
Clinical Update on Congenital Adrenal Hyperplasia: Recommendations from a Multidisciplinary Adrenal Program

Abstract
:1. Introduction
2. Epidemiology
3. Pathophysiology and Classification of Congenital Adrenal Hyperplasia Due to 21-Hydrolase Deficiency
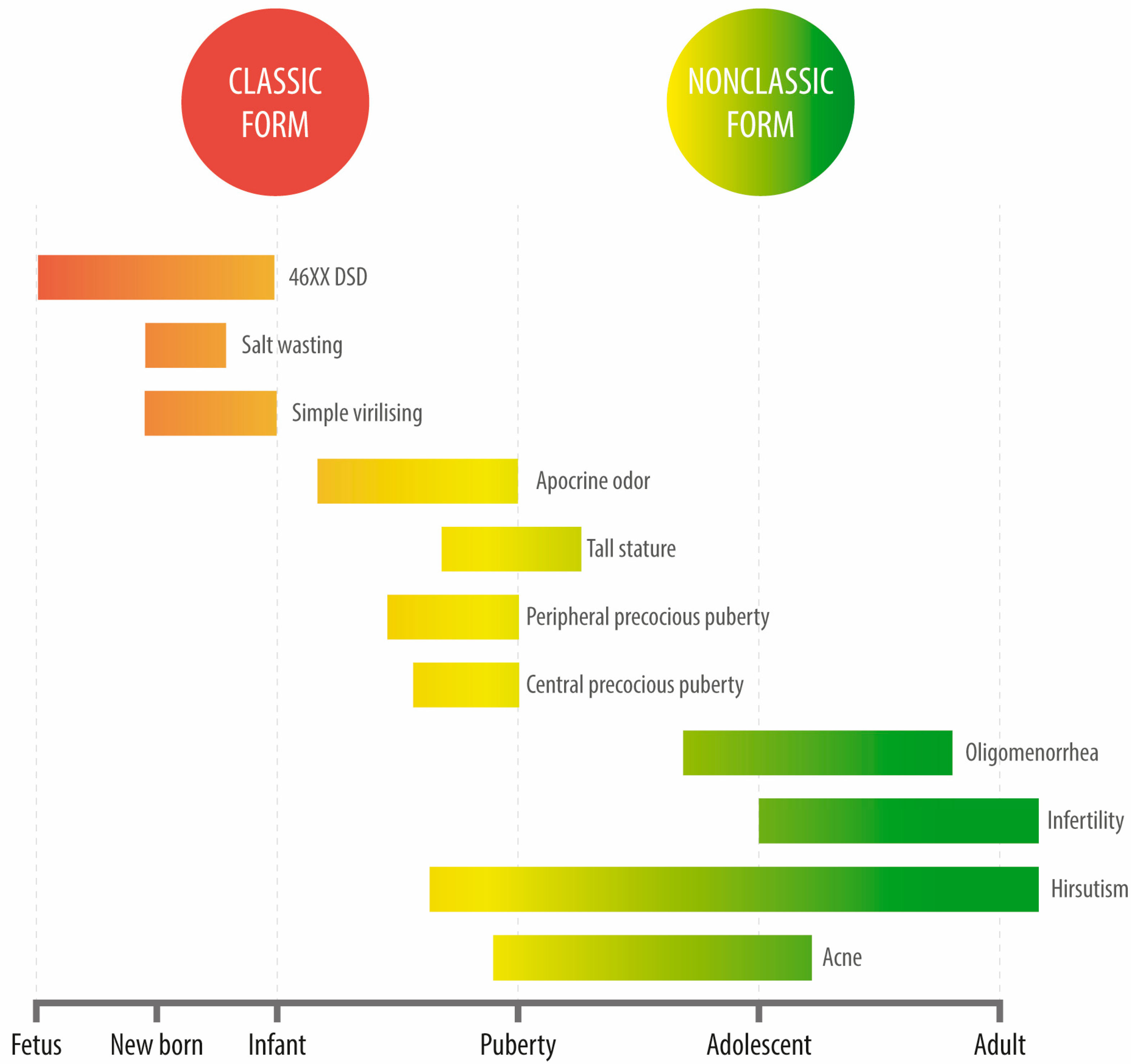
4. Clinical Features
4.1. SW Classic CAH
4.2. Simple Virilizing CAH
4.3. Non-Classical CAH
5. Initial Studies
5.1. Steroid Analyses
5.2. Genetic Aspects of Diagnosis
6. Treatment of CAH
6.1. Classic CAH
6.1.1. Treatment under Medical-Surgical Stress Conditions
6.1.2. New Drugs to Improve the Treatment of CAH Patients
6.1.3. Restoring Functional Anatomy in 46, XX Subjects Virilized by Classic CAH
6.1.4. Preventing Prenatal Virilization in At-Risk Pregnancies
6.2. NCCAH
7. Special Clinical Situations
7.1. Infertility and Pregnancy
7.1.1. Classic CAH
7.1.2. NCCAH
7.2. Genetic Counseling
7.3. Testicular Adrenal Rest Tumors (TARTs)
7.4. Behavioral Considerations in Patients with Classical CAH
8. Follow-Up
9. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Rumsby, G.; Woodward, G. Disorders of Steroidogenesis: Guide to Steroid Profiling and Biochemical Diagnosis, 1st ed.; Rumsby, G., Woodward, G., Eds.; Springer: Cham, Germany, 2019. [Google Scholar] [CrossRef]
- El-Maouche, D.; Arlt, W.; Merke, D.P. Congenital Adrenal Hyperplasia. Lancet 2017, 390, 2194–2210. [Google Scholar] [CrossRef]
- Claahsen-van der Grinten, H.L.; Speiser, P.W.; Ahmed, S.F.; Arlt, W.; Auchus, R.J.; Falhammar, H.; Flück, C.E.; Guasti, L.; Huebner, A.; Kortmann, B.B.M.; et al. Congenital Adrenal Hyperplasia-Current Insights in Pathophysiology, Diagnostics, and Management. Endocr. Rev. 2022, 43, 91–159. [Google Scholar] [CrossRef]
- Auchus, R.J. The Classic and Nonclassic Concenital Adrenal Hyperplasias. Endocr. Pract. 2015, 21, 383–389. [Google Scholar] [CrossRef]
- White, P.C. Update on Diagnosis and Management of Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 178–184. [Google Scholar] [CrossRef]
- Speiser, P.W.; Arlt, W.; Auchus, R.J.; Baskin, L.S.; Conway, G.S.; Merke, D.P.; Meyer-Bahlburg, H.F.L.; Miller, W.L.; Murad, M.H.; Oberfield, S.E.; et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2018, 103, 4043–4088. [Google Scholar] [CrossRef]
- Pignatelli, D.; Carvalho, B.L.; Palmeiro, A.; Barros, A.; Guerreiro, S.G.; Macut, D. The Complexities in Genotyping of Congenital Adrenal Hyperplasia: 21-Hydroxylase Deficiency. Front. Endocrinol. 2019, 10, 432. [Google Scholar] [CrossRef]
- Merke, D.P.; Auchus, R.J. Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. N. Engl. J. Med. 2020, 383, 1248–1261. [Google Scholar] [CrossRef]
- Nordenström, A.; Falhammar, H. Management of Endocrine Disease: Diagnosis and Management of the Patient with Non-Classic CAH Due to 21-Hydroxylase Deficiency. Eur. J. Endocrinol. 2019, 180, R127–R145. [Google Scholar] [CrossRef]
- Ueland, G.Å.; Methlie, P.; Øksnes, M.; Thordarson, H.B.; Sagen, J.; Kellmann, R.; Mellgren, G.; Ræder, M.; Dahlqvist, P.; Dahl, S.R.; et al. The Short Cosyntropin Test Revisited: New Normal Reference Range Using LC-MS/MS. J. Clin. Endocrinol. Metab. 2018, 103, 1696–1703. [Google Scholar] [CrossRef]
- Krone, N.; Arlt, W. Genetics of Congenital Adrenal Hyperplasia. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 181–192. [Google Scholar] [CrossRef]
- Krone, N.; Rose, I.T.; Willis, D.S.; Hodson, J.; Wild, S.H.; Doherty, E.J.; Hahner, S.; Parajes, S.; Stimson, R.H.; Han, T.S.; et al. Genotype-Phenotype Correlation in 153 Adult Patients with Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency: Analysis of the United Kingdom Congenital Adrenal Hyperplasia Adult Study Executive (CaHASE) Cohort. J. Clin. Endocrinol. Metab. 2013, 98, E346–E354. [Google Scholar] [CrossRef]
- Auchus, R.J.; Arlt, W. Approach to the Patient: The Adult with Congenital Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2013, 98, 2645–2655. [Google Scholar] [CrossRef]
- Auer, M.K.; Nordenström, A.; Lajic, S.; Reisch, N. Congenital Adrenal Hyperplasia. Lancet 2023, 401, 227–244. [Google Scholar] [CrossRef]
- Lajic, S.; Karlsson, L.; Zetterström, R.H.; Falhammar, H.; Nordenström, A. The Success of a Screening Program Is Largely Dependent on Close Collaboration between the Laboratory and the Clinical Follow-Up of the Patients. Int. J. Neonatal. Screen 2020, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Rivkees, S.A.; Crawford, J.D. Dexamethasone Treatment of Virilizing Congenital Adrenal Hyperplasia: The Ability to Achieve Normal Growth. Pediatrics 2000, 106, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Punthakee, Z.; Legault, L.; Polychronakos, C. Prednisolone in the Treatment of Adrenal Insufficiency: A Re-Evaluation of Relative Potency. J. Pediatr. 2003, 143, 402–405. [Google Scholar] [CrossRef] [PubMed]
- MacKay, D.; Nordenström, A.; Falhammar, H. Bilateral Adrenalectomy in Congenital Adrenal Hyperplasia: A Systematic Review and Meta-Analysis. J. Clin. Endocrinol. Metab. 2018, 103, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Hahner, S.; Ross, R.J.; Arlt, W.; Bancos, I.; Burger-Stritt, S.; Torpy, D.J.; Husebye, E.S.; Quinkler, M. Adrenal Insufficiency. Nat. Rev. Dis. Prim. 2021, 7, 19. [Google Scholar] [CrossRef]
- Sarafoglou, K.; Barnes, C.N.; Huang, M.; Imel, E.A.; Madu, I.-J.; Merke, D.P.; Moriarty, D.; Nakhle, S.; Newfield, R.S.; Vogiatzi, M.G.; et al. Tildacerfont in Adults With Classic Congenital Adrenal Hyperplasia: Results from Two Phase 2 Studies. J. Clin. Endocrinol. Metab. 2021, 106, e4666–e4679. [Google Scholar] [CrossRef]
- Mercè Fernández-Balsells, M.; Muthusamy, K.; Smushkin, G.; Lampropulos, J.F.; Elamin, M.B.; Abu Elnour, N.O.; Elamin, K.B.; Agrwal, N.; Gallegos-Orozco, J.F.; Lane, M.A.; et al. Prenatal Dexamethasone Use for the Prevention of Virilization in Pregnancies at Risk for Classical Congenital Adrenal Hyperplasia Because of 21-Hydroxylase (CYP21A2) Deficiency: A Systematic Review and Meta-Analyses. Clin. Endocrinol. 2010, 73, 436–444. [Google Scholar] [CrossRef]
- Bachelot, A.; Grouthier, V.; Courtillot, C.; Dulon, J.; Touraine, P. Management of Endocrine Disease: Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency: Update on the Management of Adult Patients and Prenatal Treatment. Eur. J. Endocrinol. 2017, 176, R167–R181. [Google Scholar] [CrossRef] [PubMed]
- Reichman, D.E.; White, P.C.; New, M.I.; Rosenwaks, Z. Fertility in Patients with Congenital Adrenal Hyperplasia. Fertil. Steril. 2014, 101, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Pavlidi, O.; Kolibianakis, E.M. Fertility Treatment in Women with Classical and Nonclassical Congenital Adrenal Hyperplasia. In Fertility and Reproductive Outcomes in Different Forms of Congenital Adrenal Hyperplasia; Ertorer, M., Ed.; Springer: Cham, Germany, 2021; pp. 115–125. [Google Scholar] [CrossRef]
- Engels, M.; Span, P.N.; van Herwaarden, A.E.; Sweep, F.C.G.J.; Stikkelbroeck, N.M.M.L.; Claahsen-van der Grinten, H.L. Testicular Adrenal Rest Tumors: Current Insights on Prevalence, Characteristics, Origin, and Treatment. Endocr. Rev. 2019, 40, 973–987. [Google Scholar] [CrossRef]
- Engberg, H.; Möller, A.; Hagenfeldt, K.; Nordenskjöld, A.; Frisén, L. Identity, Sexuality, and Parenthood in Women with Congenital Adrenal Hyperplasia. J. Pediatr. Adolesc. Gynecol. 2020, 33, 470–476. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uslar, T.; Olmos, R.; Martínez-Aguayo, A.; Baudrand, R. Clinical Update on Congenital Adrenal Hyperplasia: Recommendations from a Multidisciplinary Adrenal Program. J. Clin. Med. 2023, 12, 3128. https://doi.org/10.3390/jcm12093128
Uslar T, Olmos R, Martínez-Aguayo A, Baudrand R. Clinical Update on Congenital Adrenal Hyperplasia: Recommendations from a Multidisciplinary Adrenal Program. Journal of Clinical Medicine. 2023; 12(9):3128. https://doi.org/10.3390/jcm12093128
Chicago/Turabian StyleUslar, Thomas, Roberto Olmos, Alejandro Martínez-Aguayo, and René Baudrand. 2023. "Clinical Update on Congenital Adrenal Hyperplasia: Recommendations from a Multidisciplinary Adrenal Program" Journal of Clinical Medicine 12, no. 9: 3128. https://doi.org/10.3390/jcm12093128