Risk Stratification on Pheochromocytoma and Paraganglioma from Laboratory and Clinical Medicine

Abstract
1. Introduction
2. Pathologic Risk Stratification
2.1. GAPP
2.2. Catecholamine Type and Metastasis
3. Molecular Risk Stratification
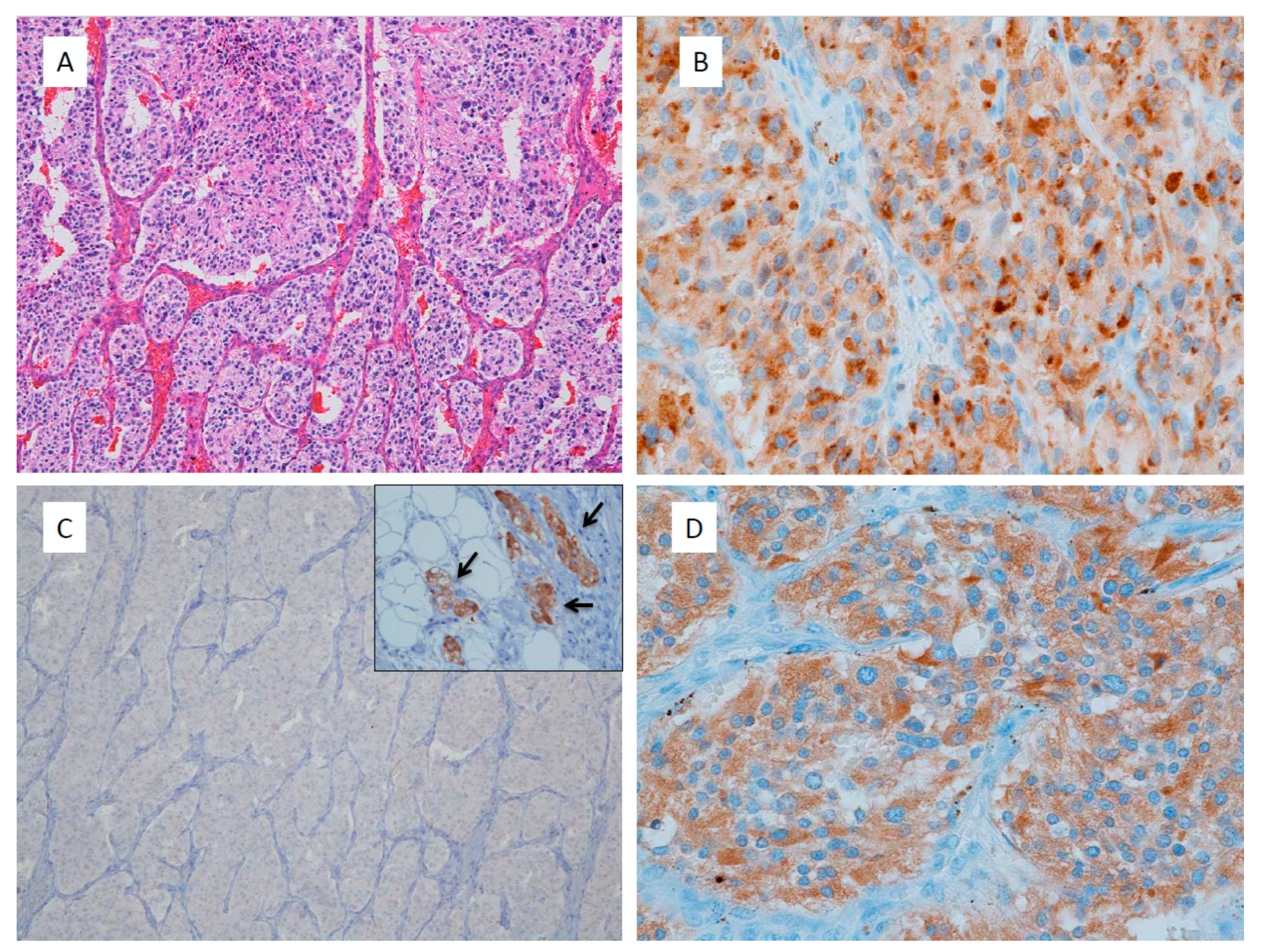
3.1. Immunohistochemistry for Gene Mutations
3.2. Differential Diagnosis and Risk Stratification
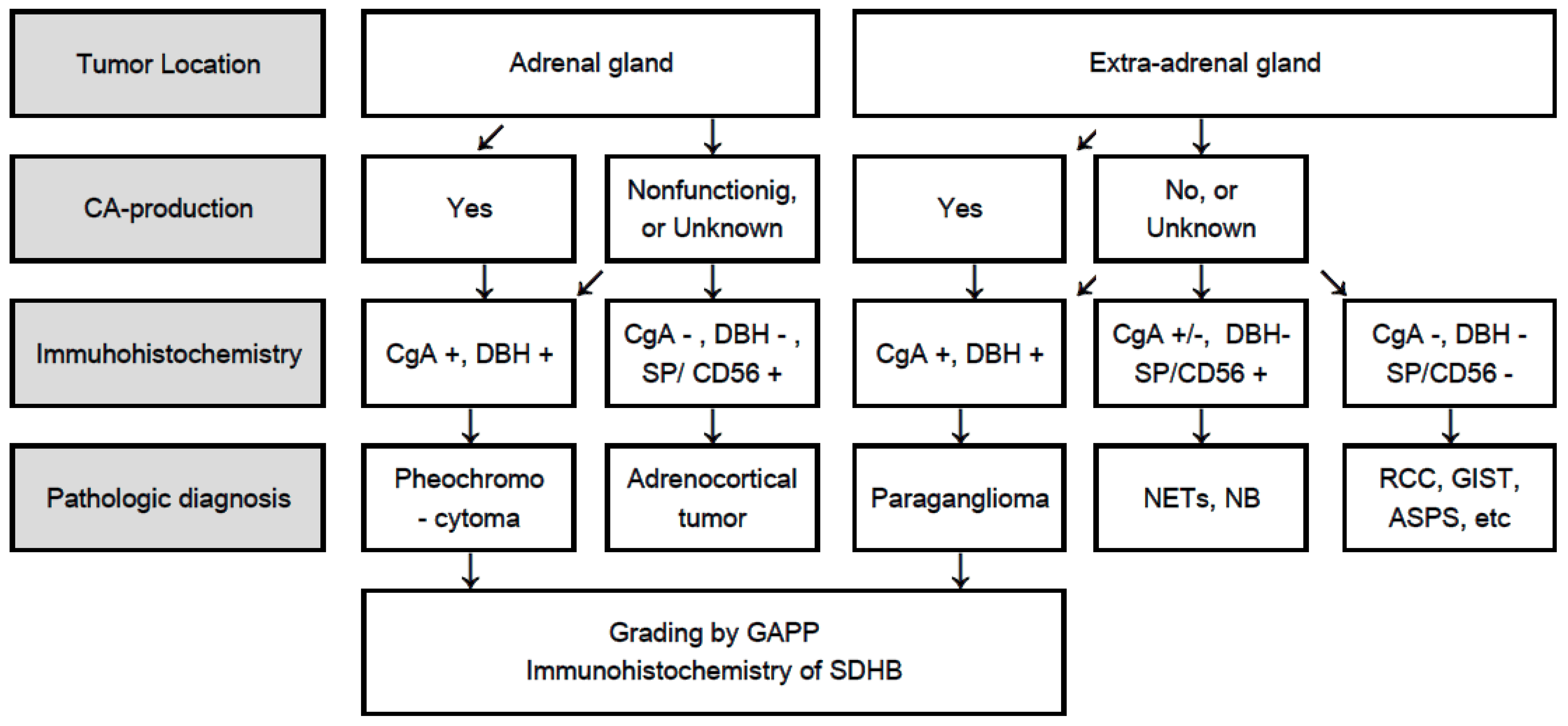
4. Flowchart for Differential Diagnosis
5. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tischler, A.S.; de Krijger, R.R. Phaeochromocytoma. In WHO Classification of Tumors of Endocrine Organs, 4th ed.; Lloyd, R.V., Osamura, R.Y., Kloppel, G., Eds.; IARC Press: Lyons, France, 2017; pp. 183–189. ISBN 978-92-832-4493-6. [Google Scholar]
- Kimura, N.; Capella, C. Extraadrenal paraganglioma. In WHO Classification of Tumors of Endocrine Organs, 4th ed.; Lloyd, R.V., Osamura, R.Y., Kloppel, G., Eds.; IARC Press: Lyons, France, 2017; pp. 190–195. ISBN 978-92-832-4493-6. [Google Scholar]
- Bellingham, G.A.; Dhir, A.K.; Luke, P.P. Case report: Retroperitoneal endooscopic pheochromocytoma removal in an adult with Eisenmenger’s syndrome. Can. J. Anaesth. 2008, 55, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Goffredo, P.; Sosa, J.A.; Roman, S.A. Malignant pheochromocytoma and paraganglioma: A population level analysis of long-term survival over two decades. J. Surg. Oncol. 2013, 107, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Harari, A.; Inabnet, W.B., 3rd. Malignant pheochromocytoma: A review. Am. J. Surg. 2011, 201, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Korevaar, T.I.; Grossman, A.B. Pheochromocytomas and paragangliomas: Assessment of malignant potential. Endocrine 2011, 40, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Neville, A.M. The adrenal medulla. In Functional Pathology of the Human Adrenal Gland; Symington, T., Ed.; E&S Livingstone Ltd.: Edinburgh, UK, 1969; pp. 219–324. [Google Scholar]
- Thompson, L.D.; Young, W.F.; Kawashima, A.; Komminoth, P. Malignant adrenal phaeochromocytoma. In World Health Organization Classification of Tumours Pathology & Genetics Tumours of Endocrine Organs, 3rd ed.; DeLellis, R.A., Lloyd, R.V., Eds.; IARC: Lyon, France, 2004; pp. 147–150. [Google Scholar]
- Roman-Gonzalez, A.; Jimenez, C. Malignant pheochromocytoma-paraganglioma: Pathogenesis, TNM staging, and current clinical trials. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Plouin, P.F.; Amar, L.; Dekkers, O.M.; Fassnacht, M.; Gimenez-Roqueplo, A.P.; Lenders, J.W.M.; Lussey-Lepoutre, C.; Steichen, O. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur. J. Endocrinol. 2016, 174, G1–G10. [Google Scholar] [CrossRef] [PubMed]
- Naruse, M.; PHEO-J Study Group. Nationwide survey and PHEO network for the study of pheochromocytoma/paraganglioma in Japan (PHEO-J). In Proceedings of the Endocrine Society’s 93rd Annual Meeting & Expo, Boston, MA, USA, 4–7 June 2011; pp. 2–631. [Google Scholar]
- Amar, L.; Lussey-Lepoutre, C.; Lenders, J.W.; Djadi-Prat, J.; Plouin, P.F.; Steichen, O. Management of endocrine disease: Recurrence or new tumors after complete resection of pheochromocytomas and paragangliomas: A systematic review and meta-analysis. Eur. J. Endocrinol. 2016, 175, R135–R145. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.D. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: A clinicopathologic and immunophenotypic study of 100 cases. Am. J. Surg. Pathol. 2002, 26, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Tischler, A.S.; Lloyd, R.V.; DeLellis, R.A.; de Krijger, R.; van Nederveen, F.; Vania, N. Observer variation in the application of the pheochromocytoma of the adrenal gland scaled score. Am. J. Surg. Pathol. 2009, 33, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Takayanagi, R.; Takizawa, N.; Itagaki, E.; Katabami, T.; Kakoi, N.; Rakugi, H.; Ikeda, Y.; Tanabe, A.; Nigawara, T.; et al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr.-Relat. Cancer 2014, 21, 405–414. [Google Scholar] [PubMed]
- Hamidi, O.; Young, W.F., Jr.; Iniguez-Ariza, N.M.; Kittah, N.E.; Gruber, L.; Bancos, C.; Tamhane, S.; Irina Bancos, I. Malignant pheochromocytoma and paraganglioma: 272 Patients Over 55 Years. J. Clin. Endocrinol. Metab. 2017, 102, 3296–3305. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.M.; Ahn, S.H.; Kim, H.; Kim, B.J.; Sung, T.Y.; Kim, Y.H.; Hong, S.J.; Song, D.E.; Lee, S.H. Validation of pathological grading systems for predicting metastatic potential in pheochromocytoma and paraganglioma. PLoS ONE 2017, 12, e0187398. [Google Scholar] [CrossRef] [PubMed]
- van der Harst, E.; de Herder, W.W.; de Krijger, R.R.; Bruining, H.A.; Bonjer, H.J.; Lamberts, S.W.; van den Meiracker, A.H.; Stijnen, T.H.; Boomsma, F. The value of plasma markers for the clinical behaviour of phaeochromocytomas. Eur. J. Endocrinol. 2002, 147, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Tischler, A.; de Krijger, R.R. Diagnostic tests and biomarkers for pheochromocytoma and extra-adrenal paraganglioma: From routine laboratory methods to disease stratification. Endocr. Pathol. 2012, 23, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Miura, Y.; Nagatsu, I.; Nagura, H. Catecholamine synthesizing enzymes in 70 cases of functioning and non-functioning phaeochromocytoma and extra-adrenal paraganglioma. Virchows Arch. 1992, 421, 25–32. [Google Scholar] [CrossRef]
- King, K.S.; Pacak, K. Familial pheochromocytomas and paragangliomas. Mol. Cell Endocrinol. 2014, 386, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Dahia, P.L.M. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat. Rev. Cancer 2014, 14, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Roqueplo, A.P.; Dahia, P.L.; Robledo, M. Update in the genetics of paraganglioma and pheochromocytoma and hereditary syndromes. Horm. Metab. Res. 2012, 44, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Baysal, B.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Drovdlic, C.M.; Savul, S.A.; McLeod, D.R.; Yee, H.A.; Brackmann, D.E.; Slattery, W.H., III; Myers, E.N.; et al. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. J. Med. Genet. 2002, 39, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.P.; Bausch, B.; McWhinney, S.R.; Bender, B.U.; Gimm, O.; Franke, G.; Schipper, J.; Klisch, J.; Altehoefer, C.; Zerres, K.; et al. Germ-line mutations in nonsyndromic pheochromocytoma. New Engl. J. Med. 2002, 346, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Benn, D.E.; Robinson, B.G.; Clifton-Bligh, R.J. 15 years of paraganglioma: Clinical manifestations of paraganglioma syndromes types 1-5. Endocr. Relat. Cancer 2015, 22, T91–T103. [Google Scholar] [CrossRef] [PubMed]
- Tischler, A.S. Familial paraganglioma-phaeochromocytoma syndromes caused by SDHB, SDHC, and SDHD mutations. In WHO Classification of Tumors of Endocrine Organs, 4th ed.; Lloyd, R.V., Osamura, R.Y., Kloppel, G., Eds.; IARC Press: Lyons, France, 2017; pp. 262–265. ISBN 978-92-832-4493-6. [Google Scholar]
- van Nederveen, F.H.; Gaal, J.; Favier, J.; Korpershoek, E.; Oldenburg, R.A.; de Bruyn, E.M.; Sleddens, H.F.; Derkx, P.; Rivière, J.; Dannenberg, H.; et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: A retrospective and prospective analysis. Lancet Oncol. 2009, 10, 764–771. [Google Scholar] [CrossRef]
- Papathomas, T.G.; Oudijk, L.; Persu, A.; Gill, A.J.; van Nederveen, F.; Tischler, A.S.; Tissier, F.; Volante, M.; Matias-Guiu, X.; Smid, M.; et al. SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: A multicenter interobserver variation analysis using virtual microscopy: A Multinational Study of the European Network for the Study of Adrenal Tumors (ENS@T). Mod. Pathol. 2015, 28, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Gill, A.J.; Benn, D.E.; Chou, A.; Clarkson, A.; Muljono, A.; Meyer-Rochow, G.Y.; Richardson, A.L.; Sidhu, S.B.; Robinson, B.G.; Clifton-Bligh, R.J. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC and SDHD in paraganglioma–pheochromocytoma. Hum. Pathol. 2010, 41, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Takekoshi, K.; Horii, A.; Morimoto, R.; Imai, T.; Oki, Y.; Saito, T.; Midorikawa, S.; Arao, T.; Sugisawa, C.; et al. Clinicopathological study of SDHB mutation-related pheochromocytoma and sympathetic paraganglioma. Endocr. Relat. Cancer 2014, 21, L13–L16. [Google Scholar] [CrossRef] [PubMed]
- Dahia, P.L.; Ross, K.N.; Wright, M.E.; Hayashida, C.Y.; Santagata, S.; Barontini, M.; Kung, A.L.; Sanso, G.; Powers, J.F.; Tischler, A.S.; et al. A HIF1a regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005, 1, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Pollard, P.J.; El-Bahrawy, M.; Poulsom, R.; Elia, G.; Killick, P.; Kelly, G.; Hunt, T.; Jeffery, R.; Seedhar, P.; Barwell, J.; et al. Expression of HIF-1, HIF-2(EPAS1), and their target genes in paraganglioma and pheochromocytoma with VHL and SDH mutations. J. Clin. Endocrinol. Metab. 2006, 91, 4593–4598. [Google Scholar] [CrossRef] [PubMed]
- Korpershoek, E.; Favier, J.; Gaal, J.; Burnichon, N.; van Gessel, B.; Oudijk, L.; Badoual, C.; Gadessaud, N.; Venisse, A.; Bayley, J.P.; et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J. Clin. Endocrinol. Metab. 2011, 96, E1472–E1476. [Google Scholar] [CrossRef] [PubMed]
- Comino-Méndez, I.; Gracia-Aznárez, F.J.; Schiavi, F.; Landa, I.; Leandro-García, L.J.; Letón, R.; Honrado, E.; Ramos-Medina, R.; Caronia, D.; Pita, G.; et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat. Genet. 2011, 43, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.A. pathologist’s view: Molecular profiles for diagnosing pheochromocytomas and paragangliomas. Int. J. Endo. Oncol. 2015, 2, 193–200. [Google Scholar] [CrossRef]
- Nolting, S.; Grossman, A.B. Signaling pathways in pheochromocytomas and paragangliomas: Prospects for future therapies. Endocr. Pathol. 2012, 23, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Favier, J.; Igaz, P.; Burnichon, N.; Amar, L.; Libé, R.; Badoual, C.; Tissier, F.; Bertherat, J.; Plouin, P.F.; Jeunemaitre, X.; et al. Rationale for anti-angiogenic therapy in pheochromocytoma and paraganglioma. Endocr. Pathol. 2012, 23, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Timmers, H.J.L.M.; Pacak, K.; Huynh, T.T.; Abu-Asab, M.; Tsokos, M.; Merino, M.J.; Baysal, B.E.; Adams, K.T.; Eisenhofer, G. Biochemically silent abdominal paragangliomas in patients with mutations in the succinate dehydrogenase subunit B gene. J. Clin. Endocrinol. Metab. 2008, 93, 4826–4832. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Kimura, N.; Yoshimoto, T.; Sekiguchi, Y.; Tomoishi, J.; Kasahara, I.; Hara, Y.; Ogawa, Y. Dopamine-secreting paraganglioma in the retroperitoneum. Endocr. Pathol. 2017, 28, 36–40. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Parameters | Score |
|---|---|
| Histological Pattern | |
| Zellballen | 0 |
| Large and irregular cell nest | 1 |
| Pseudorosette (even focal) | 1 |
| Cellularity | |
| Low (less than 150 cells/U *) | 0 |
| Moderate (150–250 cells/U *) | 1 |
| High (more than 250 cells/U *) | 2 |
| Comedo Necrosis | |
| Absence | 0 |
| Presence | 2 |
| Vascular or Capsular Invasion | |
| Absence | 0 |
| Presence | 1 |
| Ki67 Labelling Index | |
| Less than 1% | 0 |
| 1–3% | 1 |
| More than 3% | 2 |
| Catecholamine Type | |
| Adrenaline type (A **, or A + NA ***) | 0 |
| Noradrenaline type (NA, or NA + DA ****) | 1 |
| Non-functioning type | 0 |
| Total Maximum Score | 10 |
| Score | Grading |
|---|---|
| 0–2 | Well differentiated type |
| 3–6 | Moderately differentiated type |
| 7–10 | Poorly differentiated type |
| Total Score (Points) | Histological Grade (Frequency) | Metastatic Rate | 5-Year Survival (%) | Risk Stratification |
|---|---|---|---|---|
| 0–2 | Well differentiated (68%) | 3.6% | 100 | Low |
| 3–6 | Moderately differentiated (22%) | 60.0% | 66.8 | Intermediate |
| 7–10 | Poorly differentiated (10%) | 88.2% | 22.4 | High |
| Catecholamine Types | Number of Patients | Number of Metastasis | Ratio of Metastasis (%) |
|---|---|---|---|
| Epinephrine | 78 | 11 | 14.1 |
| Norepinephrine | 79 | 29 | 36.7 |
| (Adrenal) | (49) | (13) | (26.5) |
| (Extra-adrenal) | (30) | (15) | (50.0) |
| Non-functioning (Extra-adrenal) | 6 | 1 | 16.7 |
| Total Number | 163 | 41 | 25.1 |
| Syndrome | Number | Gene Mutated | Gender (Male/Female) | Age Range (Years; Mean) | Pheochromocytoma | Paraganglioma |
|---|---|---|---|---|---|---|
| NF1 | 12 | NF1 | 3/9 | 29–67 (44.2) | 12 | 0 |
| MEN2 | 24 | RET | 8/16 | 18–76 (35.6) | 24 | 0 |
| VHL | 29 | VHL | 12/13 (4 U) | 7–62 (25·6) | 21 (3 U) | 5 |
| PCC-PGL | 36 | SDHB | 13/12 (11 U) | 10–63 (34.6) | 11 (7 U) | 18 |
| PCC-PGL | 5 | SDHC | 2/3 | 15–47 (30.6) | 0 | 5 |
| PCC-PGL | 61 | SDHD | 25/35 (1 U) | 16–72 (40.9) | 5 (3 U) | 53 |
| Sporadic | 53 | None | 17/34 (2 U) | 12–79 (49.3) | 34 (1 U) | 18 |
| Cluster 1 | Cluster 1 | Cluster 2 | |
|---|---|---|---|
| Gene | SDH x (SDHA, B, C,D), SDHAF2, HIF2, KIF1B, PHD2, HRAS, FH, HIF-1 | VHL Sporadic Noradrenergic | RET, NF1, MAX, TMEM127, Sporadic adrenergic |
| Signaling pathways | Pseudo hypoxia (HIF-1a) & aberrant VEGF signaling | Kinase signalling: PI3 kinase/AKT, RAS/RAF/ERK, & mTorC1/p70S6K | |
| Catecholamine type | DA, mixed DA & NA | Noradrenaline | Adrenaline |
| Secretory phenotype | Immature | Immature | Mature |
| Tumor location | Extra-adrenal | Adrenal & Extra-adrenal | Adrenal |
| Age of presentation | Early (under 30 year-old) | Early | Late |
| Metastasis | Frequent | Occasional | Rare |
| Metastatic risk by GAPP | Intermediate–High | Low–Intermediate | Low |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimura, N.; Takekoshi, K.; Naruse, M. Risk Stratification on Pheochromocytoma and Paraganglioma from Laboratory and Clinical Medicine. J. Clin. Med. 2018, 7, 242. https://doi.org/10.3390/jcm7090242
Kimura N, Takekoshi K, Naruse M. Risk Stratification on Pheochromocytoma and Paraganglioma from Laboratory and Clinical Medicine. Journal of Clinical Medicine. 2018; 7(9):242. https://doi.org/10.3390/jcm7090242
Chicago/Turabian StyleKimura, Noriko, Kazuhiro Takekoshi, and Mitsuhide Naruse. 2018. "Risk Stratification on Pheochromocytoma and Paraganglioma from Laboratory and Clinical Medicine" Journal of Clinical Medicine 7, no. 9: 242. https://doi.org/10.3390/jcm7090242
APA StyleKimura, N., Takekoshi, K., & Naruse, M. (2018). Risk Stratification on Pheochromocytoma and Paraganglioma from Laboratory and Clinical Medicine. Journal of Clinical Medicine, 7(9), 242. https://doi.org/10.3390/jcm7090242