
Myocarditis and Chronic Inflammatory Cardiomyopathy, from Acute Inflammation to Chronic Inflammatory Damage: An Update on Pathophysiology and Diagnosis

 , , and
, , and
Abstract
:1. Introduction
2. Acute Myocarditis
2.1. Definition and Diagnosis
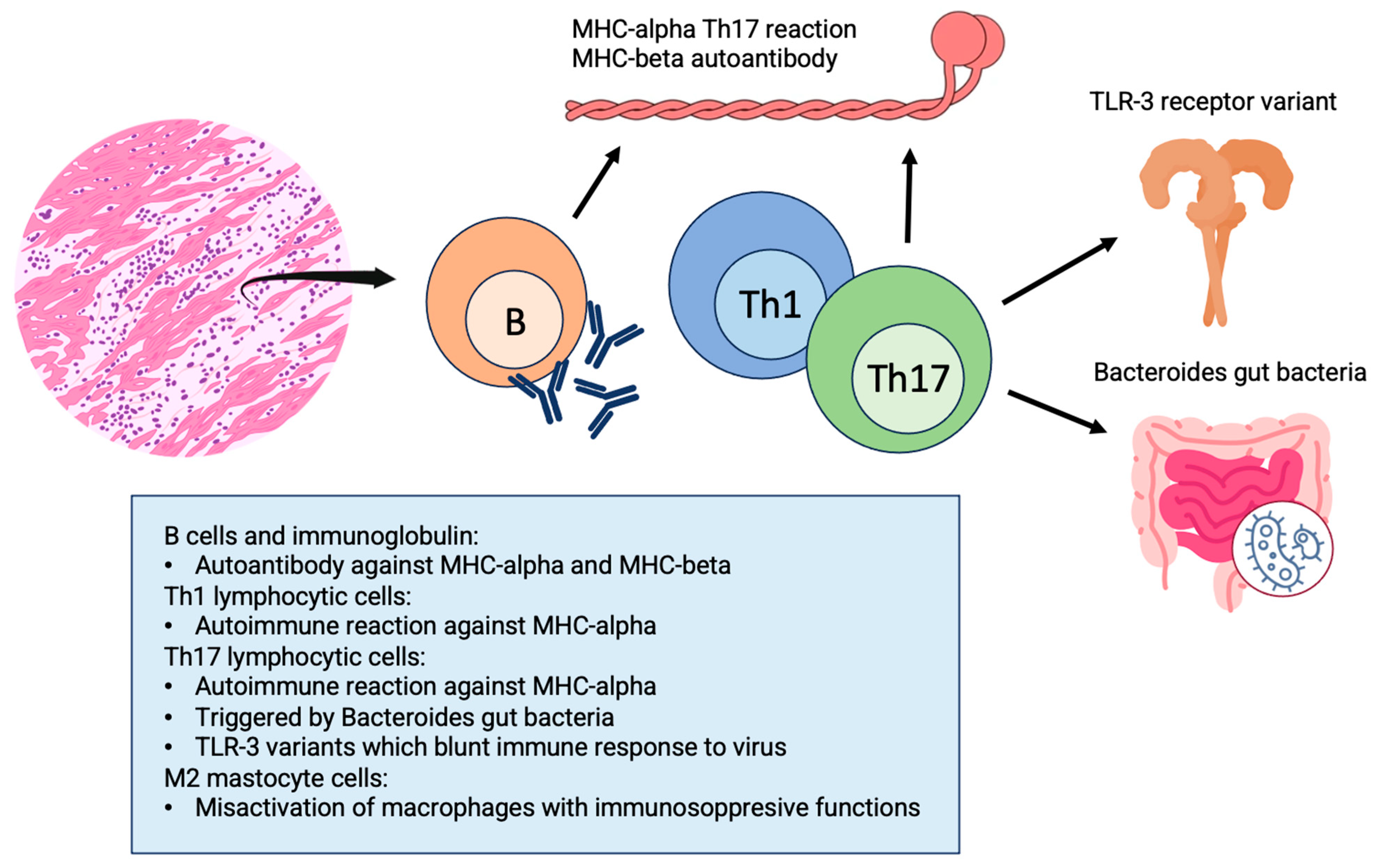
2.2. Pathophysiology
2.3. Etiologies
2.3.1. Acute Lymphocytic Myocarditis
2.3.2. COVID-19 and mRNA Vaccine-Related Myocarditis
2.3.3. Giant Cell Myocarditis
2.3.4. Check-Point Inhibitor Myocarditis
2.3.5. Eosinophilic Myocarditis
3. Chronic Inflammatory Cardiomyopathy
3.1. Etiology and Pathophysiology of Chronic infl-CMP Secondary to Acute Myocarditis
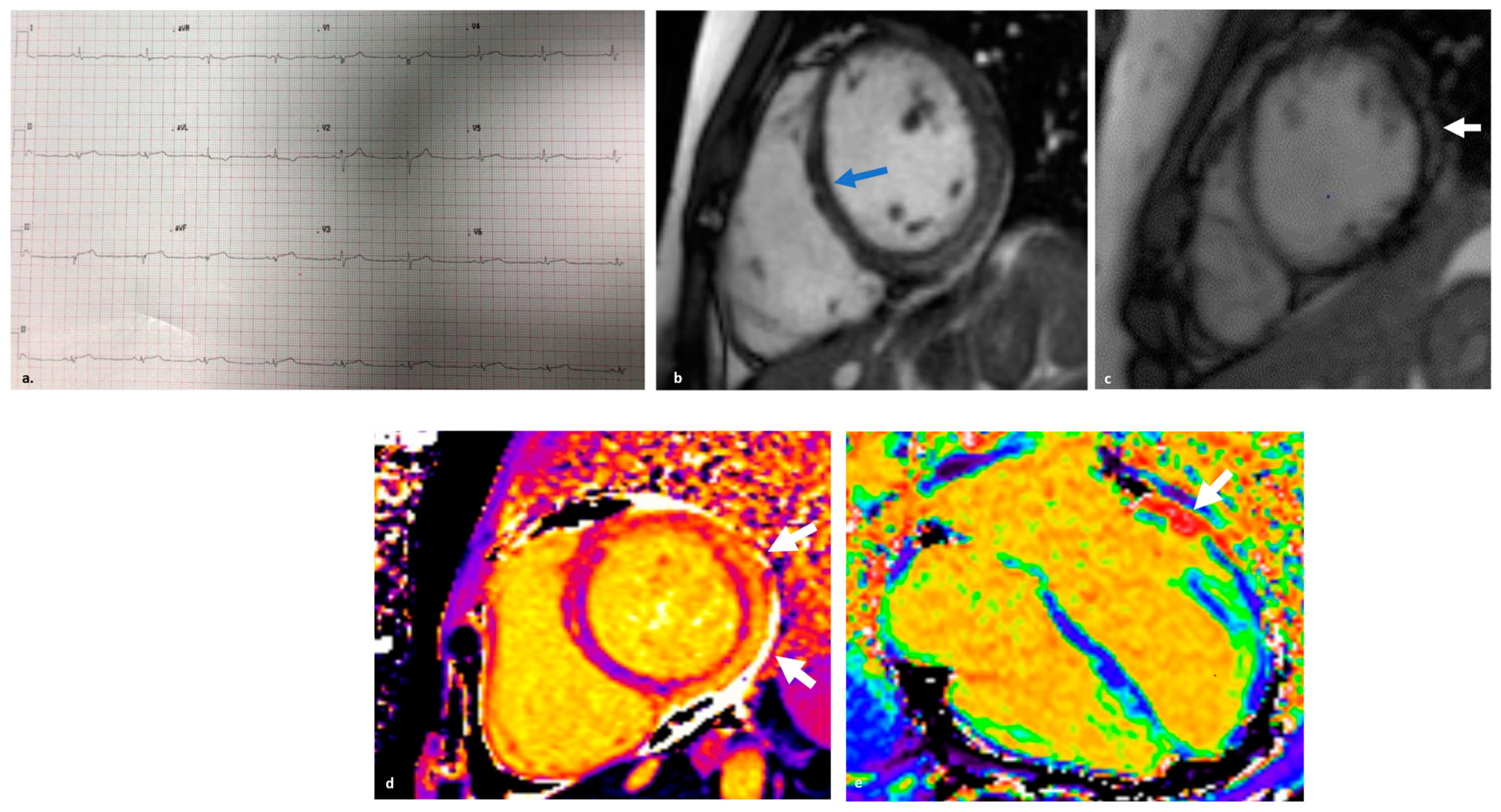
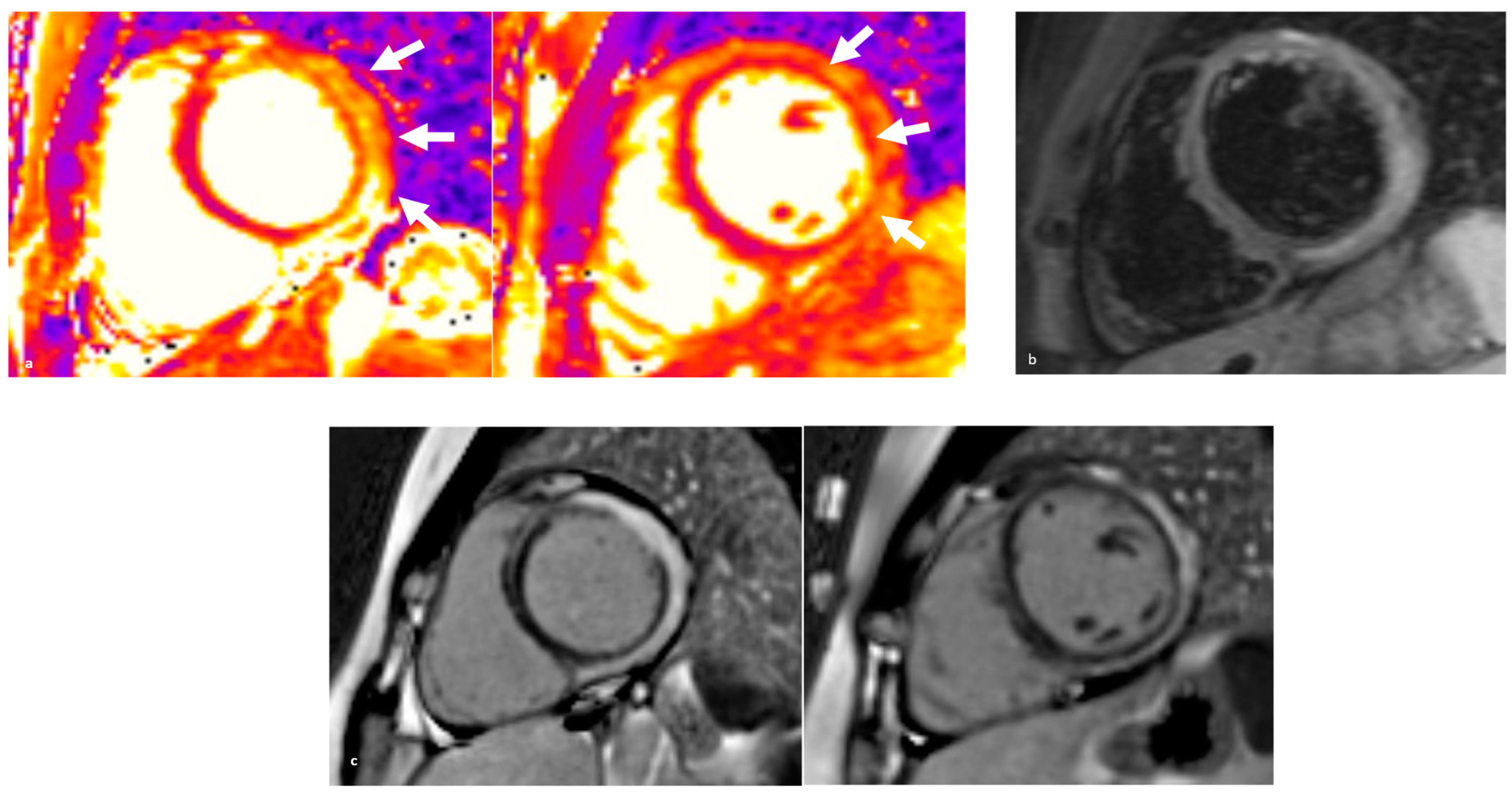
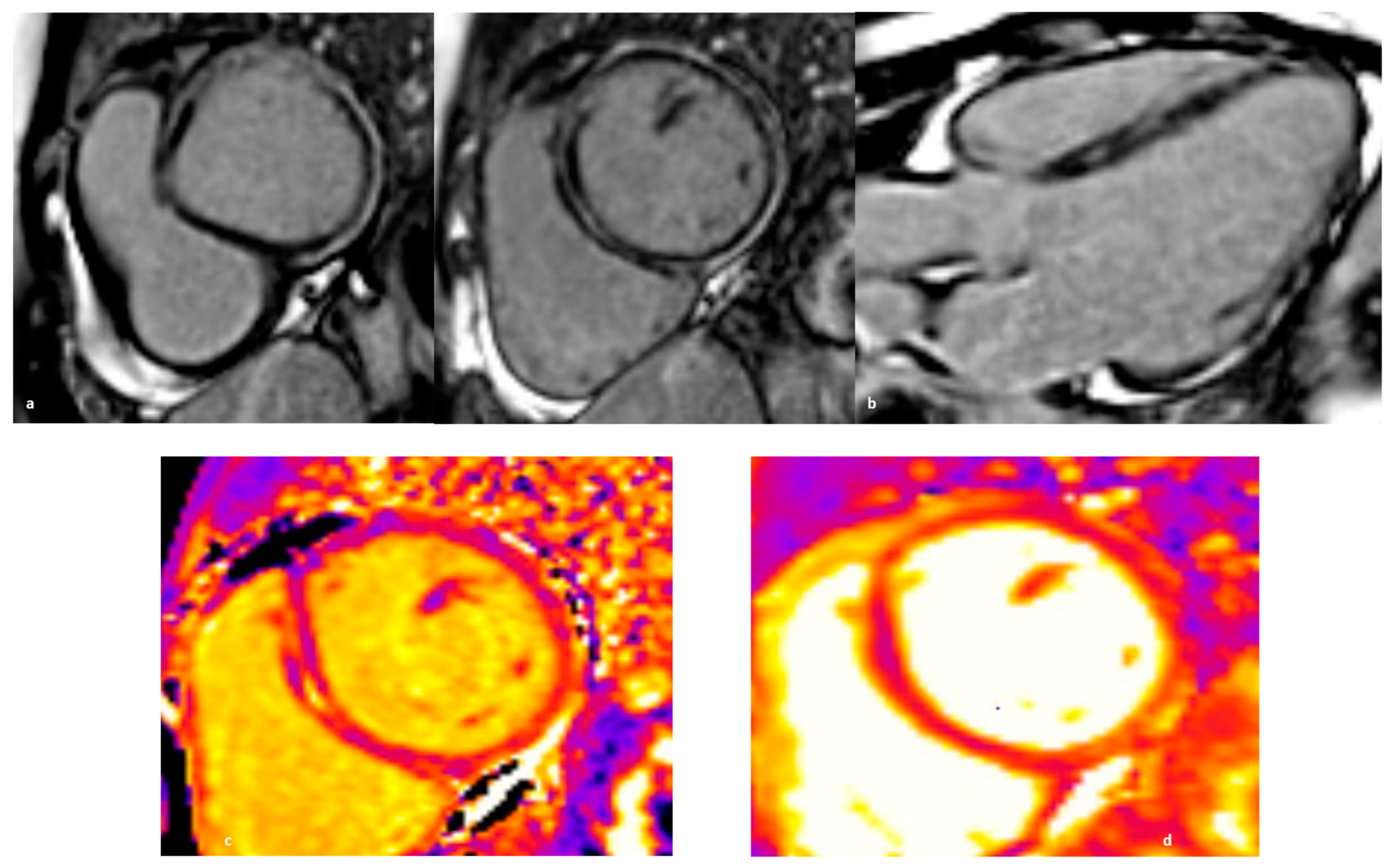
3.2. Clinical Features and Diagnosis
4. Myocardial Involvement in Systemic Inflammatory Diseases
4.1. Sarcoidosis
4.1.1. Clinical Presentation
4.1.2. Diagnostics
4.2. Systemic Sclerosis
4.2.1. Clinical Presentation
4.2.2. Diagnostics
4.3. Systemic Lupus Erythematosus
4.3.1. Clinical Presentation
4.3.2. Diagnostics
4.4. Eosinophilic Granulomatosis with Polyangiitis
4.4.1. Clinical Presentation
4.4.2. Diagnostics
4.5. Psoriasis
4.5.1. Clinical Presentation
4.5.2. Diagnostics
5. Arrhythmogenic Right Ventricular Cardiomyopathy with “Hot Phase” Episodes
5.1. Pathophysiology
5.2. Clinical Findings and Genetics
5.3. Diagnostics
6. Endomyocardial Biopsy: When and Why
7. Management
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Tschöpe, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hübner, N.; et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 2021, 18, 169–193. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Frigerio, M.; Adler, E.D.; Basso, C.; Birnie, D.H.; Brambatti, M.; Friedrich, M.G.; Klingel, K.; Lehtonen, J.; Moslehi, J.J.; et al. Management of Acute Myocarditis and Chronic Inflammatory Cardiomyopathy: An Expert Consensus Document. Circ. Heart Fail. 2020, 13, e007405. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.G.; Sechtem, U.; Schulz-Menger, J.; Holmvang, G.; Alakija, P.; Cooper, L.T.; White, J.A.; Abdel-Aty, H.; Gutberlet, M.; Prasad, S.; et al. Cardiovascular magnetic resonance in myocarditis: A JACC White Paper. J. Am. Coll. Cardiol. 2009, 53, 1475–1487. [Google Scholar] [CrossRef] [PubMed]
- Caforio, A.L.P.; Malipiero, G.; Marcolongo, R.; Iliceto, S. Myocarditis: A Clinical Overview. Curr. Cardiol. Rep. 2017, 19, 63. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Moslehi, J.J. Diagnosis and Treatment of Acute Myocarditis: A Review. JAMA 2023, 329, 1098–1113. [Google Scholar] [CrossRef] [PubMed]
- Caforio, A.L.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Heliö, T.; Heymans, S.; Jahns, R.; et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648. [Google Scholar] [CrossRef]
- Ferreira, V.M.; Schulz-Menger, J.; Holmvang, G.; Kramer, C.M.; Carbone, I.; Sechtem, U.; Kindermann, I.; Gutberlet, M.; Cooper, L.T.; Liu, P.; et al. Cardiovascular Magnetic Resonance in Nonischemic Myocardial Inflammation: Expert Recommendations. J. Am. Coll. Cardiol. 2018, 72, 3158–3176. [Google Scholar] [CrossRef]
- Trachtenberg, B.H.; Hare, J.M. Inflammatory Cardiomyopathic Syndromes. Circ. Res. 2017, 121, 803–818. [Google Scholar] [CrossRef]
- Lv, H.; Havari, E.; Pinto, S.; Gottumukkala, R.V.; Cornivelli, L.; Raddassi, K.; Matsui, T.; Rosenzweig, A.; Bronson, R.T.; Smith, R.; et al. Impaired thymic tolerance to alpha-myosin directs autoimmunity to the heart in mice and humans. J. Clin. Investig. 2011, 121, 1561–1573. [Google Scholar] [CrossRef]
- Nindl, V.; Maier, R.; Ratering, D.; De Giuli, R.; Züst, R.; Thiel, V.; Scandella, E.; Di Padova, F.; Kopf, M.; Rudin, M.; et al. Cooperation of Th1 and Th17 cells determines transition from autoimmune myocarditis to dilated cardiomyopathy. Eur. J. Immunol. 2012, 42, 2311–2321. [Google Scholar] [CrossRef]
- Frantz, S.; Falcao-Pires, I.; Balligand, J.L.; Bauersachs, J.; Brutsaert, D.; Ciccarelli, M.; Dawson, D.; de Windt, L.J.; Giacca, M.; Hamdani, N.; et al. The innate immune system in chronic cardiomyopathy: A European Society of Cardiology (ESC) scientific statement from the Working Group on Myocardial Function of the ESC. Eur. J. Heart Fail. 2018, 20, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Sinagra, G.; Porcari, A.; Gentile, P.; Artico, J.; Fabris, E.; Bussani, R.; Merlo, M. Viral presence-guided immunomodulation in lymphocytic myocarditis: An update. Eur. J. Heart Fail. 2021, 23, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Block, J.P.; Boehmer, T.K.; Forrest, C.B.; Carton, T.W.; Lee, G.M.; Ajani, U.A.; Christakis, D.A.; Cowell, L.G.; Draper, C.; Ghildayal, N.; et al. Cardiac Complications After SARS-CoV-2 Infection and mRNA COVID-19 Vaccination–PCORnet, United States, January 2021–January 2022. MMWR Morb. Mortal Wkly. Rep. 2022, 71, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Lupi, L.; Palazzini, M.; Ciabatti, M.; Rossi, V.A.; Gentile, P.; Uribarri, A.; Vecchio, C.R.; Nassiacos, D.; Cereda, A.; et al. Outcome and Morphofunctional Changes on Cardiac Magnetic Resonance in Patients with Acute Myocarditis Following mRNA COVID-19 Vaccination. Circ. Heart Fail. 2023, 16, e010315. [Google Scholar] [CrossRef] [PubMed]
- Bang, V.; Ganatra, S.; Shah, S.P.; Dani, S.S.; Neilan, T.G.; Thavendiranathan, P.; Resnic, F.S.; Piemonte, T.C.; Barac, A.; Patel, R.; et al. Management of Patients With Giant Cell Myocarditis: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2021, 77, 1122–1134. [Google Scholar] [CrossRef]
- Mahmood, S.S.; Fradley, M.G.; Cohen, J.V.; Nohria, A.; Reynolds, K.L.; Heinzerling, L.M.; Sullivan, R.J.; Damrongwatanasuk, R.; Chen, C.L.; Gupta, D.; et al. Myocarditis in Patients Treated with Immune Checkpoint Inhibitors. J. Am. Coll. Cardiol. 2018, 71, 1755–1764. [Google Scholar] [CrossRef]
- Johnson, D.B.; Balko, J.M.; Compton, M.L.; Chalkias, S.; Gorham, J.; Xu, Y.; Hicks, M.; Puzanov, I.; Alexander, M.R.; Bloomer, T.L.; et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N. Engl. J. Med. 2016, 375, 1749–1755. [Google Scholar] [CrossRef]
- Rossi, V.A.; Gawinecka, J.; Dimitriou, F.; von Eckardstein, A.; Dummer, R.; Ruschitzka, F.; Matter, C.M. Value of troponin T versus I in the diagnosis of immune checkpoint inhibitor-related myocarditis and myositis: Rechallenge? ESC Heart Fail. 2023, 10, 2680–2685. [Google Scholar] [CrossRef]
- Pöyhönen, P.; Rågback, J.; Mäyränpää, M.I.; Nordenswan, H.K.; Lehtonen, J.; Shenoy, C.; Kupari, M. Cardiac magnetic resonance in histologically proven eosinophilic myocarditis. J. Cardiovasc. Magn. Reson. 2023, 25, 79. [Google Scholar] [CrossRef]
- Noutsias, M.; Rohde, M.; Göldner, K.; Block, A.; Blunert, K.; Hemaidan, L.; Hummel, M.; Blohm, J.H.; Lassner, D.; Kühl, U.; et al. Expression of functional T-cell markers and T-cell receptor Vbeta repertoire in endomyocardial biopsies from patients presenting with acute myocarditis and dilated cardiomyopathy. Eur. J. Heart Fail. 2011, 13, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Barin, J.G.; Rose, N.R.; Ciháková, D. Macrophage diversity in cardiac inflammation: A review. Immunobiology 2012, 217, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Baldeviano, G.C.; Barin, J.G.; Talor, M.V.; Srinivasan, S.; Bedja, D.; Zheng, D.; Gabrielson, K.; Iwakura, Y.; Rose, N.R.; Cihakova, D. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ. Res. 2010, 106, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Gorbea, C.; Makar, K.A.; Pauschinger, M.; Pratt, G.; Bersola, J.L.; Varela, J.; David, R.M.; Banks, L.; Huang, C.H.; Li, H.; et al. A role for Toll-like receptor 3 variants in host susceptibility to enteroviral myocarditis and dilated cardiomyopathy. J. Biol. Chem. 2010, 285, 23208–23223. [Google Scholar] [CrossRef] [PubMed]
- Brambatti, M.; Matassini, M.V.; Adler, E.D.; Klingel, K.; Camici, P.G.; Ammirati, E. Eosinophilic myocarditis: Characteristics, treatment, and outcomes. J. Am. Coll. Cardiol. 2017, 70, 2363–2375. [Google Scholar] [CrossRef] [PubMed]
- Caforio, A.L.; Mahon, N.J.; Tona, F.; McKenna, W.J. Circulating cardiac autoantibodies in dilated cardiomyopathy and myocarditis: Pathogenetic and clinical significance. Eur. J. Heart Fail. 2002, 4, 411–417. [Google Scholar] [CrossRef]
- Caforio, A.L.; Vinci, A.; Iliceto, S. Anti-heart autoantibodies in familial dilated cardiomyopathy. Autoimmunity 2008, 41, 462–469. [Google Scholar] [CrossRef]
- Meder, B.; Rühle, F.; Weis, T.; Homuth, G.; Keller, A.; Franke, J.; Peil, B.; Lorenzo Bermejo, J.; Frese, K.; Huge, A.; et al. A genome-wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. Eur. Heart J. 2014, 35, 1069–1077. [Google Scholar] [CrossRef]
- Naruse, T.K.; Matsuzawa, Y.; Ota, M.; Katsuyama, Y.; Matsumori, A.; Hara, M.; Nagai, S.; Morimoto, S.; Sasayama, S.; Inoko, H. HLA-DQB1*0601 is primarily associated with the susceptibility to cardiac sarcoidosis. Tissue Antigens 2000, 56, 52–57. [Google Scholar] [CrossRef]
- D’Ambrosio, A.; Patti, G.; Manzoli, A.; Sinagra, G.; Di Lenarda, A.; Silvestri, F.; Di Sciascio, G. The fate of acute myocarditis between spontaneous improvement and evolution to dilated cardiomyopathy: A review. Heart 2001, 85, 499–504. [Google Scholar] [CrossRef]
- Pollack, A.; Kontorovich, A.R.; Fuster, V.; Dec, G.W. Viral myocarditis-diagnosis, treatment options, and current controversies. Nat. Rev. Cardiol. 2015, 12, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hsu, J.; Liu, X.; Lin, X.; Zhu, Y.; Jia, F.; Wu, W.; Chen, W.; Wang, Q.; Fang, L. Electrophysiological, structural, and functional disorders in patients with inflammatory cardiomyopathy secondary to inflammatory myopathy. Ann. Noninvasive Electrocardiol. 2022, 27, e12938. [Google Scholar] [CrossRef] [PubMed]
- Escher, F.; Kasner, M.; Kühl, U.; Heymer, J.; Wilkenshoff, U.; Tschöpe, C.; Schultheiss, H.P. New echocardiographic findings correlate with intramyocardial inflammation in endomyocardial biopsies of patients with acute myocarditis and inflammatory cardiomyopathy. Mediat. Inflamm. 2013, 2013, 875420. [Google Scholar] [CrossRef] [PubMed]
- Filippetti, L.; Mandry, D.; Venner, C.; Juillière, Y.; Sadoul, N.; Girerd, N.; Lamiral, Z.; Selton-Suty, C.; Marie, P.Y.; Huttin, O. Long-Term Outcome of Patients with Low/Intermediate Risk Myocarditis Is Related to the Presence of Left Ventricular Remodeling in Addition to the MRI Pattern of Delayed Gadolinium Enhancement. JACC Cardiovasc. Imaging 2018, 11, 1367–1369. [Google Scholar] [CrossRef] [PubMed]
- Aquaro, G.D.; Ghebru Habtemicael, Y.; Camastra, G.; Monti, L.; Dellegrottaglie, S.; Moro, C.; Lanzillo, C.; Scatteia, A.; Di Roma, M.; Pontone, G.; et al. Prognostic value of repeating cardiac magnetic resonance in patients with acute myocarditis. J. Am. Coll. Cardiol. 2019, 74, 2439–2448. [Google Scholar] [CrossRef] [PubMed]
- Schelbert, E.B.; Piehler, K.M.; Zareba, K.M.; Moon, J.C.; Ugander, M.; Messroghli, D.R.; Valeti, U.S.; Chang, C.C.; Shroff, S.G.; Diez, J.; et al. Myocardial Fibrosis Quantified by Extracellular Volume Is Associated with Subsequent Hospitalization for Heart Failure, Death, or Both Across the Spectrum of Ejection Fraction and Heart Failure Stage. J. Am. Heart Assoc. 2015, 4, e002613. [Google Scholar] [CrossRef] [PubMed]
- Šramko, M.; Kubánek, M.; Tintěra, J.; Kautznerová, D.; Weichet, J.; Malušková, J.; Franeková, J.; Kautzner, J. Utility of combination of cardiac magnetic resonance imaging and high-sensitivity cardiac troponin T assay in diagnosis of inflammatory cardiomyopathy. Am. J. Cardiol. 2013, 111, 258–264. [Google Scholar] [CrossRef]
- Okumura, W.; Iwasaki, T.; Toyama, T.; Iso, T.; Arai, M.; Oriuchi, N.; Endo, K.; Yokoyama, T.; Suzuki, T.; Kurabayashi, M. Usefulness of fasting 18F-FDG PET in identification of cardiac sarcoidosis. J. Nucl. Med. 2004, 45, 1989–1998. [Google Scholar]
- Nakayama, T.; Sugano, Y.; Yokokawa, T.; Nagai, T.; Matsuyama, T.A.; Ohta-Ogo, K.; Ikeda, Y.; Ishibashi-Ueda, H.; Nakatani, T.; Ohte, N.; et al. Clinical impact of the presence of macrophages in endomyocardial biopsies of patients with dilated cardiomyopathy. Eur. J. Heart Fail. 2017, 19, 490–498. [Google Scholar] [CrossRef]
- Kandolin, R.; Lehtonen, J.; Airaksinen, J.; Vihinen, T.; Miettinen, H.; Ylitalo, K.; Kaikkonen, K.; Tuohinen, S.; Haataja, P.; Kerola, T.; et al. Cardiac sarcoidosis: Epidemiology, characteristics, and outcome over 25 years in a nationwide study. Circulation 2015, 131, 624–632. [Google Scholar] [CrossRef]
- Kouranos, V.; Sharma, R. Cardiac sarcoidosis: State-of-the-art review. Heart 2021, 107, 1591–1599. [Google Scholar] [CrossRef] [PubMed]
- Trivieri, M.G.; Spagnolo, P.; Birnie, D.; Liu, P.; Drake, W.; Kovacic, J.C.; Baughman, R.; Fayad, Z.A.; Judson, M.A. Challenges in Cardiac and Pulmonary Sarcoidosis: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 76, 1878–1901. [Google Scholar] [CrossRef] [PubMed]
- Markatis, E.; Afthinos, A.; Antonakis, E.; Papanikolaou, I.C. Cardiac sarcoidosis: Diagnosis and management. Rev. Cardiovasc. Med. 2020, 21, 321–338. [Google Scholar] [CrossRef] [PubMed]
- Tavora, F.; Cresswell, N.; Li, L.; Ripple, M.; Solomon, C.; Burke, A. Comparison of necropsy findings in patients with sarcoidosis dying suddenly from cardiac sarcoidosis versus dying suddenly from other causes. Am. J. Cardiol. 2009, 104, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Ekström, K.; Lehtonen, J.; Nordenswan, H.K.; Mäyränpää, M.I.; Räisänen-Sokolowski, A.; Kandolin, R.; Simonen, P.; Pietilä-Effati, P.; Alatalo, A.; Utriainen, S.; et al. Sudden death in cardiac sarcoidosis: An analysis of nationwide clinical and cause-of-death registries. Eur. Heart J. 2019, 40, 3121–3128. [Google Scholar] [CrossRef]
- Zhou, Y.; Lower, E.E.; Li, H.P.; Costea, A.; Attari, M.; Baughman, R.P. Cardiac Sarcoidosis: The Impact of Age and Implanted Devices on Survival. Chest 2017, 151, 139–148. [Google Scholar] [CrossRef]
- Sharma, A.; Okada, D.R.; Yacoub, H.; Chrispin, J.; Bokhari, S. Diagnosis of cardiac sarcoidosis: An era of paradigm shift. Ann. Nucl. Med. 2020, 34, 87–93. [Google Scholar] [CrossRef]
- Sève, P.; Pacheco, Y.; Durupt, F.; Jamilloux, Y.; Gerfaud-Valentin, M.; Isaac, S.; Boussel, L.; Calender, A.; Androdias, G.; Valeyre, D.; et al. Sarcoidosis: A Clinical Overview from Symptoms to Diagnosis. Cells 2021, 10, 766. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Nagai, T.; Takenaka, S.; Kato, Y.; Komoriyama, H.; Nagano, N.; Kamiya, K.; Konishi, T.; Sato, T.; Omote, K.; et al. Long-Term Prognostic Significance of Ventricular Repolarization Dispersion in Patients with Cardiac Sarcoidosis. Am. J. Cardiol. 2021, 152, 125–131. [Google Scholar] [CrossRef]
- Nagao, S.; Watanabe, H.; Sobue, Y.; Kodama, M.; Tanaka, J.; Tanabe, N.; Suzuki, E.; Narita, I.; Watanabe, E.; Aizawa, Y.; et al. Electrocardiographic abnormalities and risk of developing cardiac events in extracardiac sarcoidosis. Int. J. Cardiol. 2015, 189, 1–5. [Google Scholar] [CrossRef]
- Kurmann, R.; Mankad, S.V.; Mankad, R. Echocardiography in Sarcoidosis. Curr. Cardiol. Rep. 2018, 20, 118. [Google Scholar] [CrossRef] [PubMed]
- Kouranos, V.; Tzelepis, G.E.; Rapti, A.; Mavrogeni, S.; Aggeli, K.; Douskou, M.; Prasad, S.; Koulouris, N.; Sfikakis, P.; Wells, A.; et al. Complementary Role of CMR to Conventional Screening in the Diagnosis and Prognosis of Cardiac Sarcoidosis. JACC Cardiovasc. Imaging 2017, 10, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.L.; Fong, H.K.; Birati, E.Y.; Han, Y. Cardiac Sarcoidosis. Am. J. Cardiol. 2019, 123, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Wicks, E.C.; Menezes, L.J.; Barnes, A.; Mohiddin, S.A.; Sekhri, N.; Porter, J.C.; Booth, H.L.; Garrett, E.; Patel, R.S.; Pavlou, M.; et al. Diagnostic accuracy and prognostic value of simultaneous hybrid 18F-fluorodeoxyglucose positron emission tomography/magnetic resonance imaging in cardiac sarcoidosis. Eur. Heart J. Cardiovasc. Imaging 2018, 19, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Bruni, C.; Ross, L. Cardiac involvement in systemic sclerosis: Getting to the heart of the matter. Best Pract. Res. Clin. Rheumatol. 2021, 35, 101668. [Google Scholar] [CrossRef] [PubMed]
- De Luca, G.; Cavalli, G.; Campochiaro, C.; Bruni, C.; Tomelleri, A.; Dagna, L.; Matucci-Cerinic, M. Interleukin-1 and Systemic Sclerosis: Getting to the Heart of Cardiac Involvement. Front. Immunol. 2021, 12, 653950. [Google Scholar] [CrossRef] [PubMed]
- Ross, L.; Prior, D.; Proudman, S.; Vacca, A.; Baron, M.; Nikpour, M. Defining primary systemic sclerosis heart involvement: A scoping literature review. Semin. Arthritis Rheum. 2019, 48, 874–887. [Google Scholar] [CrossRef]
- Caforio, A.L.P.; Adler, Y.; Agostini, C.; Allanore, Y.; Anastasakis, A.; Arad, M.; Böhm, M.; Charron, P.; Elliott, P.M.; Eriksson, U.; et al. Diagnosis and management of myocardial involvement in systemic immune-mediated diseases: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Disease. Eur. Heart J. 2017, 38, 2649–2662. [Google Scholar] [CrossRef]
- Tennøe, A.H.; Murbræch, K.; Andreassen, J.C.; Fretheim, H.; Garen, T.; Gude, E.; Andreassen, A.; Aakhus, S.; Molberg, Ø.; Hoffmann-Vold, A.M. Left Ventricular Diastolic Dysfunction Predicts Mortality in Patients with Systemic Sclerosis. J. Am. Coll. Cardiol. 2018, 72, 1804–1813. [Google Scholar] [CrossRef]
- Steen, V.D.; Medsger, T.A., Jr. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum. 2000, 43, 2437–2444. [Google Scholar] [CrossRef]
- Nordin, A.; Björnådal, L.; Larsson, A.; Svenungsson, E.; Jensen-Urstad, K. Electrocardiography in 110 patients with systemic sclerosis: A cross-sectional comparison with population-based controls. Scand. J. Rheumatol. 2014, 43, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Ferri, C.; Emdin, M.; Giuggioli, D.; Carpeggiani, C.; Maielli, M.; Varga, A.; Michelassi, C.; Pasero, G.; L’Abbate, A. Autonomic dysfunction in systemic sclerosis: Time and frequency domain 24 hour heart rate variability analysis. Br. J. Rheumatol. 1997, 36, 669–676. [Google Scholar] [CrossRef] [PubMed]
- van Wijngaarden, S.E.; Ben Said-Bouyeri, S.; Ninaber, M.K.; Huizinga, T.W.J.; Schalij, M.J.; Bax, J.J.; Delgado, V.; de Vries-Bouwstra, J.K.; Marsan, N.A. Progression of Left Ventricular Myocardial Dysfunction in Systemic Sclerosis: A Speckle-tracking Strain Echocardiography Study. J. Rheumatol. 2019, 46, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Allanore, Y.; Meune, C.; Vonk, M.C.; Airo, P.; Hachulla, E.; Caramaschi, P.; Riemekasten, G.; Cozzi, F.; Beretta, L.; Derk, C.T.; et al. Prevalence and factors associated with left ventricular dysfunction in the EULAR Scleroderma Trial and Research group (EUSTAR) database of patients with systemic sclerosis. Ann. Rheum. Dis. 2010, 69, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Long, A.; Duffy, G.; Bresnihan, B. Reversible myocardial perfusion defects during cold challenge in scleroderma. Br. J. Rheumatol. 1986, 25, 158–161. [Google Scholar] [CrossRef] [PubMed]
- Hachulla, A.L.; Launay, D.; Gaxotte, V.; de Groote, P.; Lamblin, N.; Devos, P.; Hatron, P.-Y.; Beregi, J.-P.; Hachulla, E. Cardiac magnetic resonance imaging in systemic sclerosis: A cross-sectional observational study of 52 patients. Ann. Rheum. Dis. 2009, 68, 1878–1884. [Google Scholar] [CrossRef] [PubMed]
- Gargani, L.; Todiere, G.; Guiducci, S.; Bruni, C.; Pingitore, A.; De Marchi, D.; Bellando Randone, S.; Aquaro, G.D.; Bazzichi, L.; Mosca, M.; et al. Early Detection of Cardiac Involvement in Systemic Sclerosis: The Added Value of Magnetic Resonance Imaging. JACC Cardiovasc. Imaging 2019, 12, 927–928. [Google Scholar] [CrossRef]
- Poindron, V.; Chatelus, E.; Canuet, M.; Gottenberg, J.E.; Arnaud, L.; Gangi, A.; Gavand, P.E.; Guffroy, A.; Korganow, A.S.; Germain, P.; et al. T1 mapping cardiac magnetic resonance imaging frequently detects subclinical diffuse myocardial fibrosis in systemic sclerosis patients. Semin. Arthritis Rheum. 2020, 50, 128–134. [Google Scholar] [CrossRef]
- Shaikh, M.F.; Jordan, N.; D’Cruz, D.P. Systemic lupus erythematosus. Clin. Med. 2017, 17, 78–83. [Google Scholar] [CrossRef]
- Bulkley, B.H.; Roberts, W.C. The heart in systemic lupus erythematosus and the changes induced in it by corticosteroid therapy. A study of 36 necropsy patients. Am. J. Med. 1975, 58, 243–264. [Google Scholar] [CrossRef]
- Doherty, N.E.; Siegel, R.J. Cardiovascular manifestations of systemic lupus erythematosus. Am. Heart J. 1985, 110, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Tincani, A.; Rebaioli, C.B.; Taglietti, M.; Shoenfeld, Y. Heart involvement in systemic lupus erythematosus, anti-phospholipid syndrome and neonatal lupus. Rheumatology 2006, 45 (Suppl. S4), iv8–iv13. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.; McGwin, G., Jr.; Vilá, L.M.; Kaslow, R.A.; Alarcón, G.S.; Reveille, J.D.; LUMINA Study Group. Associated factors and impact of myocarditis in patients with SLE from LUMINA, a multiethnic US cohort. Rheumatology 2008, 47, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Battisha, A.; Sawalha, K.; Altibi, A.M.; Madoukh, B.; Al-Akchar, M.; Patel, B. Cardiogenic shock in autoimmune rheumatologic diseases: An insight on etiologies, management, and treatment outcomes. Heart Fail. Rev. 2022, 27, 93–101. [Google Scholar] [CrossRef]
- Choe, J.Y.; Lee, S.S.; Kwak, S.G.; Kim, S.K. Anti-Sm Antibody, Damage Index, and Corticosteroid Use Are Associated with Cardiac Involvement in Systemic Lupus Erythematosus: Data from a Prospective Registry Study. J. Korean Med. Sci. 2020, 35, e139. [Google Scholar] [CrossRef] [PubMed]
- Kahl, L.E. The spectrum of pericardial tamponade in systemic lupus erythematosus. Report of ten patients. Arthritis Rheum. 1992, 35, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Katz, G.; Smilowitz, N.R.; Blazer, A.; Clancy, R.; Buyon, J.P.; Berger, J.S. Systemic Lupus Erythematosus and Increased Prevalence of Atherosclerotic Cardiovascular Disease in Hospitalized Patients. Mayo Clin. Proc. 2019, 94, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Hussain, K.; Gauto-Mariotti, E.; Cattoni, H.M.; Arif, A.W.; Richardson, C.; Manadan, A.; Yadav, N. A Meta-analysis and Systematic Review of Valvular Heart Disease in Systemic Lupus Erythematosus and Its Association with Antiphospholipid Antibodies. J. Clin. Rheumatol. 2021, 27, e525–e532. [Google Scholar] [CrossRef]
- Ruiz, D.; Oates, J.C.; Kamen, D.L. Antiphospholipid Antibodies and Heart Valve Disease in Systemic Lupus Erythematosus. Am. J. Med. Sci. 2018, 355, 293–298. [Google Scholar] [CrossRef]
- Moder, K.G.; Miller, T.D.; Tazelaar, H.D. Cardiac involvement in systemic lupus erythematosus. Mayo Clin. Proc. 1999, 74, 275–284. [Google Scholar] [CrossRef]
- Zagelbaum Ward, N.K.; Linares-Koloffon, C.; Posligua, A.; Gandrabur, L.; Kim, W.Y.; Sperber, K.; Wasserman, A.; Ash, J. Cardiac Manifestations of Systemic Lupus Erythematous: An Overview of the Incidence, Risk Factors, Diagnostic Criteria, Pathophysiology and Treatment Options. Cardiol. Rev. 2022, 30, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ge, L.; Li, Y. Value of Autostrain LV in the study of left ventricular systolic function and synchronization in patients with systemic lupus erythematosus. Echocardiography 2023, 40, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Joyce, E.; Ninaber, M.K.; Katsanos, S.; Debonnaire, P.; Kamperidis, V.; Bax, J.J.; Taube, C.; Delgado, V.; Ajmone Marsan, N. Subclinical left ventricular dysfunction by echocardiographic speckle-tracking strain analysis relates to outcome in sarcoidosis. Eur. J. Heart Fail. 2015, 17, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Mavrogeni, S.; Bratis, K.; Markussis, V.; Spargias, C.; Papadopoulou, E.; Papamentzelopoulos, S.; Constadoulakis, P.; Matsoukas, E.; Kyrou, L.; Kolovou, G. The diagnostic role of cardiac magnetic resonance imaging in detecting myocardial inflammation in systemic lupus erythematosus. Differentiation from viral myocarditis. Lupus 2013, 22, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Robson, J.C.; Grayson, P.C.; Ponte, C.; Suppiah, R.; Craven, A.; Judge, A.; Khalid, S.; Hutchings, A.; Watts, R.A.; Merkel, P.A.; et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology classification criteria for granulomatosis with polyangiitis. Ann. Rheum. Dis. 2022, 81, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Neumann, T.; Manger, B.; Schmid, M.; Kroegel, C.; Hansch, A.; Kaiser, W.A.; Reinhardt, D.; Wolf, G.; Hein, G.; Mall, G.; et al. Cardiac involvement in Churg-Strauss syndrome: Impact of endomyocarditis. Medicine 2009, 88, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, M.; Emmi, G.; Beltrami, M.; Fumagalli, C.; Urban, M.L.; Dei, L.L.; Marchi, A.; Berteotti, M.; Tomberli, A.; Baldini, K.; et al. Cardiac involvement in eosinophilic granulomatosis with polyangiitis (formerly Churg-Strauss syndrome): Prospective evaluation at a tertiary referral centre. Eur. J. Intern. Med. 2021, 85, 68–79. [Google Scholar] [CrossRef]
- Miszalski-Jamka, T.; Szczeklik, W.; Sokołowska, B.; Karwat, K.; Belzak, K.; Mazur, W.; Kereiakes, D.J.; Musiał, J. Standard and feature tracking magnetic resonance evidence of myocardial involvement in Churg-Strauss syndrome and granulomatosis with polyangiitis (Wegener’s) in patients with normal electrocardiograms and transthoracic echocardiography. Int. J. Cardiovasc. Imaging 2013, 29, 843–853. [Google Scholar] [CrossRef]
- Eliakim-Raz, N.; Shuvy, M.; Lotan, C.; Planer, D. Psoriasis and dilated cardiomyopathy: Coincidence or associated diseases? Cardiology 2008, 111, 202–206. [Google Scholar] [CrossRef]
- Frustaci, A.; Galea, N.; Dominici, L.; Verardo, R.; Alfarano, M.; Scialla, R.; Richetta, A.G. Interleukin-17A-Correlated Myocarditis in Patients with Psoriasis: Cardiac Recovery following Secukinumab Administration. J. Clin. Med. 2023, 12, 4010. [Google Scholar] [CrossRef]
- Alshami, A.; Alfraji, N.; Douedi, S.; Patel, S.; Hossain, M.; Alpert, D.; Calderon, D. Psoriasis as risk factor for non-ischemic dilated cardiomyopathy: A population-based cross-sectional study. BMC Cardiovasc. Disord. 2021, 21, 161. [Google Scholar] [CrossRef]
- Domenico, C.; Aris, A.; Cristina, B.; Barbara, B.; Carina, B.L.; Chiara, B.D.; Alberto, C.; Carlo, A.; Estelle, G.; Juan, J.I.; et al. Proposed diagnostic criteria for arrhythmogenic cardiomyopathy. European Task Force consensus report. Int. J. Cardiol. 2023, 395, 131447. [Google Scholar] [CrossRef]
- Boogerd, C.J.; Lacraz, G.P.A.; Vértesy, Á.; van Kampen, S.J.; Perini, I.; de Ruiter, H.; Versteeg, D.; Brodehl, A.; van der Kraak, P.; Giacca, M.; et al. Spatial transcriptomics unveils ZBTB11 as a regulator of cardiomyocyte degeneration in arrhythmogenic cardiomyopathy. Cardiovasc. Res. 2023, 119, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Weiss, J.; Debus, J.D.; Stanasiuk, C.; Klauke, B.; Deutsch, M.A.; Fox, H.; Bax, J.; Ebbinghaus, H.; Gärtner, A.; et al. A homozygous DSC2 deletion associated with arrhythmogenic cardiomyopathy is caused by uniparental isodisomy. J. Mol. Cell Cardiol. 2020, 141, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef]
- Basso, C.; Thiene, G.; Corrado, D.; Angelini, A.; Nava, A.; Valente, M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation 1996, 94, 983–991. [Google Scholar] [CrossRef]
- Bariani, R.; Cipriani, A.; Rizzo, S.; Celeghin, R.; Bueno Marinas, M.; Giorgi, B.; De Gaspari, M.; Rigato, I.; Leoni, L.; Zorzi, A.; et al. ‘Hot phase’ clinical presentation in arrhythmogenic cardiomyopathy. Europace 2021, 23, 907–917. [Google Scholar] [CrossRef]
- Kontorovich, A.R.; Patel, N.; Moscati, A.; Richter, F.; Peter, I.; Purevjav, E.; Selejan, S.R.; Kindermann, I.; Towbin, J.A.; Bohm, M.; et al. Myopathic Cardiac Genotypes Increase Risk for Myocarditis. JACC Basic Transl. Sci. 2021, 6, 584–592. [Google Scholar] [CrossRef]
- Martins, D.; Ovaert, C.; Khraiche, D.; Boddaert, N.; Bonnet, D.; Raimondi, F. Myocardial inflammation detected by cardiac MRI in Arrhythmogenic right ventricular cardiomyopathy: A paediatric case series. Int. J. Cardiol. 2018, 271, 81–86. [Google Scholar] [CrossRef]
- Asatryan, B.; Asimaki, A.; Landstrom, A.P.; Khanji, M.Y.; Odening, K.E.; Cooper, L.T.; Marchlinski, F.E.; Gelzer, A.R.; Semsarian, C.; Reichlin, T.; et al. Inflammation and Immune Response in Arrhythmogenic Cardiomyopathy: State-of-the-Art Review. Circulation 2021, 144, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Bariani, R.; Rigato, I.; Cipriani, A.; Bueno Marinas, M.; Celeghin, R.; Basso, C.; Corrado, D.; Pilichou, K.; Bauce, B. Myocarditis-like Episodes in Patients with Arrhythmogenic Cardiomyopathy: A Systematic Review on the So-Called Hot-Phase of the Disease. Biomolecules 2022, 12, 1324. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Raimondi, F.; Piriou, N.; Sardo Infirri, L.; Mohiddin, S.A.; Mazzanti, A.; Shenoy, C.; Cavallari, U.A.; Imazio, M.; Aquaro, G.D.; et al. Acute Myocarditis Associated with Desmosomal Gene Variants. JACC Heart Fail. 2022, 10, 714–727. [Google Scholar] [CrossRef] [PubMed]
- Guan, F.; Wolber, T.; Saguner, A.M.; Medeiros, A.; Müggler, O.; Berger, F.; Gass, M.; Molitor, N.; Ruschitzka, F.; Brunckhorst, C.; et al. A desmoplakin variant associated with isolated arrhythmogenic left ventricular cardiomyopathy with rapid monomorphic ventricular tachycardia at first presentation. HeartRhythm Case Rep. 2023, 9, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Piriou, N.; Marteau, L.; Kyndt, F.; Serfaty, J.M.; Toquet, C.; Le Gloan, L.; Warin-Fresse, K.; Guijarro, D.; Le Tourneau, T.; Conan, E.; et al. Familial screening in case of acute myocarditis reveals inherited arrhythmogenic left ventricular cardiomyopathies. ESC Heart Fail. 2020, 7, 1520–1533. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez-Jiménez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.Á.; Álvarez, M.; López-Fernández, S.; et al. Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Rezazadeh, S.; Williams, T.; Munsie, N.M.; Liedtke, D.; Oh, T.; Ferrier, R.; Shen, Y.; Jones, S.J.M.; Stiegler, A.L.; et al. Mutations in ILK, encoding integrin-linked kinase, are associated with arrhythmogenic cardiomyopathy. Transl. Res. 2019, 208, 15–29. [Google Scholar] [CrossRef]
- van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef]
- Brodehl, A.; Belke, D.D.; Garnett, L.; Martens, K.; Abdelfatah, N.; Rodriguez, M.; Diao, C.; Chen, Y.X.; Gordon, P.M.; Nygren, A.; et al. Transgenic mice overexpressing desmocollin-2 (DSC2) develop cardiomyopathy associated with myocardial inflammation and fibrotic remodeling. PLoS ONE 2017, 12, e0174019. [Google Scholar] [CrossRef]
- Lopez-Ayala, J.M.; Pastor-Quirante, F.; Gonzalez-Carrillo, J.; Lopez-Cuenca, D.; Sanchez-Munoz, J.J.; Oliva-Sandoval, M.J.; Gimeno, J.R. Genetics of myocarditis in arrhythmogenic right ventricular dysplasia. Heart Rhythm 2015, 12, 766–773. [Google Scholar] [CrossRef]
- Perazzolo Marra, M.; Rizzo, S.; Bauce, B.; De Lazzari, M.; Pilichou, K.; Corrado, D.; Thiene, G.; Iliceto, S.; Basso, C. Arrhythmogenic right ventricular cardiomyopathy. Contribution of cardiac magnetic resonance imaging to the diagnosis. Herz 2015, 40, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Murray, B.; Tichnell, C.; Gilotra, N.A.; Zimmerman, S.L.; Gasperetti, A.; Scheel, P.; Tandri, H.; Calkins, H.; James, C.A. Clinical characteristics and risk stratification of desmoplakin cardiomyopathy. Europace 2022, 24, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Protonotarios, A.; Wicks, E.; Ashworth, M.; Stephenson, E.; Guttmann, O.; Savvatis, K.; Sekhri, N.; Mohiddin, S.A.; Syrris, P.; Menezes, L.; et al. Prevalence of 18F-fluorodeoxyglucose positron emission tomography abnormalities in patients with arrhythmogenic right ventricular cardiomyopathy. Int. J. Cardiol. 2019, 284, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Divakaran, S.; Stewart, G.C.; Lakdawala, N.K.; Padera, R.F.; Zhou, W.; Desai, A.S.; Givertz, M.M.; Mehra, M.R.; Kwong, R.Y.; Hedgire, S.S.; et al. Diagnostic Accuracy of Advanced Imaging in Cardiac Sarcoidosis. Circ. Cardiovasc. Imaging 2019, 12, e008975. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Buono, A.; Moroni, F.; Gigli, L.; Power, J.R.; Ciabatti, M.; Garascia, A.; Adler, E.D.; Pieroni, M. State-of-the-Art of Endomyocardial Biopsy on Acute Myocarditis and Chronic Inflammatory Cardiomyopathy. Curr. Cardiol. Rep. 2022, 24, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Kociol, R.D.; Cooper, L.T.; Fang, J.C.; Moslehi, J.J.; Pang, P.S.; Sabe, M.A.; Shah, R.V.; Sims, D.B.; Thiene, G.; Vardeny, O.; et al. Recognition and Initial Management of Fulminant Myocarditis: A Scientific Statement from the American Heart Association. Circulation 2020, 141, e69–e92. [Google Scholar] [CrossRef]
- Bennett, M.K.; Gilotra, N.A.; Harrington, C.; Rao, S.; Dunn, J.M.; Freitag, T.B.; Halushka, M.K.; Russell, S.D. Evaluation of the role of endomyocardial biopsy in 851 patients with unexplained heart failure from 2000–2009. Circ. Heart Fail. 2013, 6, 676–684. [Google Scholar] [CrossRef]
- Casella, M.; Dello Russo, A.; Bergonti, M.; Catto, V.; Conte, E.; Sommariva, E.; Gasperetti, A.; Vettor, G.; Tundo, F.; Sicuso, R.; et al. Diagnostic Yield of Electroanatomic Voltage Mapping in Guiding Endomyocardial Biopsies. Circulation 2020, 142, 1249–1260. [Google Scholar] [CrossRef]
- Terasaki, F.; Azuma, A.; Anzai, T.; Ishizaka, N.; Ishida, Y.; Isobe, M.; Inomata, T.; Ishibashi-Ueda, H.; Eishi, Y.; Kitakaze, M.; et al. JCS 2016 Guideline on diagnosis and treatment of cardiac sarcoidosis―Digest version―. Circ. J. 2019, 83, 2329–2388. [Google Scholar] [CrossRef]
- Birnie, D.H.; Sauer, W.H.; Bogun, F.; Cooper, J.M.; Culver, D.A.; Du-vernoy, C.S.; Judson, M.A.; Kron, J.; Mehta, D.; Cosedis Nielsen, J.; et al. HRS expert consensus statement on the diagnosis and management of arrhythmias associated with cardiac sarcoidosis. Heart Rhythm. 2014, 11, 1304–1323. [Google Scholar] [CrossRef]
- Groh, M.; Masciocco, G.; Kirchner, E.; Kristen, A.; Pellegrini, C.; Varnous, S.; Bortman, G.; Rosenberg, M.; Brucato, A.; Waterworth, P.; et al. Heart transplantation in patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome). J. Heart Lung Transplant. 2014, 33, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Mueller, K.A.; Mueller, I.I.; Eppler, D.; Zuern, C.S.; Seizer, P.; Kramer, U.; Koetter, I.; Roecken, M.; Kandolf, R.; Gawaz, M.; et al. Clinical and histopathological features of patients with systemic sclerosis undergoing endomyocardial biopsy. PLoS ONE 2015, 10, e0126707. [Google Scholar] [CrossRef] [PubMed]
- Krueger, G.R.; Ablashi, D.V. Human herpesvirus-6: A short review of its biological behavior. Intervirology 2003, 46, 257–269. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2022, 24, 4–131. [Google Scholar] [CrossRef] [PubMed]
- McNamara, D.M.; Holubkov, R.; Starling, R.C.; Dec, G.W.; Loh, E.; Torre-Amione, G.; Gass, A.; Janosko, K.; Tokarczyk, T.; Kessler, P.; et al. Controlled trial of intravenous immune globulin in recent-onset dilated cardiomyopathy. Circulation 2001, 103, 2254–2259. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.W.; O’Connell, J.B.; Herskowitz, A.; Rose, N.R.; McManus, B.M.; Billingham, M.E.; Moon, T.E. A clinical trial of immunosuppressive therapy for myocarditis. The Myocarditis Treatment Trial Investigators. N. Engl. J. Med. 1995, 333, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; Wang, W.; Wu, S.N.; Liu, J.P. Corticosteroids for viral myocarditis. Cochrane Database Syst. Rev. 2013, 10, CD004471. [Google Scholar] [CrossRef]
- Wojnicz, R.; Nowalany-Kozielska, E.; Wojciechowska, C.; Glanowska, G.; Wilczewski, P.; Niklewski, T.; Zembala, M.; Polonski, L.; Rozek, M.M.; Wodniecki, J. Randomized, placebo-controlled study for immunosuppressive treatment of inflammatory dilated cardiomyopathy: Two-year follow-up results. Circulation 2001, 104, 39–45. [Google Scholar] [CrossRef]
- Cooper, L.T., Jr.; Hare, J.M.; Tazelaar, H.D.; Edwards, W.D.; Starling, R.C.; Deng, M.C.; Menon, S.; Mullen, G.M.; Jaski, B.; Bailey, K.R.; et al. Usefulness of immunosuppression for giant cell myocarditis. Am. J. Cardiol. 2008, 102, 1535–1539. [Google Scholar] [CrossRef]
- Frustaci, A.; Russo, M.A.; Chimenti, C. Randomized study on the efficacy of immunosuppressive therapy in patients with virus-negative inflammatory cardiomyopathy: The TIMIC study. Eur. Heart J. 2009, 30, 1995–2002. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Clinical Presentation | ECG | Echocardiography | CMR | Characteristic Systemic Manifestation for Differential Diagnosis |
|---|---|---|---|---|---|
| Sarcoidosis | Advanced AVB VA and SCD HF SVA (AF, AFl) Pericardial effusion | PR elongation Branch block and fascicular block PVCs Fragmented QRS Infero-lateral infarct pattern ST segment and T wave abnormalities | Non-ischemic wall motion abnormality IVS thinning (chronic phase) or hypertrophy (acute phase) LV and RV systolic dysfunction | Subendocardial, mid-wall, subepicardial LGE Patchy distribution | Pulmonary involvement Uveitis Skin lesions (Lupus Pernio, erythema nodosum) Central and peripheral nervous system involvement |
| Systemic sclerosis | VA Diastolic dysfunction AM HF SVA Pulmonary Hypertension | Multiform PVCs Branch block AVB Pseudo-infarct Q | Diastolic dysfunction Pericardial effusion LV and RV systolic dysfunction Pulmonary Hypertension | Subepicardial and IVS mid-wall LGE | Skin ulcers and diffuse fibrosis Raynaud phenomenon Pulmonary fibrosis Scleroderma renal crisis |
| Systemic lupus erythematosus | Pericarditis AM, fulminant myocarditis HF CS (vasculitis, accelerated atherosclerosis) Hypertensive cardiomyopathy Valvular heart disease | PR depression ST segment and T wave abnormalities | Pericardial effusion LV Systolic dysfunction with or without ischemic wall motion abnormalities Libman-Sacks endocarditis | Subendocardial, mid-wall, subepicardial LGE | Photosensitive malar rash Arthritis and myositis Glomerulonephritis Gastroenterological involvement Neuropsychiatric lupus |
| Eosinophilic granulomatosis polyangioiitis | Eosinophilic myocarditis, fulminant myocarditis HF RCM or DCM Pericarditis, pericardial tamponade or constriction Microvascular angina | ST segment and T wave abnormalities | LV systolic disfunction RCM or DCM phenotype Peff Ventricular thrombus | Subendocardial LGE Patchy distribution | Asthma or chronic sinusitis Peripheral blood eosinophilia Gastroenterological involvement Skin hemorrhagic lesions (Palpable purpura) Peripheral neuropathy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uccello, G.; Bonacchi, G.; Rossi, V.A.; Montrasio, G.; Beltrami, M. Myocarditis and Chronic Inflammatory Cardiomyopathy, from Acute Inflammation to Chronic Inflammatory Damage: An Update on Pathophysiology and Diagnosis. J. Clin. Med. 2024, 13, 150. https://doi.org/10.3390/jcm13010150
Uccello G, Bonacchi G, Rossi VA, Montrasio G, Beltrami M. Myocarditis and Chronic Inflammatory Cardiomyopathy, from Acute Inflammation to Chronic Inflammatory Damage: An Update on Pathophysiology and Diagnosis. Journal of Clinical Medicine. 2024; 13(1):150. https://doi.org/10.3390/jcm13010150
Chicago/Turabian StyleUccello, Giuseppe, Giacomo Bonacchi, Valentina Alice Rossi, Giulia Montrasio, and Matteo Beltrami. 2024. "Myocarditis and Chronic Inflammatory Cardiomyopathy, from Acute Inflammation to Chronic Inflammatory Damage: An Update on Pathophysiology and Diagnosis" Journal of Clinical Medicine 13, no. 1: 150. https://doi.org/10.3390/jcm13010150