Targeting Immune Signaling Checkpoints in Acute Myeloid Leukemia

1
Department of Experimental Hematooncology, Medical University of Lublin, 20-093 Lublin, Poland
2
Department of Hematology, St John’s Cancer Centre, 20-093 Lublin, Poland
J. Clin. Med. 2019, 8(2), 236; https://doi.org/10.3390/jcm8020236
Submission received: 15 January 2019
/
Revised: 26 January 2019
/
Accepted: 5 February 2019
/
Published: 12 February 2019
(This article belongs to the Special Issue Immunotherapies for Acute Myeloid Leukemia)
Abstract
:The modest successes of targeted therapies along with the curative effects of allogeneic hematopoietic stem cell transplantation (alloHSCT) in acute myeloid leukemia (AML) stimulate the development of new immunotherapies. One of the promising methods of immunotherapy is the activation of immune response by the targeting of negative control checkpoints. The two best-known inhibitory immune checkpoints are cytotoxic T-lymphocyte antigen-4 (CTLA-4) and the programmed cell death protein 1 receptor (PD-1). In AML, PD-1 expression is observed in T-cell subpopulations, including T regulatory lymphocytes. Increased PD-1 expression on CD8+ T lymphocytes may be one of the factors leading to dysfunction of cytotoxic T cells and inhibition of the immune response during the progressive course of AML. Upregulation of checkpoint molecules was observed after alloHSCT and therapy with hypomethylating agents, pointing to a potential clinical application in these settings. Encouraging results from recent clinical trials (a response rate above 50% in a relapsed setting) justify further clinical use. The most common clinical trials employ two PD-1 inhibitors (nivolumab and pembrolizumab) and two anti-PD-L1 (programmed death-ligand 1) monoclonal antibodies (atezolizumab and durvalumab). Several other inhibitors are under development or in early phases of clinical trials. The results of these clinical trials are awaited with great interest in, as they may allow for the established use of checkpoint inhibitors in the treatment of AML.
1. Introduction
The modest successes of targeted therapies in acute myeloid leukemia (AML) and the proven power of the immune system to fight effectively against leukemic blasts in the context of the graft-versus-leukemia effect, stimulate the development of new immunotherapies. One of the promising methods of immunotherapy is the activation of immune response by the targeting of negative control checkpoints on the surface of immune cells or by eliminating the regulatory proteins present in the tumor microenvironment [1]. The homeostasis of the stimulating and inhibiting signals of the immune response is regulated by immunological control checkpoints, which allow the activation of cytotoxic response against pathogens, while maintaining a tolerance to the organism’s own cells.
To avoid autoimmunity, the process of T-cell activation must be strictly regulated. For stimulation, T cells require at least two different signals from antigen presenting cells (APC). The first signal is the recognition of the antigen, and more specifically its immunogenic part—an epitope presented by the major histocompatibility complex (MHC), located on the APC and recognized by the T-cell receptor (TCR) (Figure 1a). The second signal is the result of costimulation, mainly by the interaction of the CD28 molecule of the T-lymphocyte with CD80 (B7.1) or CD86 (B7.2) molecules, on the APC. The stimulation of T cells is also closely related to other stimulatory signals sent to the cell as a result of the combination of specific receptor pairs and their ligands, including the glucocorticoid-induced TNF (tumor necrosis factor) receptor and its specific ligand, and also the interaction between the transmembrane 4-1BB receptor (CD137) and its ligand, 4-1BBL (CD137L) [2,3,4].
The two best-known inhibitory immune checkpoints are cytotoxic T-lymphocyte antigen-4 (CTLA-4) and the programmed cell death protein 1 receptor(PD-1) (Figure 1a) [1]. On T cells, the CTLA-4 receptor inhibits T cell maturation and differentiation by competing with the costimulatory receptor CD28, for CD80 (B7.1) and CD86 (B7.2) [5]. Although the increased CD80 and CD86 expressions that are associated with poor outcome were reported in AML, treatment with the anti-CTLA-4 monoclonal antibody ipilimumab, proved limited clinical activity [6,7,8]. Encouraging results from trials on solid tumors have turned the attention of researchers to the potential that blocking the PD-1 signaling pathway may have potential applications in the field of hematology–oncology. Those new therapies might target the immune synapse of patients irrespective of the PD-1 expression and thereby could be proposed for a majority of AML patients (Figure 1b).
2. The Role of PD-1/PD-L1 Signaling Pathway
PD-1 is a surface glycoprotein cell receptor that belongs to the CD28 family. PD-1 is composed of 288 amino acids, and its molecular weight ranges from 50 to 55 kDa2. PD-1 exhibits approximately 31–33% homology with CTLA-4, CD28, and Inducible T-cell COStimulator (ICOS) molecules. PD-1 interactions with ligands prevent autoimmunity on the one hand, by inducing apoptosis of autoantigen-specific T cells and on the other hand, by inhibiting regulatory T cell (Treg) apoptosis. The expressions of PD-1 on T and B cells is a consequence of the activation of the signaling pathway TCR or the B-cell receptor (BCR), respectively [9].
The PD-1 protein is encoded by the PDCD-1 gene, located on chromosome 2 (2q.37.3) [10]. PDCD-1 consists of five exons. Exon 1 encodes a leader peptide that is extracellular. Exon 2 encodes the immunoglobulin (Ig) variable (V-like domain. Amino acid fragments (ca. 20) are located at the IgV-like domain, that separates it from the cell membrane. A transmembrane domain encapsulated by exon 3 is anchored within the cell membrane. Exons 4 and 5 encode an intracellular domain, in which we distinguish two tyrosines, located in two amino acid motifs—proximal (tyrosine-based motif inhibitors—ITIM) and distal (a tyrosine immunoreceptor-based switch motif—ITSM) [11]. The tyrosines mentioned above play a fundamental role in the function of PD-1 as an inhibitor [12]. Under physiological conditions, PD-1 is expressed on the cells of the immune system, including mature CD4+ and CD8+ T cells, as well as on B cells and T cells during their thymus development [13,14]. In addition, PD-1 expression is found on natural killer (NK) cells, some dendritic cell (DC) subpopulations, and monocytes [15,16]. In a form unrelated to the cell membrane, PD-1 may be present in the cytoplasm of Treg and naїve CD4+ cells. PD-1 can be regulated by various factors, including hormones, cytokines or suppressor genes, such as Phosphatase and tensin homolog (PTEN) and liver kinase B1 (LKB1) [17]. The cytokines that stimulate the expression of PD-1 are interleukin 2 (IL-2), IL-7, IL-15 and IL-21. It has been shown that in the induction of PD-1 expression in T cells, there is a significant role played by the nuclear factor of stimulated Tc1 cells (NF-ATc1). It has also been proven that the specific inhibition of this factor, consisting in the abolition of its translocation to the nucleus, results in the reduction of PD-1 expression, and the mutation of the gene encoding NF-ATc1 results in the complete lack of receptor expression [18]. The transmission of the signal through TCR after its stimulation leads to the binding of NFAT to the promoter region of the PDCD1 gene [18]. PD-1 expression in B-lymphocytes is induced by the molecules that stimulate the activation and the proliferation of these lymphocytes, including anti-IgM, anti-CD40 and lipopolysaccharide (LPS) [9]. The interaction with toll-like receptors (TLRs) such as TLR2, TLR3, TLR4 and the nucleotide-binding oligomerization domain (NOD) has a stimulating effect on the expression of PD-1 in DC. In turn, IL-4 and TLR9 act to inhibit the expression of PD-1 in DC [19]. In macrophages, PD-1 expression is stimulated by an interferon-stimulated response element (ISRE), signal transducers and activators of transcription (STAT), including STAT1 and STAT2, and interferon α (IFNα), through ISRE [20].
The programmed death-ligand 1 (PD-L1), also referred to as B7-H1 or CD274, is a transmembrane type I glycoprotein, made up of 290 amino acids, belonging to the B7 family. This protein has two extracellular IgV- and Ig constant (C)-like domains, wherein the IgV-like domain allows for interaction with the analogous domain of the PD-1 receptor. The cytoplasmic domain of the PD-L1 ligand is short, and its exact role in the transmission of intracellular signals has not yet been determined [21]. The expression of PD-L1 at the mRNA level is detected in almost all cells. The expression of the PD-L1 protein on hematopoietic cells is limited primarily to antigen-presenting cells, such as dendritic cells, macrophages, and B28 lymphocytes. PD-L1 is also expressed in activated T cells [12]. PD-L1 is also found in tissues not belonging to the immune system, including pancreatic islet cells, hepatic stellate cells, vascular endothelial cells and placental trophoblast cells [18,22]. The expression of PD-L1 on B cells is stimulated by anti-IgM antibodies, LPS, type I and II IFNs, TNF and IL-21. In the case of T cells, the inducers of PD-L1 expression are anti-CD3 antibodies or cytokines, such as IL-2, IL-7, IL-15, IFN and TNF. The expression of PD-L1 on macrophages is stimulated by a granulocyte-macrophage-colony-stimulating factor (GM-CSF), monocytes by IL-10, and on DC by IFN-γ, IL-4, IL-12 and GM-CSF [23].
Programmed death-ligand 2 (PD-L2), also referred to as B7-DC and CD273, is the second ligand able to attach to the PD-1 receptor [24]. PD-L2 has extracellular Ig-V- and IgC-like domains, and a short intracellular domain. The expression of PD-L2, as compared with PD-L1, is not as common and is limited to macrophages, DC, and some B-cell subpopulations [25,26,27]. The partial presence of PD-L2 was also demonstrated on mast cells of myeloid origin, T-lymphocytes and vascular endothelial cells [28,29]. The PD-1 receptor interacts with its specific ligands—PD-L1 and PD-L2. Ligands compete with each other for binding to PD-1, but PD-L1 plays a major role in regulating the PD-1/PD-L1/PD-L2 pathway. Although PD-L2 has a stronger affinity for PD-1 compared to PD-L1, the extent of expression of this molecule is limited [30]. The expression of PD-1 on lymphocytes may be induced by the contact of the lymphocyte receptor with an antigen [1,31]. The interaction of PD-1 with ligands results in the activation of phosphotyrosine phosphatase, containing the SH2 domain (SHP2) and the decrease in Bcl-xL expression, leading to the inhibition of phosphatidylinositol 3-kinase/serine-threonine protein kinase—(PI3K/AKT). Functionally, elevated levels of PD-1 expression are observed on tumor-infiltrating lymphocytes (TILs) that interact with tumor cells by linking to the PD-L1 and PD-L2 ligands present on them, which in turn could lead to a lymphocyte depletion. This phenomenon leads to the inhibition of the effector functions of T cells, which lose the cytotoxic ability to kill tumor cells. PD-1 also plays a significant role in T-cell adhesion, which is activated upon contact with APC. The interaction of these cells may be impaired by PD-1-derived inhibitory signals that are necessary for its interaction with PD-L1. This hypothesis is reinforced by experimental in vitro studies showing lower T-cell mobility and improved T-cell interactions with APC after blocking with PD-1 or PD-L1 antibodies [32]. Furthermore, PD-1 could inhibit T-cell adhesion and the formation of the immunological synapse [33,34].
3. PD-1/PD-L1 Expression in Leukemias
The expression of PD-1 in hematological malignancies has been the subject of many studies in recent years. In patients with chronic lymphocytic leukemia (CLL), PD-1 expression is observed on T lymphocytes, but also on leukemic cells [35,36,37]. In addition, we proved earlier that PD-1 expression on leukemic cells in CLL patients was higher compared to the healthy group, both at the level of the transcript and in the form of membrane protein. However, the significance of PD-1 and PD-L1 expression in the prognostic context has not been confirmed [35]. In addition, in CLL patients with an advanced disease (stage III and IV, according to the Rai classification), a higher percentage of CD4+PD-1+ T lymphocytes was observed than in patients with a less advanced disease [38].
In AML, PD-1 expression was observed in T-cell subpopulations, including CD4+ T-effector cells, Tregs and CD8+ T cells, both in untreated patients and in patients with a recurrent disease [39]. An increased PD-1 expression on CD8+ T cells may be one of the factors leading to the dysfunction of cytotoxic T cells and the inhibition of the immune response during the progressive course of AML [40]. Knaus et al. [41] characterized the T-cell exhaustion in AML at diagnosis, that diverged between responders and non-responders upon treatment. Response to therapy correlated with the upregulation of costimulatory T-cell signaling pathways, and the downregulation of inhibitory T-cell signaling pathways, indicative of the restoration of T-cell function. Notably, CD8+ T-cell dysfunction was, in part, reversible upon PD-1 blockade in vitro. By contrast, a similar expression of inhibitory molecules on T cells from patients at AML diagnosis and from age-matched healthy controls were observed [42]. However, when observed at relapse after allogeneic hematopoietic stem-cell transplantation (alloHSCT), the PD-1 expression was significantly increased, compared with its expression at diagnosis, in both CD4+ and CD8+ T cells. Notably, bone marrow CD8 T cells consisted of a higher frequency of PD-1+ cells compared with those from peripheral blood [43]. These cells were also functionally deficient, as was the case in the functional model of WT-1-specific leukemia-reactive CD8+ cells from bone marrow that released lower levels of IFN-γ, granzyme B and TNF, when compared with those from peripheral blood.
Most studies suggest that in newly diagnosed AML patients, PD-L1 expression on blasts is usually not observed [44]. However, it might depend on the detection method, since in one study the PD-L1 was expressed in 24 out of 75 AML patients [45]. The appearance of PD-L1 on AML blasts was associated with the negative course of the disease [44]. The PD-L1 overexpression in AML usually occurred during therapy, after alloHSCT and at the relapse of the disease. The PD-L1 positive rate in the relapsed/refractory group was higher than that in the de novo patient group (56.3% vs. 25.4%, p = 0.019). In 59 de novo patients, the complete remission (CR) rate of the PD-L1 positive group after one course of chemotherapy, was lower than that of the PD-L1 negative group (66.7% vs. 71.4%); the CR rate of PD-L1 positive group after 2 two courses of chemotherapy, was also lower than that of PD-L1 negative group (70% vs. 88.6%). The relapse rate and the proportion of refractory patients in PD-L1 positive group were higher than those in the PD-L1 negative group [45].
The factors that stimulate the expression of PD-L1 in AML were cytokines, particularly IFN-γ [46,47]. In addition, we reported that TP53 might specifically modulate the immune response to tumor antigens by regulating PD-L1 via miR-34 and blocking its expression [48]. The PD-L1 expression was elevated in the AML group with TP53 mut, compared with the TP53 wt group, with a median expression of 9.1 vs. 8.3, p < 0.001. In line with this finding, Goltz et al. [49], analyzing gene methylation status in AML patients, found that low PD-L1 methylation was found in the TP53 mut group. Wang et al. [50] have shown that PD-L1 was overexpressed in the AML samples and that the expression level was reversely correlated with miR-34a expression, that directly targets the 3′ untranslated region of PD-L1, thereby modulating PD-L1 expression. We also found that the highest expression of PD-L1 was in a group with a poor prognosis, compared with favorable and intermediate groups, as defined by The Cancer Genome Atlas research network’s risk stratification. The expression of PD-L1 was also associated with the number of recurrent mutations. Possibly, an increased number of driver mutations created more neoantigens, which in turn, modified the immune microenvironment and caused an increase in PD-L1 expression [51]. The expression of PD-L1 in AML is therefore associated with adverse gene mutations that affect the microenvironment of the tumor and may lead to an unfavorable clinical course of the disease [45].
4. Inhibition of PD-1/PD-L1 in AML
The PD-1/PD-L1/PD-L2 pathway may be inhibited by blocking the PD-1 receptor or its ligands. Blocking the PD-1 molecule itself prevents its interaction with PD-L1 and PD-L2, which is considered the most effective activation of the immune response (Figure 1b). By contrast, PD-L1 blockade affects only the PD-1/PD-L1 axis, and considering its widespread expression, the activation of the immune response might be significant. Due to the limited expression of the PD-L2 ligand, it is not suitable target for therapies that use monoclonal antibodies [52]. Thus, in actions directed toward the PD-1/PD-L1 pathway, anti-PD-1 antibodies, as well as anti-PD-L1, are used [30]. The most common clinical trials employ two PD-1 inhibitors (nivolumab and pembrolizumab) and two anti-PD-L1 monoclonal antibodies (atezolizumab and durvalumab) (Table 1). Several others are under development or are in early phases of clinical trials. The key mechanism of action in anti-PD-1 and anti-PD-L1 monoclonal antibodies, relies on blocking PD-1 interaction with PD-L1 and/or PD-L2 ligands. To minimalize the side effects mediated by the recognition of fragment crystallizable (Fc) region, atezolizumab and durvalumab have a point mutation in the Fc domain; thus, they did not induce the cytotoxicity of the antibody-dependent cellular cytotoxicity, nor the complement-dependent cytotoxicity. The upregulation of checkpoint molecules was observed after alloHSCT and therapy with hypomethylating agents, pointing to a potential clinical application in these settings [53,54]. Moreover, a higher PD-1 expression on T cells was strongly associated with leukemia relapse, post-alloHSCT [55]. This was especially the case in the subpopulation of CD8+ T cells that, characterized by the expression of two exhaustion markers, PD-1 and the T-cell immunoglobulin domain and mucin domain 3 (TIM-3), presented the strongest predictive value for leukemia relapse, post-alloHSCT. The median frequencies of CD8+ PD-1+TIM-3+ in relapsed AML were 8.6%, compared with 0.5% in patients maintaining remission. Notably, the increase of PD-1+ TIM-3+ CD8+ T cells occurred before a clinical diagnosis of leukemia relapse, suggesting their predictive value. This study might provide an early diagnostic approach and a therapeutic target for leukemia relapse, post-transplantation.
The expression of PD-1 is regulated by DNA methylation. The demethylation of the PD-1 promoter correlated with an increase in PD-1 expression. The demethylation of the PD-1 promoter correlated with a significantly worse overall response rate (8% vs. 60%, p = 0.014), and a trend towards a shorter overall survival (OS) (p = 0.11) was observed [56]. In a cohort of patients treated with hypomethylating therapy, PD-L1, PD-L2, PD-1 and CTLA4 expressions were upregulated [54]. The treatment of leukemia cells with a hypomethylating agent, decitabine, resulted in the dose-dependent upregulation of PD-L1, PD-L2, PD-1 and CTLA4. Decitabine could also increase the expression of genes involved in antigen processing and presentation by the respective promoter, demethylation. In a mouse model of colorectal cancer, a significantly larger inhibition of tumor growth and a prolongation of survival were observed after treatment with a combination of PD-1 blockade and decitabine, than in mice treated with decitabine or PD-1 blockade alone [57]. These results suggest that PD-1 signaling may be involved in resistance mechanisms to hypomethylating agents, and provide evidence that checkpoint inhibition could be a potential therapy for treating AML.
In recently published results from a phase II study, relapsed/refractory AML patients were treated with nivolumab and azacytidine [58]. The overall response rate (ORR) was 33%, including 15 (22%) patients with complete remission/complete remission with insufficient count recovery (CRi), one patient with a partial response, and seven patients with hematologic improvements (HI) that were maintained for >6 months. Six patients (9%) exhibited a stable disease for >6 months. The highest ORR (58%) was observed in hypomethylating agent-naive patients. Grades 2–4 immune-related adverse events (irAE) occurred in 16 (23%) patients. Fourteen of the 16 (88%) patients with toxicities responded to steroids, and these 14 patients were safely rechallenged with nivolumab. In this study, a total of 13% of the patients had to discontinue nivolumab (all discontinuations were due to grades 3/4 irAE; no discontinuations were due to grade 2 irAE) and were subsequently kept only on azacitidine.
The preliminary results of a phase-II study (NCT02532231) of nivolumab maintenance in high-risk AML patients, who have achieved CR following induction and consolidation chemotherapy, also showed promising results, with the rates for a 6- and a 12-month CR duration being 79% and 71%, respectively [59]. In a series of case reports, Albring et al. [60] reported three AML patients who, at relapse after alloHSCT, were treated with nivolumab. In one patient, the therapy led to CR; in another, it led to disease stabilization; a third patient failed to respond to nivolumab. The only side effects were the irAE of pancytopenia and a graft-versus-host disease of the skin in one patient, as well as muscle and joint pain in another patient.
5. Future Treatment Modalities with Checkpoint Inhibitors in AML
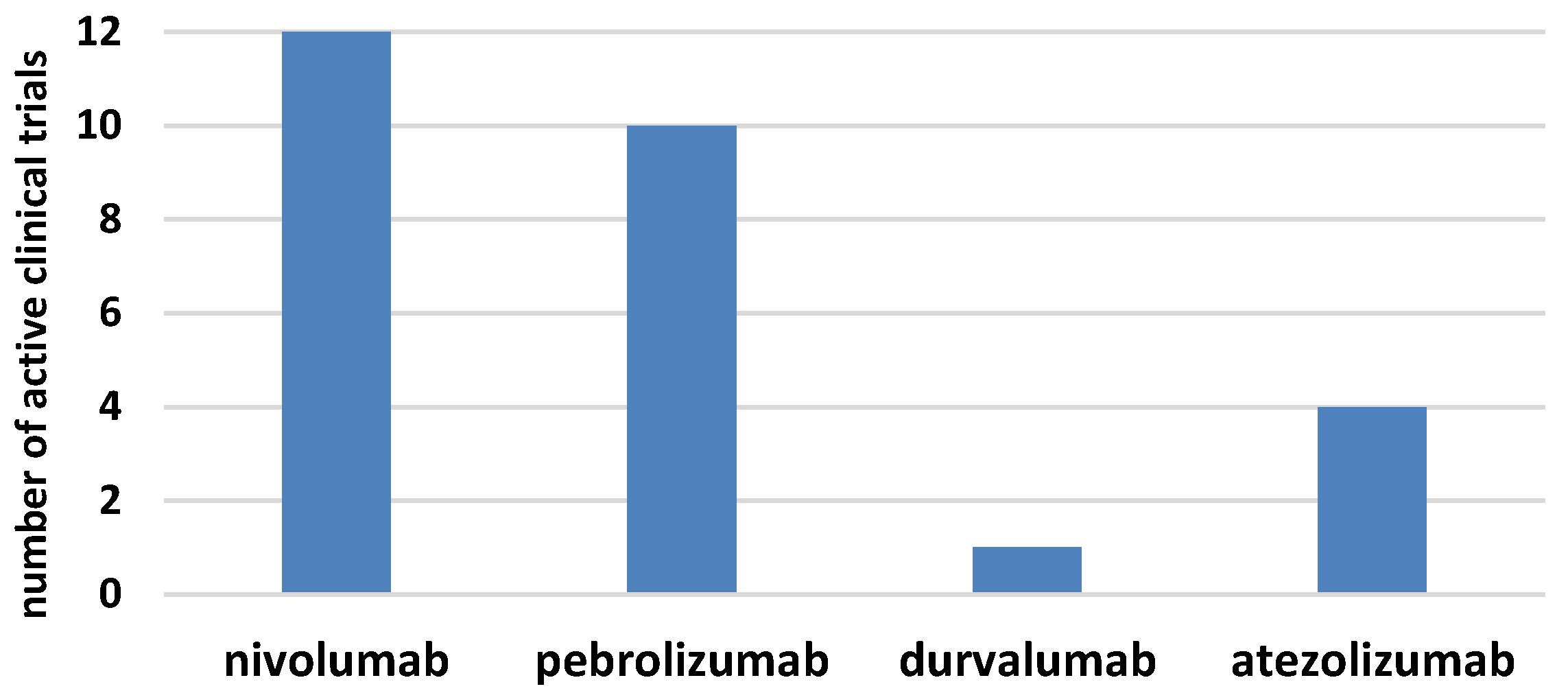
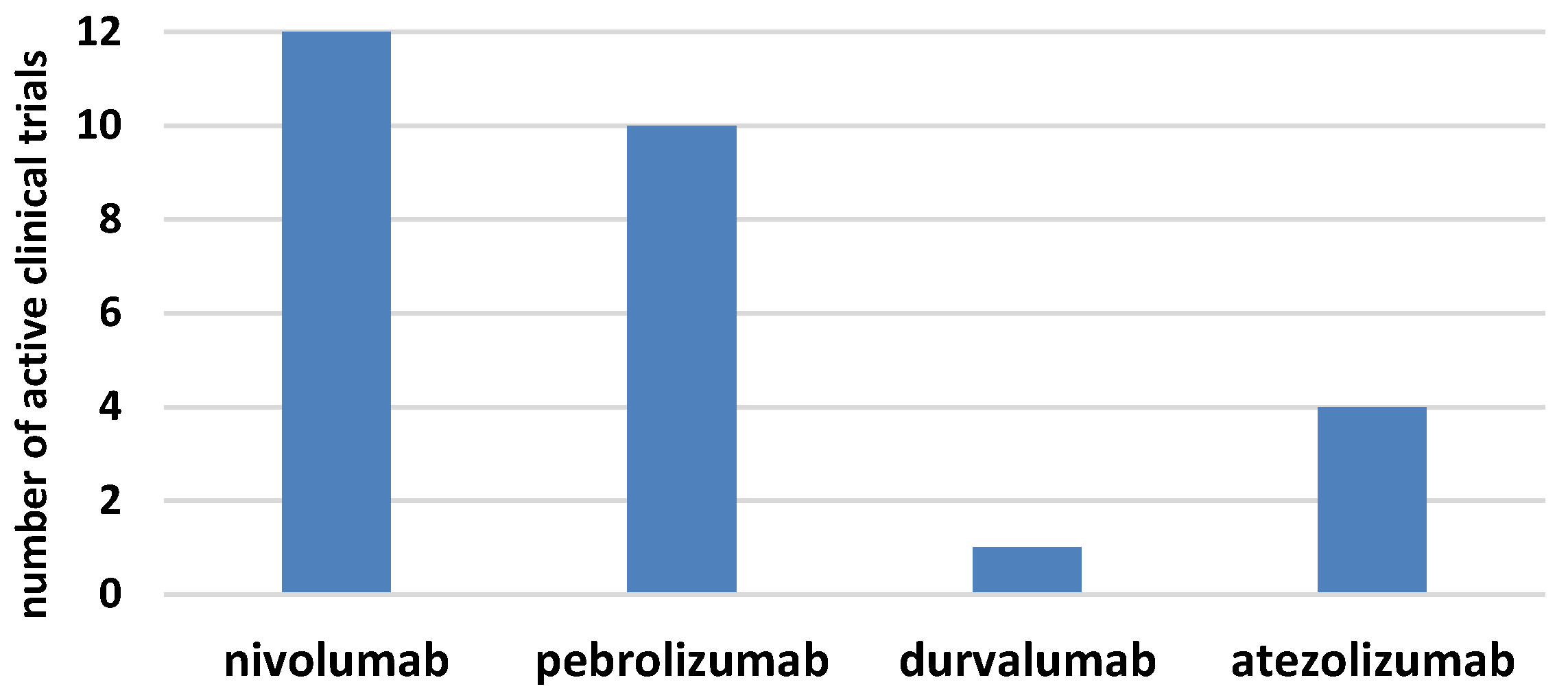
The era of immunotherapy in AML started several years ago when the curative potential of alloHSCT, due to the graft-versus-leukemia effect, was discovered. Nowadays, chimeric antigen receptor (CAR)T-cell therapy replaces the defective immune system and the checkpoint inhibitors restore function to the antileukemia immune response (Figure 1b). Several phases I/II clinical trials for checkpoint inhibitors in monotherapy or combined treatment for AML have started in the last few years (Figure 2) [61,62]. The results of larger clinical trials are needed to determine the role that checkpoint inhibition plays in AML. It seems that the clinical setting can be complex, starting from combined therapy with hypomethylating drugs for patients who are ineligible for transplantation, followed by combined therapy with other modifiers of the immune system and finally, to the augmentation of the graft-versus-leukemia effect, post-alloHSCT, in either monotherapy or polytherapy. In this regard, the double blockade of CTLA-4 and PD-1/PD-L1 presents an interesting treatment option, in boosting antileukemic immunity. Recent results in solid tumors proved that in advanced melanoma, combined therapy was effective in 57% of patients, compared with 43% for patients treated with nivolumab monotherapy and 19% for patients in the ipilimumab monotherapy arm [63]. The first clinical trials on the ipilimumab and nivolumab combined treatment in patients with relapsed AML, post-alloHSCT, have been initiated. Another approach is a double blockade of TIM-3 and PD-1 that is being evaluated in an ongoing clinical trial. Furthermore, checkpoint inhibitors might be effective in combination with cellular therapies, including chimeric antigen receptor T cells (CART) or vaccination strategies [64,65]. This is an evolving field since anti-PD1/PD-L1 therapy along with vaccination could expand the pre-existing specific T cells and induce functionally active antileukemic cytotoxic T cells. The results from clinical trials in melanoma have not provided clear support for this approach, since similar clinical activity was observed irrespective of vaccination [66]. Similarly, PD-1 inhibition in combination with CART did not further enhance the expansion or persistence of CART [64]. Further results from these clinical trials and others are awaited with great interest, as they may allow for the established use of checkpoint inhibitors in the treatment of AML.
Figure 2 displays the number of active clinical trials for checkpoint inhibitors in AML both in monotherapy and in combination with chemotherapy. The histogram presents trials registered on clinicaltrial.gov [62].
Funding
The studies on PD-1/PD-L1 function in AML were supported by the Polish National Science Centre, grant no 2013/10/M/NZ5/00313. The APC was funded by Medical University of Lublin grant No. 462.
Acknowledgments
Author would like to thank Dominika Iwanek and Jacek Przesmycki for technical support in the preparation of figures.
Conflicts of Interest
The author declares no conflict of interest.
References
- Pianko, M.J.; Liu, Y.; Bagchi, S.; Lesokhin, A.M. Immune checkpoint blockade for hematologic malignancies: A review. Stem Cell Investig. 2017, 4, 32. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, C.C.; Jin, L.; Zhang, X.D. Regulation of PD-L1: A novel role of pro-survival signalling in cancer. Ann. Oncol. 2016, 27, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Park, S.J.; Choi, B.K.; Kim, H.H.; Nam, K.O.; Kwon, B.S. 4-1BB promotes the survival of CD8+ T lymphocytes by increasing expression of Bcl-xL and Bfl-1. J. Immunol. 2002, 169, 4882–4888. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, S.; Zollo, O.; Bruscoli, S.; Agostini, M.; Bianchini, R.; Nocentini, G.; Ayroldi, E.; Riccardi, C. GITR, a member of the TNF receptor superfamily, is costimulatory to mouse T lymphocyte subpopulations. Eur. J. Immunol. 2004, 34, 613–622. [Google Scholar] [CrossRef]
- Sehgal, A.; Whiteside, T.L.; Boyiadzis, M. Programmed death-1 checkpoint blockade in acute myeloid leukemia. Expert Opin. Biol. Ther. 2015, 15, 1191–1203. [Google Scholar] [CrossRef] [PubMed]
- Costello, R.T.; Mallet, F.; Sainty, D.; Maraninchi, D.; Gastaut, J.A.; Olive, D. Regulation of CD80/B7-1 and CD86/B7-2 molecule expression in human primary acute myeloid leukemia and their role in allogenic immune recognition. Eur. J. Immunol. 1998, 28, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Graf, M.; Reif, S.; Hecht, K.; Pelka-Fleischer, R.; Kroell, T.; Pfister, K.; Schmetzer, H. High expression of costimulatory molecules correlates with low relapse-free survival probability in acute myeloid leukemia (AML). Ann. Hematol. 2005, 84, 287–297. [Google Scholar] [CrossRef]
- Alatrash, G.; Daver, N.; Mittendorf, E.A. Targeting immune checkpoints in hematologic malignancies. Pharmacol. Rev. 2016, 68, 1014–1025. [Google Scholar] [CrossRef]
- Yamazaki, T.; Akiba, H.; Iwai, H.; Matsuda, H.; Aoki, M.; Tanno, Y.; Shin, T.; Tsuchiya, H.; Pardoll, D.M.; Okumura, K.; et al. Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol. 2002, 169, 5538–5545. [Google Scholar] [CrossRef]
- Shinohara, T.; Taniwaki, M.; Ishida, Y.; Kawaichi, M.; Honjo, T. Structure and chromosomal localization of the human PD-1 gene (PDCD1). Genomics 1994, 23, 704–706. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Dougall, W.C.; Kurtulus, S.; Smyth, M.J.; Anderson, A.C. TIGIT and CD96: New checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 2017, 276, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Latchman, Y.E.; Buhlmann, J.E.; Tomczak, M.F.; Horwitz, B.H.; Freeman, G.J.; Sharpe, A.H. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur. J. Immunol. 2003, 33, 2706–2716. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, H.; Agata, Y.; Kawasaki, A.; Sato, M.; Imamura, S.; Minato, N.; Yagita, H.; Nakano, T.; Honjo, T. Developmentally regulated expression of the PD-1 protein on the surface of double-negative (CD4-CD8-) thymocytes. Int. Immunol. 1996, 8, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Kawano, S.; Hatachi, S.; Kurimoto, C.; Okazaki, T.; Iwai, Y.; Honjo, T.; Tanaka, Y.; Minato, N.; Komori, T.; et al. Enhanced expression of programmed death-1 (PD-1)/PD-L1 in salivary glands of patients with Sjögren’s syndrome. J. Rheumatol. 2005, 32, 2156–2163. [Google Scholar] [PubMed]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef] [Green Version]
- Oestreich, K.J.; Yoon, H.; Ahmed, R.; Boss, J.M. NFATc1 regulates PD-1 expression upon T cell activation. J. Immunol. 2008, 181, 4832–4839. [Google Scholar] [CrossRef]
- Yao, S.; Wang, S.; Zhu, Y.; Luo, L.; Zhu, G.; Flies, S.; Xu, H.; Ruff, W.; Broadwater, M.; Choi, I.H.; et al. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood 2009, 113, 5811–5818. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.Y.; Lee, S.W.; Seo, S.K.; Choi, I.W.; Choi, I.; Lee, S.W. Interferon-sensitive response element (ISRE) is mainly responsible for IFN-alpha-induced upregulation of programmed death-1 (PD-1) in macrophages. Biochim. Biophys. Acta 2008, 1779, 811–819. [Google Scholar] [CrossRef]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 2017, 24, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, R.; Morrow, M.; Hammond, S.A.; Mulgrew, K.; Marcus, D.; Poon, E.; Watkins, A.; Mullins, S.; Chodorge, M.; Andrews, J.; et al. Identification and Characterization of MEDI4736, an Antagonistic Anti-PD-L1 Monoclonal Antibody. Cancer Immunol. Res. 2015, 3, 1052–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinter, A.L.; Godbout, E.J.; McNally, J.P.; Sereti, I.; Roby, G.A.; O’Shea, M.A.; Fauci, A.S. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J. Immunol. 2008, 181, 6738–6746. [Google Scholar] [CrossRef] [PubMed]
- Messal, N.; Serriari, N.E.; Pastor, S.; Nunès, J.A.; Olive, D. PD-L2 is expressed on activated human T cells and regulates their function. Mol. Immunol. 2011, 48, 2214–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkes, E.A.; Grigg, A.; Chong, G. Programmed cell death-1 inhibition in lymphoma. Lancet Oncol. 2015, 16, e234–e245. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Tumang, J.R.; Gao, W.; Bai, C.; Rothstein, T.L. PD-L2 expression extends beyond dendritic cells/macrophages to B1 cells enriched for V(H)11/V(H)12 and phosphatidylcholine binding. Eur. J. Immunol. 2007, 37, 2405–2410. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Salmaninejad, A.; Khoramshahi, V.; Azani, A.; Soltaninejad, E.; Aslani, S.; Zamani, M.R.; Zal, M.; Nesaei, A.; Hosseini, S.M. PD-1 and cancer: Molecular mechanisms and polymorphisms. Immunogenetics 2018, 70, 73–86. [Google Scholar] [CrossRef]
- Ohaegbulam, K.C.; Assal, A.; Lazar-Molnar, E.; Yao, Y.; Zang, X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol. Med. 2015, 21, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Pauken, K.E.; Eagar, T.N.; Obu, T.; Wu, J.; Tang, Q.; Azuma, M.; Krummel, M.F.; Bluestone, J.A. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immunol. 2009, 10, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Sari, D.; Boussiotis, V.A. PD-1 inhibits T cell proliferation by upregulating p27 and p15 and suppressing Cdc25A. Cell Cycle 2012, 11, 4305–4309. [Google Scholar] [CrossRef] [Green Version]
- Zinselmeyer, B.H.; Heydari, S.; Sacristán, C.; Nayak, D.; Cammer, M.; Herz, J.; Cheng, X.; Davis, S.J.; Dustin, M.L.; McGavern, D.B. PD-1 promotes immune exhaustion by inducing antiviral T cell motility paralysis. J. Exp. Med. 2013, 210, 757–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzywnowicz, M.; Zaleska, J.; Mertens, D.; Tomczak, W.; Wlasiuk, P.; Kosior, K.; Piechnik, A.; Bojarska-Junak, A.; Dmoszynska, A.; Giannopoulos, K. Programmed death-1 and its ligand are novel immunotolerant molecules expressed on leukemic B cells in chronic lymphocytic leukemia. PLoS ONE 2012, 7, e35178. [Google Scholar] [CrossRef] [PubMed]
- Xerri, L.; Chetaille, B.; Serriari, N.; Attias, C.; Guillaume, Y.; Arnoulet, C.; Olive, D. Programmed death 1 is a marker of angioimmunoblastic T-cell lymphoma and B-cell small lymphocytic lymphoma/chronic lymphocytic leukemia. Hum. Pathol. 2008, 39, 1050–1058, Erratum in 2010, 41, 1655. [Google Scholar] [CrossRef] [PubMed]
- Grzywnowicz, M.; Karabon, L.; Karczmarczyk, A.; Zajac, M.; Skorka, K.; Zaleska, J.; Wlasiuk, P.; Chocholska, S.; Tomczak, W.; Bojarska-Junak, A.; et al. The function of a novel immunophenotype candidate molecule PD-1 in chronic lymphocytic leukemia. Leuk. Lymphoma 2015, 56, 2908–2913. [Google Scholar] [CrossRef] [PubMed]
- Rusak, M.; Eljaszewicz, A.; Bołkun, Ł.; Łuksza, E.; Łapuć, I.; Piszcz, J.; Singh, P.; Dąbrowska, M.; Bodzenta-Łukaszyk, A.; Kłoczko, J.; et al. Prognostic significance of PD-1 expression on peripheral blood CD4+ T cells in patients with newly diagnosed chronic lymphocytic leukemia. Pol. Arch. Med. Wewn. 2015, 125, 553–559. [Google Scholar] [CrossRef]
- Williams, P.; Basu, S.; Garcia-Manero, G.; Hourigan, C.S.; Oetjen, K.A.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Al-Hamal, Z.; Konopleva, M.; et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer 2018. [Google Scholar] [CrossRef]
- Tan, J.; Chen, S.; Lu, Y.; Yao, D.; Xu, L.; Zhang, Y.; Yang, L.; Chen, J.; Lai, J.; Yu, Z.; et al. Higher PD-1 expression concurrent with exhausted CD8+ T cells in patients with de novo acute myeloid leukemia. Chin. J. Cancer Res. 2017, 29, 463–470. [Google Scholar] [CrossRef] [Green Version]
- Knaus, H.A.; Berglund, S.; Hackl, H.; Blackford, A.L.; Zeidner, J.F.; Montiel-Esparza, R.; Mukhopadhyay, R.; Vanura, K.; Blazar, B.R.; Karp, J.E.; et al. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI Insight. 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Schnorfeil, F.M.; Lichtenegger, F.S.; Emmerig, K.; Schlueter, M.; Neitz, J.S.; Draenert, R.; Hiddemann, W.; Subklewe, M. T cells are functionally not impaired in AML: Increased PD-1 expression is only seen at time of relapse and correlates with a shift towards the memory T cell compartment. J. Hematol. Oncol. 2015, 8, 93. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Wang, L.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; Mineishi, S.; Rizvi, S.; Shike, H.; Bayerl, M.; Schell, T.D.; et al. Bone marrow CD8 T cells express high frequency of PD-1 and exhibit reduced anti-leukemia response in newly diagnosed AML patients. Blood Cancer J. 2018, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Annibali, O.; Crescenzi, A.; Tomarchio, V.; Pagano, A.; Bianchi, A.; Grifoni, A.; Avvisati, G. PD-1 /PD-L1 checkpoint in hematological malignancies. Leuk. Res. 2018, 67, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.F.; Zhang, Q.T.; Xin, H.Z.; Gan, S.L.; Ma, J.; Liu, Y.F.; Xie, X.S.; Sun, H. Expression of Programmed Death Ligand-1 (PD-L1) in Human Acute Leukemia and Its Clinical Significance. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2015, 23, 930–934. [Google Scholar] [CrossRef] [PubMed]
- Krönig, H.; Kremmler, L.; Haller, B.; Englert, C.; Peschel, C.; Andreesen, R.; Blank, C.U. Interferon-induced programmed death-ligand 1 (PD-L1/B7-H1) expression increases on human acute myeloid leukemia blast cells during treatment. Eur. J. Haematol. 2014, 92, 195–203. [Google Scholar] [CrossRef]
- Jelinek, T.; Mihalyova, J.; Kascak, M.; Duras, J.; Hajek, R. PD-1/PD-L1 inhibitors in haematological malignancies: Update 2017. Immunology 2017, 152, 357–371. [Google Scholar] [CrossRef]
- Zajac, M.; Zaleska, J.; Dolnik, A.; Bullinger, L.; Giannopoulos, K. Expression of CD274 (PD-L1) is associated with unfavourable recurrent mutations in AML. Br. J. Haematol. 2018, 183, 822–825. [Google Scholar] [CrossRef]
- Goltz, D.; Gevensleben, H.; Grünen, S.; Dietrich, J.; Kristiansen, G.; Landsberg, J.; Dietrich, D. PD-L1 (CD274) promoter methylation predicts survival in patients with acute myeloid leukemia. Leukemia 2017, 31, 738–743. [Google Scholar] [CrossRef]
- Wang, X.; Li, J.; Dong, K.; Lin, F.; Long, M.; Ouyang, Y.; Wei, J.; Chen, X.; Weng, Y.; He, T.; et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal 2015, 27, 443–452. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. D-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A.; Jin, J.Y.; Lemaire, V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann. Oncol. 2018, 29, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chang, Y.J.; Xu, L.P.; Zhang, X.H.; Wang, Y.; Liu, K.Y.; Huang, X.J. Reversal of T Cell Exhaustion by the First Donor Lymphocyte Infusion Is Associated with the Persistently Effective Antileukemic Responses in Patients with Relapsed AML after Allo-HSCT. Biol. Blood Marrow Transplant. 2018, 24, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Bueso-Ramos, C.; DiNardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Zhang, J.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; Zhu, L.; Zeng, H.; Schell, T.D.; Zheng, H. PD-1(hi)TIM-3(+) T cells associate with and predict leukemia relapse in AML patients post allogeneic stem cell transplantation. Blood Cancer J. 2015, 5, e330. [Google Scholar] [CrossRef] [PubMed]
- Ørskov, A.D.; Treppendahl, M.B.; Skovbo, A.; Holm, M.S.; Friis, L.S.; Hokland, M.; Grønbæk, K. Hypomethylation and up-regulation of PD-1 in T cells by azacytidine in MDS/AML patients: A rationale for combined targeting of PD-1 and DNA methylation. Oncotarget. 2015, 6, 9612–9626. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wu, Y.; Wang, W.; Xu, J.; Lv, X.; Cao, X.; Wan, T. Low-dose decitabine enhances the effect of PD-1 blockade in colorectal cancer with microsatellite stability by re-modulating the tumor microenvironment. Cell Mol. Immunol. 2018. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.M.; et al. Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Non-randomized, Open-label, Phase 2 Study. Cancer Discov. 2018. [Google Scholar] [CrossRef]
- Kadia, T.M.; Cortes, J.E.; Ghorab, A.; Ravandi, F.; Jabbour, E.; Daver, N.G.; Alvarado, Y.; Ohanian, M.; Konopleva, M.; Kantarjian, H.M. Nivolumab (Nivo) maintenance (maint) in high-risk (HR) acute myeloid leukemia (AML) patients. J. Clin. Oncol. 2018, 36 (Suppl. 15), 7014. [Google Scholar] [CrossRef]
- Albring, J.C.; Inselmann, S.; Sauer, T.; Schliemann, C.; Altvater, B.; Kailayangiri, S.; Rössig, C.; Hartmann, W.; Knorrenschild, J.R.; Sohlbach, K.; et al. PD-1 checkpoint blockade in patients with relapsed AML after allogeneic stem cell transplantation. Bone Marrow Transplant. 2017, 52, 317–320. [Google Scholar] [CrossRef]
- Liu, Y.; Bewersdorf, J.P.; Stahl, M.; Zeidan, A.M. Immunotherapy in acute myeloid leukemia and myelodysplastic syndromes: The dawn of a new era? Blood Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 11 January 2019).
- Larkin, J.; Hodi, F.S.; Wolchok, J.D. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 1270–1271. [Google Scholar] [CrossRef] [PubMed]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Kudchadkar, R.R.; DeConti, R.C.; Thebeau, M.S.; Czupryn, M.P.; Tetteh, L.; Eysmans, C.; Richards, A.; Schell, M.J.; Fisher, K.J.; et al. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin. Cancer Res. 2015, 21, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Kudchadkar, R.R.; Yu, B.; Gallenstein, D.; Horak, C.E.; Inzunza, H.D.; Zhao, X.; Martinez, A.J.; Wang, W.; Gibney, G.; et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J. Clin. Oncol. 2013, 31, 4311–4318. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(a) The regulation of antileukemic immune response; the activation of an immune response requires two signals. The first is responsible for the specific recognition of antigen (peptide) located in the great groove of the major histocompatibility complex (MHC) by the T-cell receptor (TCR). The second is the costimulatory signal that is transmitted by the interaction of CD28 and B7 molecules. These signals are regulated by many negative receptors including the lymphocyte-activation gene 3 (LAG3), OX40, programmed cell death protein 1 receptor (PD-1), CD43 as well as T-cell immunoglobulin domain and mucin domain 3 (TIM3) on the T cell that interacts with ligands on acute myeloid leukemia (AML) blasts, i.e., CD47, programmed death-ligand 1 (PD-L1) and Cytotoxic T-lymphocyte Antigen-4 (CTLA-4). (b) The restoration of antileukemic immune response by the targeting of the negative control checkpoints. In order to restore the antileukemic immune response, two inhibitory immune checkpoint molecules might be targeted by treatment with specific monoclonal antibodies directed against the cytotoxic T-lymphocyte antigen-4 (CTLA-4), ipilimumab, and the programmed cell death protein 1, nivolumab and pembrolizumab as well as PD-L1, atezolizumab and durvalumab.
Figure 1.
(a) The regulation of antileukemic immune response; the activation of an immune response requires two signals. The first is responsible for the specific recognition of antigen (peptide) located in the great groove of the major histocompatibility complex (MHC) by the T-cell receptor (TCR). The second is the costimulatory signal that is transmitted by the interaction of CD28 and B7 molecules. These signals are regulated by many negative receptors including the lymphocyte-activation gene 3 (LAG3), OX40, programmed cell death protein 1 receptor (PD-1), CD43 as well as T-cell immunoglobulin domain and mucin domain 3 (TIM3) on the T cell that interacts with ligands on acute myeloid leukemia (AML) blasts, i.e., CD47, programmed death-ligand 1 (PD-L1) and Cytotoxic T-lymphocyte Antigen-4 (CTLA-4). (b) The restoration of antileukemic immune response by the targeting of the negative control checkpoints. In order to restore the antileukemic immune response, two inhibitory immune checkpoint molecules might be targeted by treatment with specific monoclonal antibodies directed against the cytotoxic T-lymphocyte antigen-4 (CTLA-4), ipilimumab, and the programmed cell death protein 1, nivolumab and pembrolizumab as well as PD-L1, atezolizumab and durvalumab.
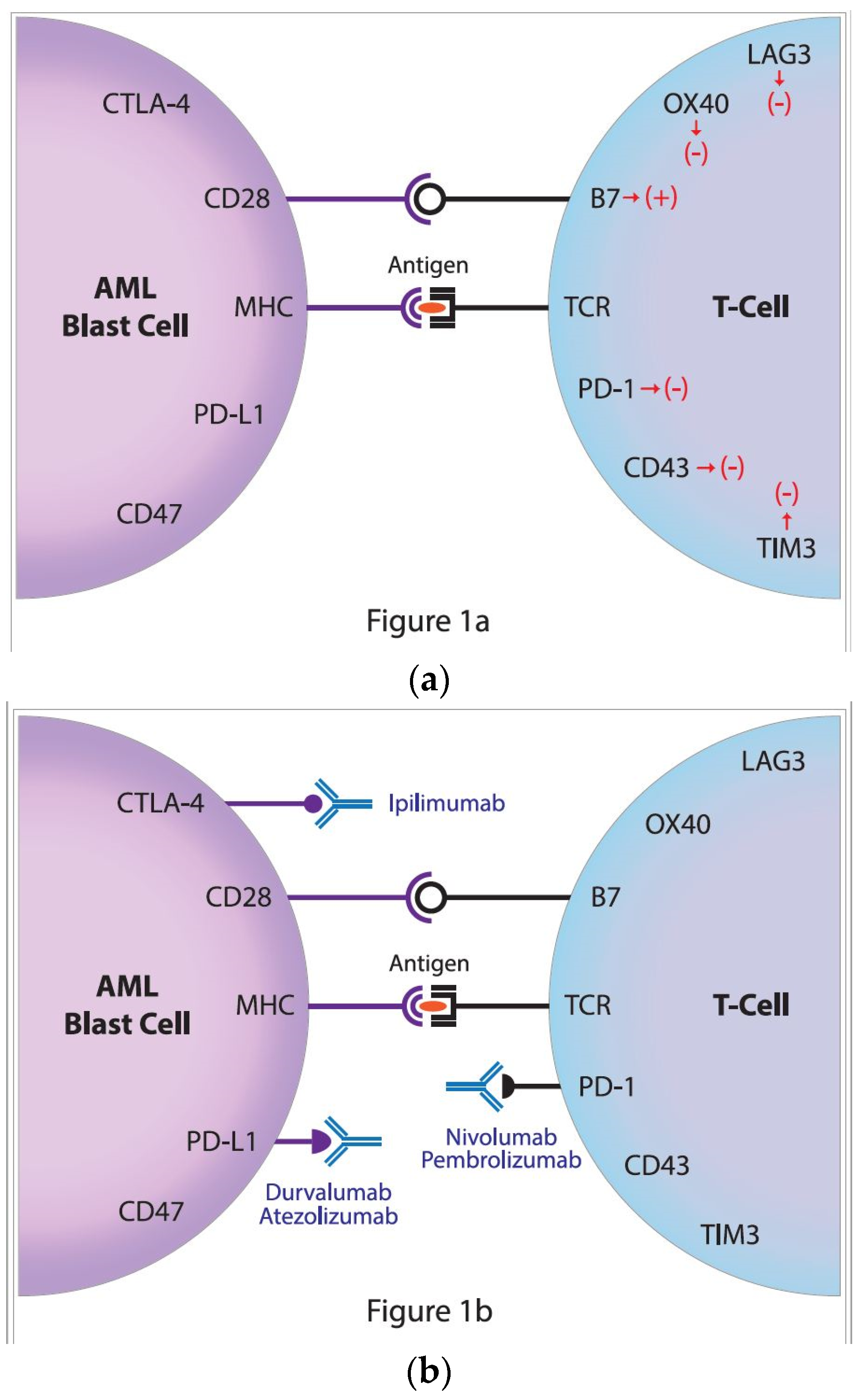
Figure 2.
The numbers of active clinical trials for checkpoints inhibitors in acute myeloid leukemia.
Figure 2.
The numbers of active clinical trials for checkpoints inhibitors in acute myeloid leukemia.

{kind=link}
{kind=link}
Table 1.
The first agents for PD-1/PD-L1 inhibition in AML.
| Monoclonal Antibody | Target/Type | First Registration |
|---|---|---|
| Nivolumab | PD-1, IgG4 | Melanoma—06.2015 |
| Pembrolizumab | PD-1, IgG4 | Melanoma—08.2014 |
| Atezolizumab | PD-L1, IgG1 modified Fc region | Urothelial carcinoma—05.2016 |
| Durvalumab | PD-L1, IgG1 modified Fc region | Urothelial carcinoma—05.2017 |
PD-1: programmed cell death protein 1 receptor; PD-L1: programmed death-ligand 1; AML: acute myeloid leukemia; IgG: Intravenous Gamma Globulin.
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Giannopoulos, K. Targeting Immune Signaling Checkpoints in Acute Myeloid Leukemia. J. Clin. Med. 2019, 8, 236. https://doi.org/10.3390/jcm8020236
AMA Style
Giannopoulos K. Targeting Immune Signaling Checkpoints in Acute Myeloid Leukemia. Journal of Clinical Medicine. 2019; 8(2):236. https://doi.org/10.3390/jcm8020236
Chicago/Turabian StyleGiannopoulos, Krzysztof. 2019. "Targeting Immune Signaling Checkpoints in Acute Myeloid Leukemia" Journal of Clinical Medicine 8, no. 2: 236. https://doi.org/10.3390/jcm8020236
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.