C.; Hsieh, H.L.; Chi, P.L.; Hsiao, L.D. c-Src-dependent transactivation of EGFR mediates CORM-2-induced HO-1 expression in human tracheal smooth muscle cells. J. Cell. Physiol. 2015, 230, 2351–2361. [Google Scholar] [CrossRef]
- Lee, I.T.; Lin, C.C.; Lee, C.Y.; Hsieh, P.W.; Yang, C.M. Protective effects of (−)-epigallocatechin-3-gallate against TNF-α-induced lung inflammation via ROS-dependent ICAM-1 inhibition. J. Nutr. Biochem. 2013, 24, 124–136. [Google Scholar] [CrossRef]
- Hsieh, H.L.; Wang, H.H.; Wu, C.Y.; Yang, C.M. Reactive Oxygen Species-Dependent c-Fos/Activator Protein 1 Induction Upregulates Heme Oxygenase-1 Expression by Bradykinin in Brain Astrocytes. Antioxid. Redox Signal. 2010, 13, 1829–1844. [Google Scholar] [CrossRef]
- Lee, C.W.; Lin, C.C.; Lin, W.N.; Liang, K.C.; Luo, S.F.; Wu, C.B.; Wang, S.W.; Yang, C.M. TNF-α induces MMP-9 expression via activation of Src/EGFR, PDGFR/PI3K/Akt cascade and promotion of NF-κB/p300 binding in human tracheal smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L799–L812. [Google Scholar] [CrossRef] [PubMed]
- Tung, W.H.; Hsieh, H.L.; Lee, I.T.; Yang, C.M. Enterovirus 71 modulates a COX-2/PGE2/cAMP-dependent viral replication in human neuroblastoma cells: Role of the c-Src/EGFR/p42/p44 MAPK/CREB signaling pathway. J. Cell. Biochem. 2011, 112, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wang, D.; Xu, M.; Liu, Y.; Wang, X.; Liu, J.; Yang, X.; Yao, P.; Yan, H.; Liu, L. Quinocetone-induced Nrf2/HO-1 pathway suppression aggravates hepatocyte damage of Sprague-Dawley rats. Food Chem. Toxicol. 2014, 69, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Kamata, H.; Hirata, H. Redox regulation of cellular signalling. Cell. Signal. 1999, 11, 1–14. [Google Scholar] [CrossRef]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Asp. Med. 2011, 32, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Mulier, B.; Rahman, I.; Watchorn, T.; Donaldson, K.; MacNee, W.; Jeffery, P.K. Hydrogen peroxide-induced epithelial injury: The protective role of intracellular nonprotein thiols (NPSH). Eur. Respir. J. 1998, 11, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, S.S.; Cavalca, V.; Eligini, S.; Brambilla, M.; Caiani, A.; Tremoli, E.; Colli, S. Apocynin prevents cyclooxygenase 2 expression in human monocytes through NADPH oxidase and glutathione redox-dependent mechanisms. Free Radic. Biol. Med. 2004, 37, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Stanley, A.; Hynes, A.; Brakebusch, C.; Quondamatteo, F. Rho GTPases and Nox dependent ROS production in skin. Is there a connection? Histol. Histopathol. 2012, 27, 1395–1406. [Google Scholar] [PubMed]
- Tavares, A.F.; Teixeira, M.; Romao, C.C.; Seixas, J.D.; Nobre, L.S.; Saraiva, L.M. Reactive oxygen species mediate bactericidal killing elicited by carbon monoxide-releasing molecules. J. Biol. Chem. 2011, 286, 26708–26717. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R. NO and CO as second messengers in oxygen sensing in the carotid body. Respir. Physiol. 1999, 115, 161–168. [Google Scholar] [CrossRef]
- Choi, Y.K.; Maki, T.; Mandeville, E.T.; Koh, S.H.; Hayakawa, K.; Arai, K.; Kim, Y.M.; Whalen, M.J.; Xing, C.H.; Wang, X.Y.; et al. Dual effects of carbon monoxide on pericytes and neurogenesis in traumatic brain injury. Nat. Med. 2016, 22, 1335–1341. [Google Scholar] [CrossRef]
- Byeon, S.E.; Yu, T.; Yang, Y.; Lee, Y.G.; Kim, J.H.; Oh, J.; Jeong, H.Y.; Hong, S.; Yoo, B.C.; Cho, W.J.; et al. Hydroquinone regulates hemeoxygenase-1 expression via modulation of Src kinase activity through thiolation of cysteine residues. Free Radic. Biol. Med. 2013, 57, 105–118. [Google Scholar] [CrossRef]
- Yang, C.M.; Lee, I.T.; Lin, C.C.; Yang, Y.L.; Luo, S.F.; Kou, Y.R.; Hsiao, L.D. Cigarette smoke extract induces COX-2 expression via a PKCalpha/c-Src/EGFR, PDGFR/PI3K/Akt/NF-kappaB pathway and p300 in tracheal smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L892–L902. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wu, J.; Zhang, L.; Ai, Y. Post-conditioning with sevoflurane induces heme oxygenase-1 expression via the PI3K/Akt pathway in lipopolysaccharide-induced acute lung injury. Mol. Med. Rep. 2014, 9, 2435–2440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.; Rojo, A.I.; Salinas, M.; Diaz, R.; Gallardo, G.; Alam, J.; De Galarreta, C.M.; Cuadrado, A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J. Biol. Chem. 2004, 279, 8919–8929. [Google Scholar] [CrossRef] [PubMed]
- Salinas, M.; Diaz, R.; Abraham, N.G.; Ruiz de Galarreta, C.M.; Cuadrado, A. Nerve growth factor protects against 6-hydroxydopamine-induced oxidative stress by increasing expression of heme oxygenase-1 in a phosphatidylinositol 3-kinase-dependent manner. J. Biol. Chem. 2003, 278, 13898–13904. [Google Scholar] [CrossRef]
- Busserolles, J.; Megias, J.; Terencio, M.C.; Alcaraz, M.J. Heme oxygenase-1 inhibits apoptosis in Caco-2 cells via activation of Akt pathway. Int. J. Biochem. Cell Biol. 2006, 38, 1510–1517. [Google Scholar] [CrossRef]
- Sen, C.K.; Packer, L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996, 10, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef]
- Muller, R.M.; Taguchi, H.; Shibahara, S. Nucleotide sequence and organization of the rat heme oxygenase gene. J. Biol. Chem. 1987, 262, 6795–6802. [Google Scholar] [PubMed]
- Wagener, F.A.; Volk, H.D.; Willis, D.; Abraham, N.G.; Soares, M.P.; Adema, G.J.; Figdor, C.G. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol. Rev. 2003, 55, 551–571. [Google Scholar] [CrossRef] [PubMed]
- Almolki, A.; Taille, C.; Martin, G.F.; Jose, P.J.; Zedda, C.; Conti, M.; Megret, J.; Henin, D.; Aubier, M.; Boczkowski, J. Heme oxygenase attenuates allergen-induced airway inflammation and hyperreactivity in guinea pigs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L26–L34. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Sun, H.; Liu, C.; Shen, J.; Chen, Z.; Chen, X. Role of CO-releasing molecules liberated CO in attenuating leukocytes sequestration and inflammatory responses in the lung of thermally injured mice. J. Surg. Res. 2007, 139, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Urquhart, P.; Rosignoli, G.; Cooper, D.; Motterlini, R.; Perretti, M. Carbon monoxide-releasing molecules modulate leukocyte-endothelial interactions under flow. J. Pharmacol. Exp. Ther. 2007, 321, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Wu, S.; Li, J.; Fu, W.; He, W.; Liu, X.; Slutsky, A.S.; Zhang, H.; Li, Y. Human alveolar epithelial type II cells in primary culture. Physiol. Rep. 2015, 3, e12288. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-C.; Hsiao, L.-D.; Cho, R.-L.; Yang, C.-M. CO-Releasing Molecule-2 Induces Nrf2/ARE-Dependent Heme Oxygenase-1 Expression Suppressing TNF-α-Induced Pulmonary Inflammation. J. Clin. Med. 2019, 8, 436. https://doi.org/10.3390/jcm8040436
Lin C-C, Hsiao L-D, Cho R-L, Yang C-M. CO-Releasing Molecule-2 Induces Nrf2/ARE-Dependent Heme Oxygenase-1 Expression Suppressing TNF-α-Induced Pulmonary Inflammation. Journal of Clinical Medicine. 2019; 8(4):436. https://doi.org/10.3390/jcm8040436
Chicago/Turabian StyleLin, Chih-Chung, Li-Der Hsiao, Rou-Ling Cho, and Chuen-Mao Yang. 2019. "CO-Releasing Molecule-2 Induces Nrf2/ARE-Dependent Heme Oxygenase-1 Expression Suppressing TNF-α-Induced Pulmonary Inflammation" Journal of Clinical Medicine 8, no. 4: 436. https://doi.org/10.3390/jcm8040436