Bevacizumab-Based Chemotherapy Triggers Immunological Effects in Responding Multi-Treated Recurrent Ovarian Cancer Patients by Favoring the Recruitment of Effector T Cell Subsets

 , , , ,
, , , ,  , and
, and
Abstract
:1. Introduction
2. Experimental Section
2.1. Patient Selection
2.2. PBMC Purification
2.3. Cell Phenotype
2.4. Intracellular Cytokine Staining
2.5. Statistical Analysis
3. Results
3.1. Patients’ Characteristics and Clinical Response
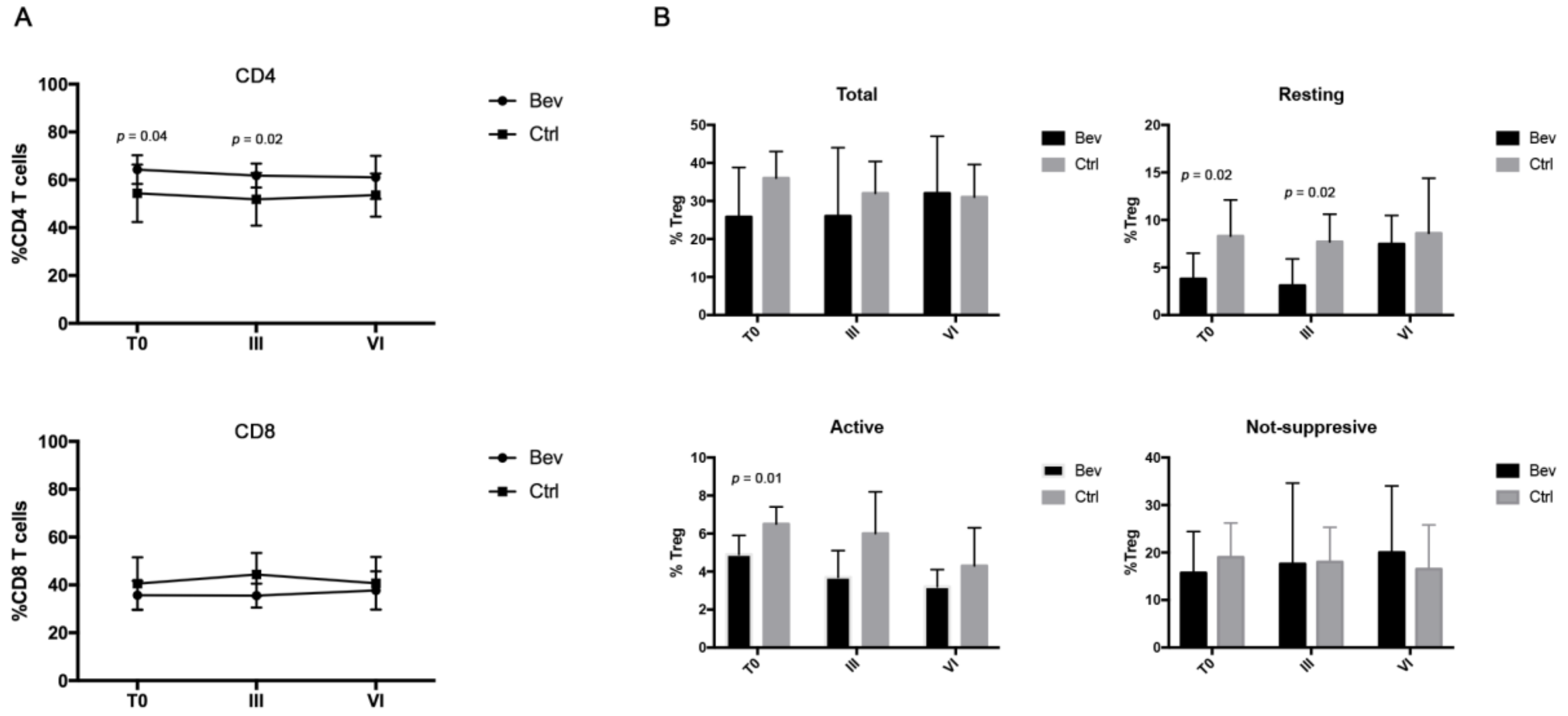
3.2. Bevacizumab-Treated Patients Showed a Different Immunological Signature Compared with the Control Group
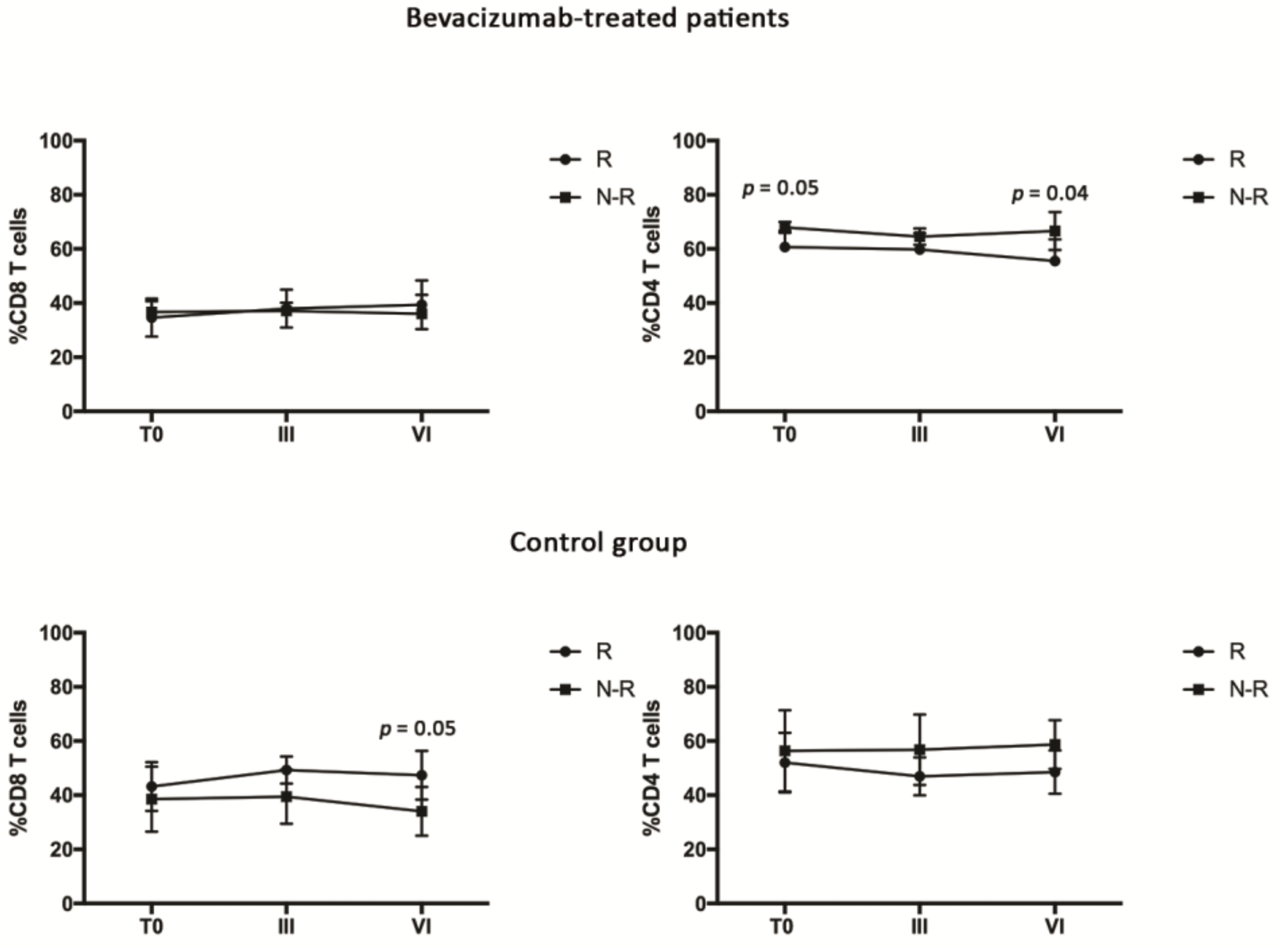
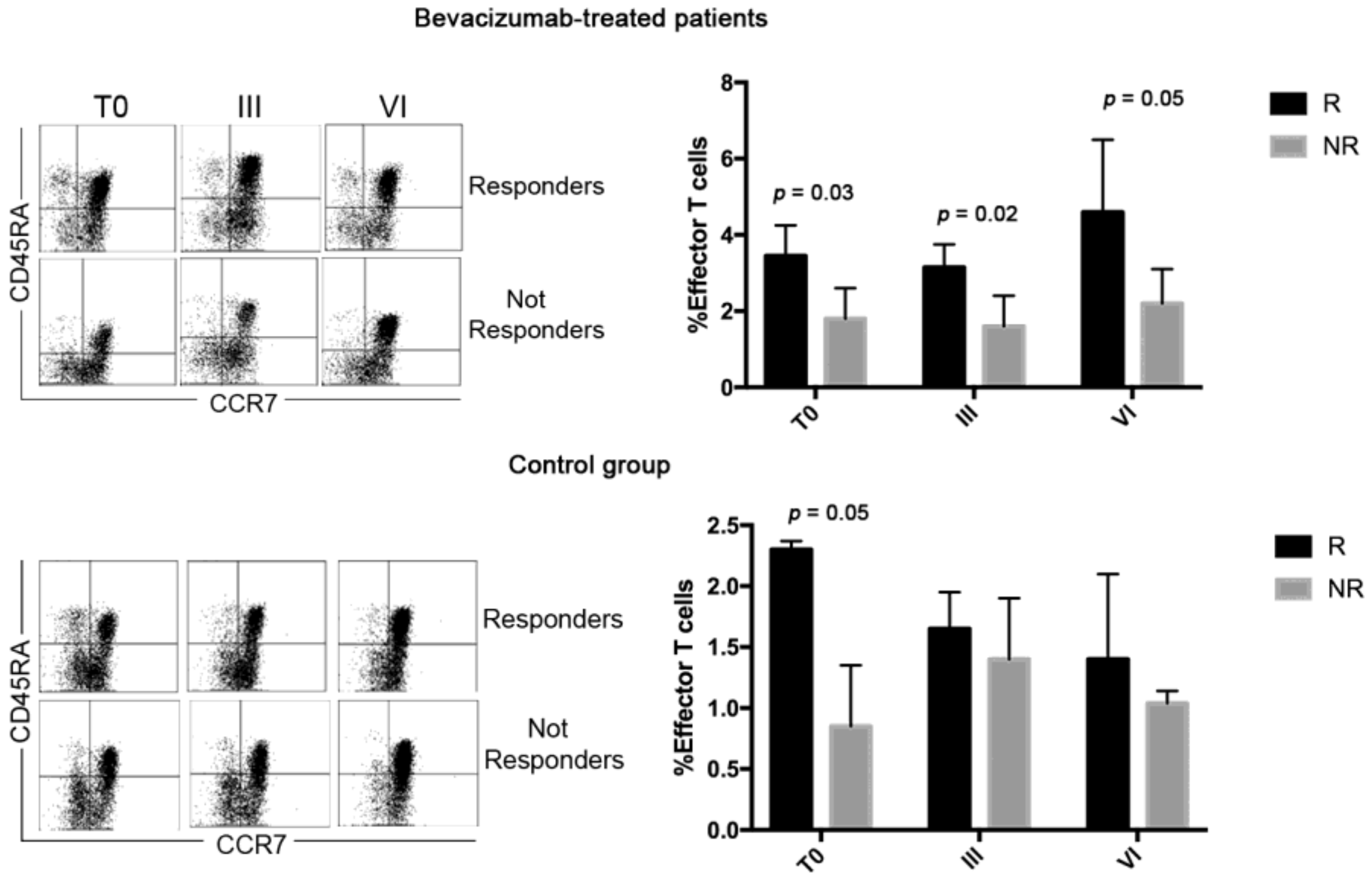
3.3. Bevacizumab-Treated Patients Showed a Discrete CD4 Effector T Cell Population throughout the Treatment
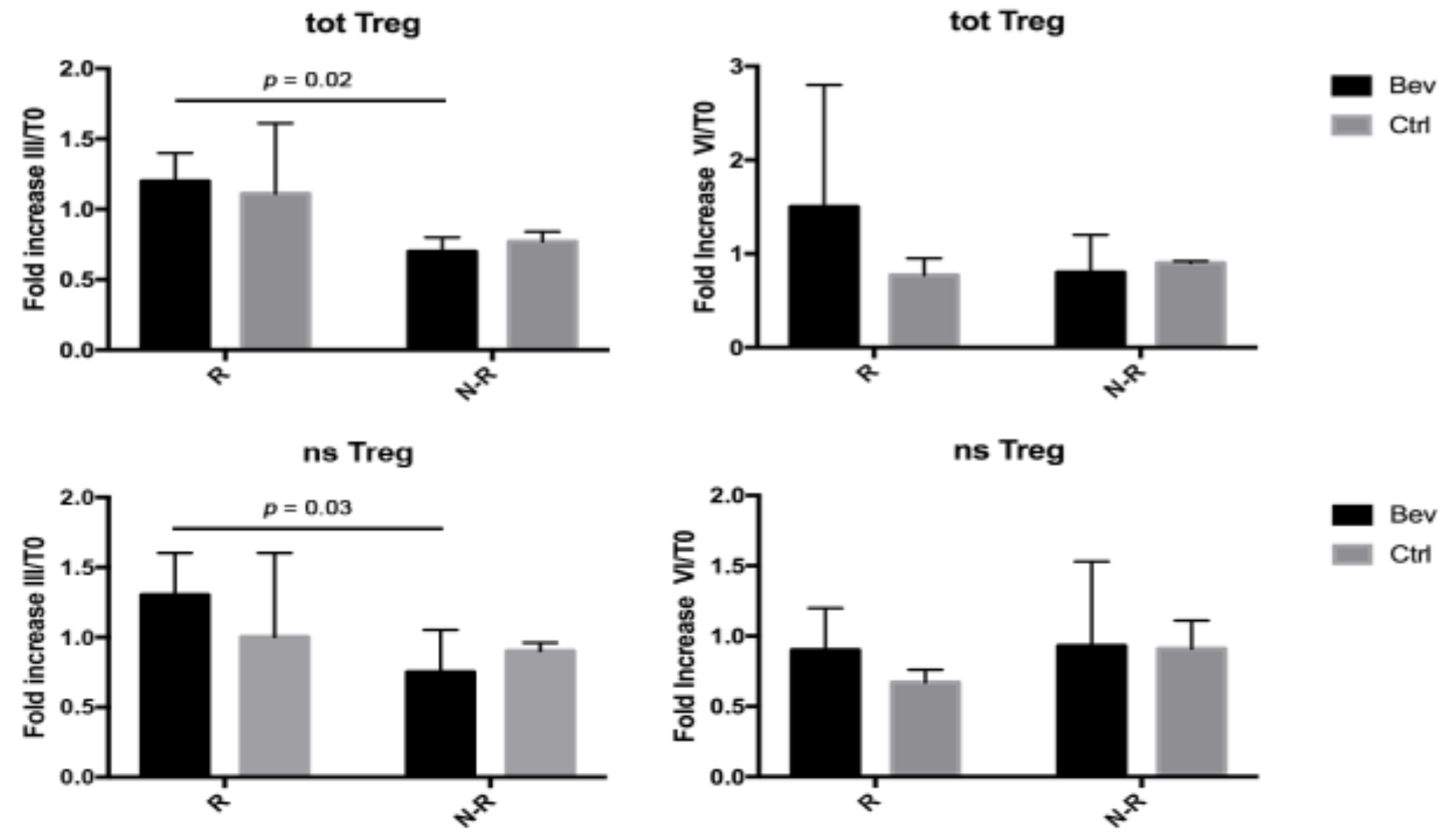
3.4. Tregs Were Modulated in Bevacizumab-Treated Patients during Therapies
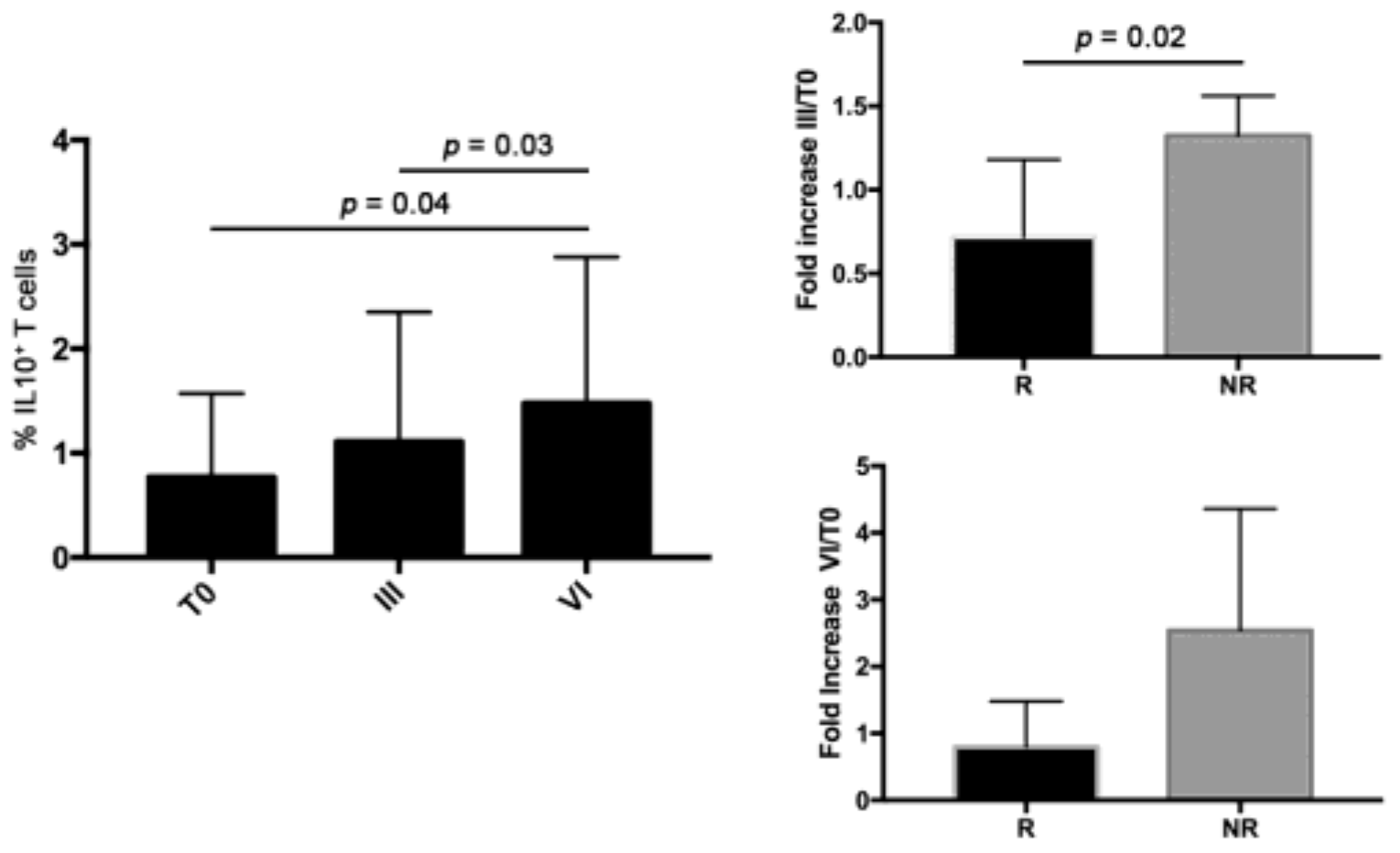
3.5. Bevacizumab-Treated N-R Patients Had Higher Level of IL10+ T Cells Compared to R Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, M.; Rovati, B.; Ronzoni, M.; Loupakis, F.; Mariucci, S.; Ricci, V.; Gattoni, E.; Salvatore, L.; Tinelli, C.; Villa, E.; et al. Immunological effects of bevacizumab-based treatment in metastatic colorectal cancer. Oncology 2010, 79, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Wada, J.; Suzuki, H.; Fuchino, R.; Yamasaki, A.; Nagai, S.; Yanai, K.; Koga, K.; Nakamura, M.; Tanaka, M.; Morisaki, T.; et al. The contribution of vascular endothelial growth factor to the induction of regulatory T-cells in malignant effusions. Anticancer Res. 2009, 29, 881–888. [Google Scholar]
- Terme, M.; Pernot, S.; Marcheteau, E.; Sandoval, F.; Benhamouda, N.; Colussi, O.; Dubreuil, O.; Carpentier, A.F.; Tartour, E.; Taieb, J. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013, 73, 539–549. [Google Scholar] [CrossRef]
- Kobold, S.; Hegewisch-Becker, S.; Oechsle, K.; Jordan, K.; Bokemeyer, C.; Atanackovic, D. Intraperitoneal VEGF inhibition using bevacizumab: A potential approach for the symptomatic treatment of malignant ascites? Oncologist 2009, 14, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Bellati, F.; Napoletano, C.; Ruscito, I.; Pastore, M.; Pernice, M.; Antonilli, M.; Nuti, M.; Benedetti Panici, P. Complete remission of ovarian cancer induced intractable malignant ascites with intraperitoneal bevacizumab. Immunological observations and a literature review. Investig. New Drugs 2010, 28, 887–894. [Google Scholar] [CrossRef]
- Shamsunder, S.; Kumar, L.; Gupta, S.; Kumar, S.; Bhatla, N.; Singh, R.; Kochupillai, V. Chemotherapy in recurrent epithelial ovarian cancer (EOC): An analysis of prognostic factors. J. Obstet. Gynaecol. Res. 2000, 26, 215–222. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumors: Revised RECIST guideline (version 1.1). Eur. J. Cancers 2009, 2, 228–247. [Google Scholar] [CrossRef]
- Lanzavecchia, A.; Sallusto, F. Dynamics of T lymphocyte responses: Intermediates, effectors, and memory cells. Science 2000, 290, 92–97. [Google Scholar] [CrossRef]
- Miyara, M.; Yoshioka, Y.; Kitoh, A.; Shima, T.; Wing, K.; Niwa, A.; Parizot, C.; Taflin, C.; Heike, T.; Valeyre, D.; et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009, 30, 899–911. [Google Scholar] [CrossRef]
- Shou, J.; Zhang, Z.; Lai, Y.; Chen, Z.; Huang, J. Worse outcome in breast cancer with higher tumor-infiltrating FOXP3+ Tregs: A systematic review and meta-analysis. BMC Cancer 2016, 16, 687. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Jiang, T.; Zhang, L.; Yang, H.; Liu, X.; Jia, Y.; Zhou, C. Clinicopathological and prognostic significance of regulatory T cells in patients with non-small cell lung cancer: A systematic review with meta-analysis. Oncotarget 2016, 7, 36065–36073. [Google Scholar] [CrossRef]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Du, Y.; Lin, X.; Qian, Y.; Zhou, T.; Huang, Z. CD4+CD25+ regulatory T cells in tumor immunity. Int. Immunopharmacol. 2016, 34, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Chen, Y.H. Nuclear factor-κB in immunity and inflammation: The Treg and Th17 connection. Adv. Exp. Med. Biol. 2012, 946, 207–221. [Google Scholar] [PubMed]
- Segev, Y.; Segev, L.; Schmidt, M.; Auslender, R.; Lavie, O. Palliative care in ovarian carcinoma patients-a personalized approach of a team work: A review. Arch. Gynecol. Obstet. 2017, 296, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Joly, F.; Hilpert, F.; Okamoto, A.; Stuart, G.; Ochiai, K.; Friedlander, M. Fifth Ovarian Cancer Consensus Conference of the Gynecologic Cancer InterGroup: Recommendations on incorporating patient-reported outcomes in clinical trials in epithelial ovarian cancer. Eur. J. Cancer 2017, 78, 133–138. [Google Scholar] [CrossRef]
- Lapeyre-Prost, A.; Terme, M.; Pernot, S.; Pointet, A.L.; Voron, T.; Tartour, E.; Taieb, J. Immunomodulatory Activity of VEGF in Cancer. In International Review of Cell and Molecular Biology; Academic Press: Cambridge, MA, USA, 2017; Volume 330, pp. 295–342. [Google Scholar]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.; Ishida, T.; Oyama, T.; Ran, S.; Kravtsov, V.; Nadaf, S.; Carbone, D.P. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood 1998, 92, 4150–4166. [Google Scholar] [PubMed]
- Oyama, T.; Ran, S.; Ishida, T.; Nadaf, S.; Kerr, L.; Carbone, D.P.; Gabrilovich, D.I. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J. Immunol. 1998, 160, 1224–1232. [Google Scholar]
- Li, B.; Lalani, A.S.; Harding, T.C.; Luan, B.; Koprivnikar, K.; Huan Tu, G.; Prell, R.; VanRoey, M.J.; Simmons, A.D.; Jooss, K. Vascular endothelial growth factor blockade reduces intratumoral regulatory T cells and enhances the efficacy of a GM-CSF-secreting cancer immunotherapy. Clin. Cancer. Res. 2006, 12, 6808–6816. [Google Scholar] [CrossRef] [PubMed]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Prigerson, H.G.; Bao, Y.; Shah, M.A.; Paulk, M.E.; LeBlanc, T.W.; Schneider, B.J.; Garrido, M.M.; Reid, M.C.; Berlin, D.A.; Adelson, K.B.; et al. Chemotherapy Use, Performance Status, and Quality of Life at the End of Life. JAMA Oncol. 2015, 1, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.A.; Fisher, J.L.; Hampton, T.H.; Christensen, B.C.; Tsongalis, G.J.; Rahme, G.J.; Whipple, C.A.; Steel, S.E.; Davis, M.C.; Gaur, A.B.; et al. Immune modulation associated with vascular endothelial growth factor (VEGF) blockade in patients with glioblastoma. Cancer Immunol. Immunother. 2017, 66, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Wallin, J.J.; Bendell, J.C.; Funke, R.; Sznol, M.; Korski, K.; Jones, S.; Hernandez, G.; Mier, J.; He, X.; Hodi, F.S.; et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat. Commun. 2016, 7, 12624. [Google Scholar] [CrossRef] [PubMed]
- Jabeen, S.; Zucknick, M.; Nome, M.; Dannenfelser, R.; Fleischer, T.; Kumar, S.; Lüders, T.; von der Lippe Gythfeldt, H.; Troyanskaya, O.; Kyte, J.A.; et al. Serum cytokine levels in breast cancer patients during neoadjuvant treatment with bevacizumab. Oncoimmunology 2018, 7, e1457598. [Google Scholar] [CrossRef]
- Abajo, A.; Boni, V.; Lopez, I.; Gonzalez-Huarriz, M.; Bitarte, N.; Rodriguez, J.; Zarate, R.; Bandres, E.; Garcia-Foncillas, J. Identification of predictive circulating biomarkers of bevacizumab-containing regimen efficacy in pre-treated metastatic colorectal cancer patients. Br. J. Cancer 2012, 107, 287–290. [Google Scholar] [CrossRef]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Cropet, C.; Van Glabbeke, M.; Sebban, C.; Le Cesne, A.; Judson, I.; Tredan, O.; Verweij, J.; Biron, P.; Labidi, I.; et al. Lymphopenia as a prognostic factor for overall survival in advanced carcinomas, sarcomas, and lymphomas. Cancer Res. 2009, 69, 5383–5391. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bevacizumab-Treated Patients | Control Group | p-Value | |
|---|---|---|---|
| Patient n° | 10 | 10 | |
| Age (median, range) | 54 years (42y–67y) | 48.5 years (45y–71y) | 0.845 |
| ECOG Performance Status | |||
| 1 | 1/10 (10%) | 2/10 (20%) | |
| 2 | 7/10 (70%) | 5/10 (50%) | |
| 3 | 2/10 (20%) | 3/10 (30%) | |
| Tumor Grading at primary diagnosis | 0.628 | ||
| I | 0 | 0 | |
| II | 4/10 (40%) | 2/10 (20%) | |
| III | 6/10 (60%) | 8/10 (80%) | |
| FIGO stage at primary diagnosis | 1 | ||
| IIIC | 8/10 (80%) | 7/10 (70%) | |
| IV | 2/10 (20%) | 3/10 (30%) | |
| PDS NACT | 5/10 (50%) 5/10 (50%) | 6/10 (60%) 4/10 (40%) | 1 |
| RT at first surgery (cm) | 1 | ||
| =0 | 9/10 (90%) | 8/10 (80%) | |
| >0 | 1/10 (10%) | 2/10 (20%) | |
| Type of recurrence at the time of blood sampling | 0.061 | ||
| Intraperitoneal only | 7/10 (70%) | 5/10 (50%) | |
| intraperitoneal + retroperitoneal | 1/10 (10%) | 2/10 (20%) | |
| widespread | 2/10 (20%) | 3/10 (30%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Napoletano, C.; Ruscito, I.; Bellati, F.; Zizzari, I.G.; Rahimi, H.; Gasparri, M.L.; Antonilli, M.; Panici, P.B.; Rughetti, A.; Nuti, M. Bevacizumab-Based Chemotherapy Triggers Immunological Effects in Responding Multi-Treated Recurrent Ovarian Cancer Patients by Favoring the Recruitment of Effector T Cell Subsets. J. Clin. Med. 2019, 8, 380. https://doi.org/10.3390/jcm8030380
Napoletano C, Ruscito I, Bellati F, Zizzari IG, Rahimi H, Gasparri ML, Antonilli M, Panici PB, Rughetti A, Nuti M. Bevacizumab-Based Chemotherapy Triggers Immunological Effects in Responding Multi-Treated Recurrent Ovarian Cancer Patients by Favoring the Recruitment of Effector T Cell Subsets. Journal of Clinical Medicine. 2019; 8(3):380. https://doi.org/10.3390/jcm8030380
Chicago/Turabian StyleNapoletano, Chiara, Ilary Ruscito, Filippo Bellati, Ilaria Grazia Zizzari, Hassan Rahimi, Maria Luisa Gasparri, Morena Antonilli, Pierluigi Benedetti Panici, Aurelia Rughetti, and Marianna Nuti. 2019. "Bevacizumab-Based Chemotherapy Triggers Immunological Effects in Responding Multi-Treated Recurrent Ovarian Cancer Patients by Favoring the Recruitment of Effector T Cell Subsets" Journal of Clinical Medicine 8, no. 3: 380. https://doi.org/10.3390/jcm8030380